

Abstract
An exposure of the yeast Saccharomyces cerevisiae to exogenous palmitoleic acid (POA) elicits “liponecrosis," a mode of programmed cell death (PCD) which differs from the currently known PCD subroutines. Here, we report the following mechanism for liponecrotic PCD. Exogenously added POA is incorporated into POA-containing phospholipids that then amass in the endoplasmic reticulum membrane, mitochondrial membranes and the plasma membrane. The buildup of the POA-containing phospholipids in the plasma membrane reduces the level of phosphatidylethanolamine in its extracellular leaflet, thereby increasing plasma membrane permeability for small molecules and committing yeast to liponecrotic PCD. The excessive accumulation of POA-containing phospholipids in mitochondrial membranes impairs mitochondrial functionality and causes the excessive production of reactive oxygen species in mitochondria. The resulting rise in cellular reactive oxygen species above a critical level contributes to the commitment of yeast to liponecrotic PCD by: (1) oxidatively damaging numerous cellular organelles, thereby triggering their massive macroautophagic degradation; and (2) oxidatively damaging various cellular proteins, thus impairing cellular proteostasis. Several cellular processes in yeast exposed to POA can protect cells from liponecrosis. They include: (1) POA oxidation in peroxisomes, which reduces the flow of POA into phospholipid synthesis pathways; (2) POA incorporation into neutral lipids, which prevents the excessive accumulation of POA-containing phospholipids in cellular membranes; (3) mitophagy, a selective macroautophagic degradation of dysfunctional mitochondria, which sustains a population of functional mitochondria needed for POA incorporation into neutral lipids; and (4) a degradation of damaged, dysfunctional and aggregated cytosolic proteins, which enables the maintenance of cellular proteostasis.
Keywords: apoptosis; autophagy; cellular proteostasis; lipid metabolism in cellular organelles; mitophagy; mitochondria,; plasma membrane; mechanisms of programmed cell death; signal transduction; yeast
Abbreviations
- CFU
colony forming units
- CL
cardiolipin
- Cvt
cytoplasm-to-vacuole pathway
- ER
endoplasmic reticulum
- IMM
inner mitochondrial membrane
- LD
lipid droplets
- NL
neutral lipids
- PA
phosphatidic acid
- PC
phosphatidylcholine
- PCD
programmed cell death
- PE
phosphatidylethanolamine
- PI
phosphatidylinositol
- PL
phospholipids
- PM
plasma membrane
- POA
palmitoleic acid
- PS
phosphatidylserine
- ROS
reactive oxygen species
- TAG
triacylglycerols
- WT
wild-type
Introduction
We recently identified a form of programmed cell death (PCD) which differs from such presently known PCD subroutines as apoptosis, autophagic cell death and necrosis. We named this previously unknown PCD subroutine “liponecrosis”; it can be instigated by a short-term exposure of the yeast Saccharomyces cerevisiae to exogenous palmitoleic acid (POA).1 We demonstrated that the liponecrotic mode of cell death exhibits all 3 key features that have been recommended as mandatory for a cell death mode to be called “PCD”.2-4 In fact, we found that liponecrotic PCD: (1) is a genetically programmed, regulated cellular process which can be accelerated or decelerated by genetic manipulations impairing functionality of only certain proteins - in particular proteins enabling the maintenance of functional mitochondria, metabolism of certain lipid classes or autophagic degradation of some cellular constituents; (2) operates as a series of consecutive cellular events triggered by POA and following each other in a certain order; and (3) is likely to provide some benefits for the survival of a population of yeast cells because it represents an age-related mode of PCD - i.e., the degree to which it reduces cell viability following an exposure to POA appears to progress with the chronological age of a yeast cell.1 Importantly, yeast cells committed to liponecrotic PCD do not exhibit a combination of morphological traits and biochemical features characteristic of cells committed to any of the presently known apoptotic, autophagic or necrotic subroutines of PCD.1 Moreover, yeast cells committed to liponecrotic PCD display an excessive accumulation of lipid droplets where non-esterified fatty acids are deposited in the form of neutral lipids; this hallmark feature of liponecrotic PCD has not been reported for any of the presently known PCD modalities.1 Collectively, these findings imply that liponecrosis in yeast is a previously unknown mode of PCD. In this study, we investigated a mechanism underlying liponecrotic PCD subroutine.
Results
The extent of POA incorporation into neutral lipids and phospholipids correlates with the susceptibility of yeast to liponecrotic PCD
We recently analyzed how the fox1Δ, dga1Δ and are2Δ mutations, which impair metabolism of some lipid classes, influence the susceptibility of yeast to POA-induced liponecrosis.1 Our analysis suggested the following hypothesis: (1) exogenous POA can be incorporated into phospholipids (PL) that accumulate within cellular membranes and into neutral lipids (NL) that amass in lipid droplets (LD); (2) an excessive accumulation of POA-containing PL within cellular membranes is a pro-death process which triggers liponecrosis by creating the extreme cellular stress; (3) POA incorporation into NL is a pro-survival process which prevents the buildup of POA-containing PL within cellular membranes by reducing the flow of POA into PL synthesis; and (4) mitophagy, an Atg32p-driven selective autophagic degradation of dysfunctional mitochondria,5,6 is a pro-survival process which sustains a population of functional mitochondria needed for POA incorporation into NL deposited within LD.1
As a first step toward verifying this hypothesis, we compared lipidomes of wild-type (WT) and mitophagy-deficient atg32Δ cells exposed to various concentrations of POA. We found that an exposure of WT cells to POA used at a low final concentration of 0.05 mM causes: (1) an increase in the cellular levels of triacylglycerols (TAG), a major class of NL that are synthesized in the endoplasmic reticulum (ER) and then deposited within LD; and (2) a decrease in the cellular levels of PL (Fig. S1). Furthermore, an exposure of WT cells to POA used at a higher final concentration of 0.1 mM or 0.15 mM reduced the extent to which such exposure increased the cellular levels of TAG and elevated the extent to which it decreased the cellular levels of PL (Fig. S1). Moreover, after an exposure of WT cells to POA, the major portion of molecular species of TAG and PL undergoing such changes were POA-containing C16:1 species (compare Figs. 1 and S1). The above trend of the concentration-dependent effect of exogenous POA on the cellular levels of PL (including their most abundant POA-containing species) was observed for all major classes of PL, such as phosphatidic acid (PA), phosphatidylserine (PS), phosphatidylethanolamine (PE), phosphatidylcholine (PC), phosphatidylinositol (PtdIns) and cardiolipin (CL) (Fig. 2; Fig. S2). Some of these PL classes (such as PA, PS, PC and PI) are known to be synthesized exclusively in the ER and to be then transported to mitochondria via mitochondria-ER junctions and to the plasma membrane (PM) via PM-ER junctions (Fig. S3).7-15 Other PL classes (such as PE and CL) are known to be formed only in the inner mitochondrial membrane (IMM); PE is then transported from mitochondria to the ER via mitochondria-ER junctions and from the ER to the PM via PM-ER junctions (Fig. S3).7-15
Figure 1.
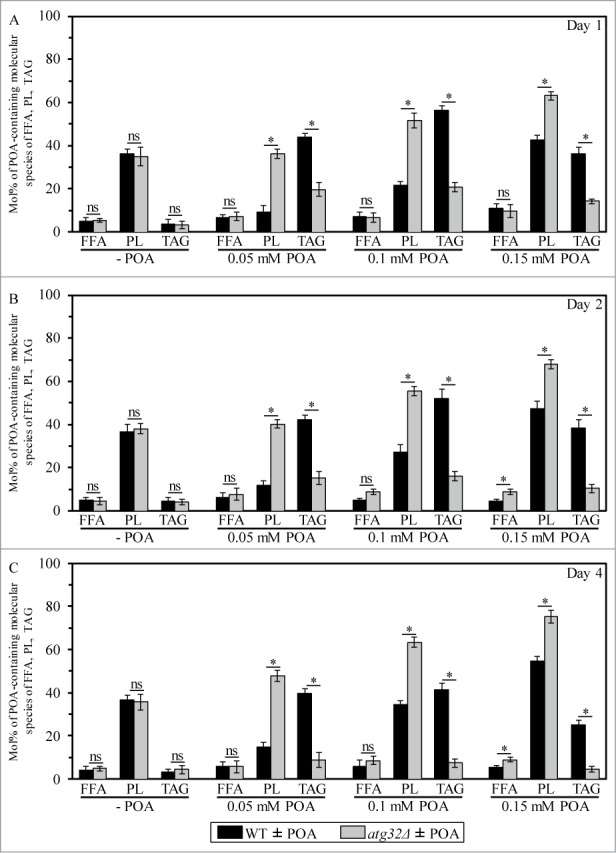
An exposure of WT cells to various concentrations of exogenous POA elicits differential effects on the relative levels of C16:1 molecular species (i.e., POA-containing species) of PL and TAG, and the atg32Δ-dependent mutational block of mitophagy alters these effects. WT and atg32Δ cells were recovered at days 1, 2 and 4 of culturing in YP medium initially containing 0.2% glucose as carbon source. Extraction of cellular lipids, and mass spectrometric identification and quantitation of various molecular species of non-esterified (“free”) fatty acids (FFA), phospholipids (PL) and triacylglycerols (TAG) were carried out as described in Materials and Methods. The relative level of POA-containing molecular species for each lipid class (i.e., FFA, PL and TAG) was calculated as mol% of all these lipid classes. Data are presented as means ± SEM (n = 3; *P < 0.01; ns, not significant). For FFA, POA-containing molecular species are their C16:1 species. For the PA, PS, PE, PC and PI classes of PL, POA-containing molecular species are the C32:1, C32:2, C34:1 and C34:2 species of each of them. For the CL class of PL, POA-containing molecular species are its C64:4, C66:4, C68:4, C70:4 species. For TAG, POA-containing molecular species are their C48:2, C48:3, C50:2, C50:3, C52:2, C52:3 species.
Figure 2.
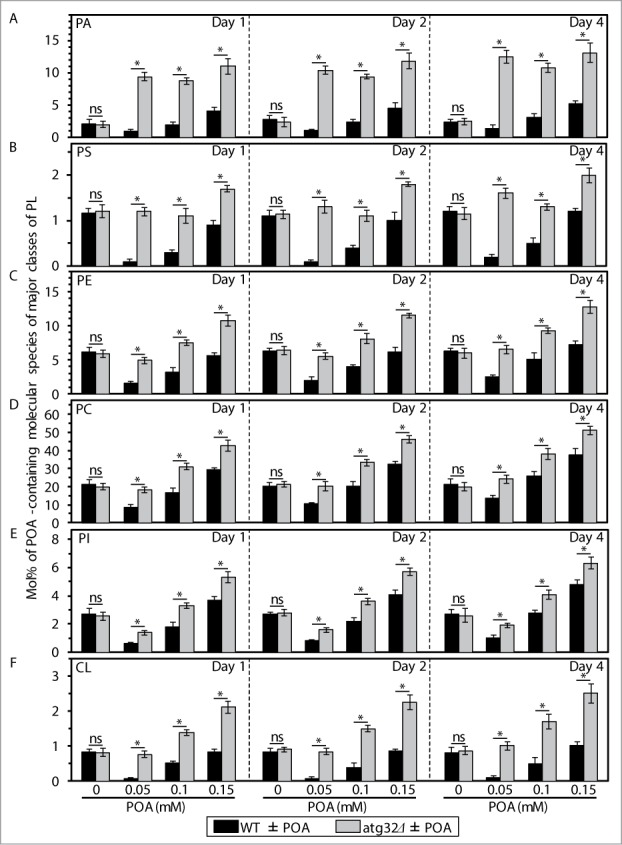
An exposure of WT cells to various concentrations of exogenous POA elicits differential effects on the relative levels of C16:1 molecular species (i.e., POA-containing species) of all major classes of PL, and the atg32Δ-dependent mutational block of mitophagy alters these effects. WT and atg32Δ cells were recovered at days 1, 2 and 4 of culturing in YP medium initially containing 0.2% glucose as carbon source. Extraction of cellular lipids, and mass spectrometric identification and quantitation of various molecular species of phospholipids (PL) were carried out as described in Materials and Methods. The relative level of POA-containing molecular species for each class of PL (i.e., PA, PS, PE, PC, PtdIns and CL) was calculated as mol% of all these PL classes. Data are presented as means ± SEM (n = 3; *P < 0.01; ns, not significant). For the PA, PS, PE, PC and PI classes of PL, POA-containing molecular species are the C32:1, C32:2, C34:1 and C34:2 species of each of them. For the CL class of PL, POA-containing molecular species are its C64:4, C66:4, C68:4, C70:4 species.
Our mass spectrometric analysis of yeast exposed to exogenous POA also revealed that the atg32Δ-dependent mutational block of mitophagy: (1) substantially reduces the extent to which such exposure increases the cellular levels of TAG (including their POA-containing species) (Fig. 1; Fig. S1); and (2) significantly elevates the degree to which such exposure increases the cellular levels of various classes of PL (including their POA-containing species) (Fig. 2; Fig. S2). As we recently reported, the atg32Δ mutation substantially increases the susceptibility of yeast to POA-induced liponecrotic PCD.1
To further verify our hypothesis, we examined how various single-gene-deletion mutations (each eliminating an enzyme involved in POA transport to the ER or in POA incorporation into PL and/or NL within the ER; see refs.7-18 and Fig. S3) influence the susceptibility of yeast to POA-induced liponecrotic PCD. Importantly, all these enzymes are redundant and none of them is an essential protein in yeast.9,16 Thus, although none of these single-gene-deletion mutations completely abolishes the incorporation of POA into PL and/or NL, each of them causes significant changes in the cellular levels of PL and/or NL.9,16 We found that the fat1Δ mutation reduces the susceptibility of yeast to POA-induced liponecrosis (Fig. 3A); this mutation decreases cellular uptake of POA and, thus, mitigates the incorporation of POA into both PL and NL (Fig. S3).18 Furthermore, the gpt2Δ, ayr1Δ and ale1Δ mutations make yeast less susceptible to POA-induced liponecrosis (Figs. 3B–D); all these mutations attenuate POA incorporation into PL (Fig. S3).9,16 Moreover, the dga1Δ and lro1Δ mutations increase the susceptibility of yeast to POA-induced liponecrosis (Figs. 3E and F); both these mutations decrease the incorporation of POA into TAG, a major class of NL (Fig. S3).9,16 In addition, we recently reported that the are2Δ mutation makes yeast more susceptible to POA-induced liponecrotic PCD;1 this mutation is known to mitigate POA incorporation into ergosteryl esters, a class of NL.9,16
Figure 3.
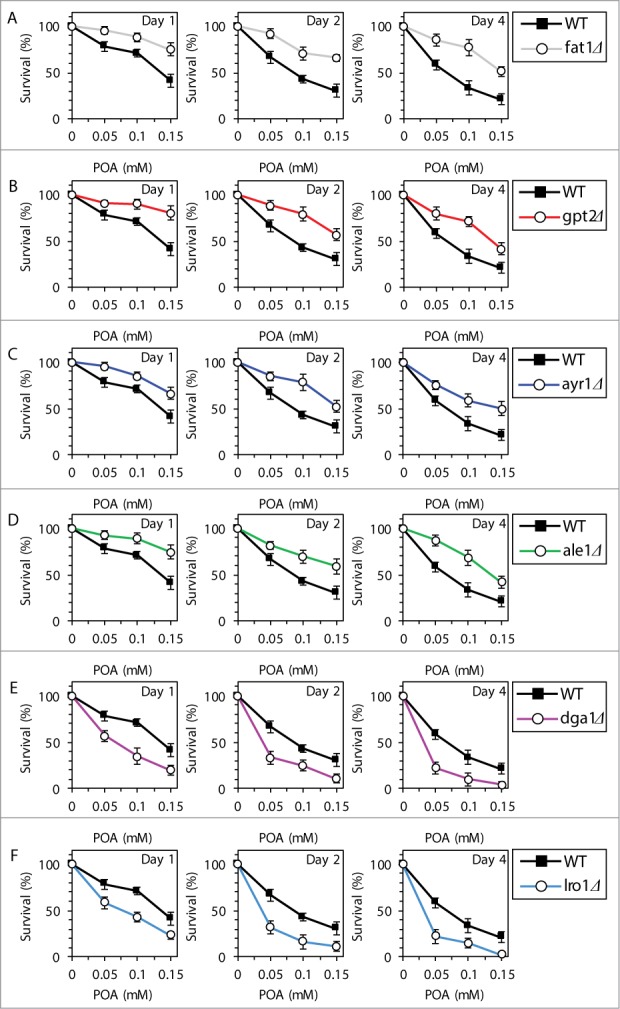
Various single-gene-deletion mutations (each eliminating an enzyme involved in POA transport to the ER or in POA incorporation into PL and/or NL within the ER) elicit differential effects on the susceptibility of yeast to POA-induced liponecrotic PCD. WT and mutant cells were recovered at days 1, 2 and 4 of culturing in YP medium initially containing 0.2% glucose as carbon source. Cell survival was assessed by measuring the clonogenicity of cells after 2 h of treatment with various concentrations of exogenous POA. Data for the effect of the dga1Δ mutation on the susceptibility of yeast to POA-induced liponecrosis are from a different series of experiments than the ones reported in our recent publication [1]. Data are presented as means ± SEM (n = 12–19).
In sum, the data of the lipidomic analysis and cell survival assays of various mutant strains revealed that the concentration of exogenous POA added to yeast cells: (1) is inversely proportional to the levels of TAG (including their POA-containing species) in these cells (Figs. 1; Fig. S1); (2) positively correlates with the levels of all major classes of PL (including their POA-containing species) in these cells (Figs. 2; Fig. S2); and (3) is in a positive correlation with the susceptibility of these cells to liponecrosis (Fig. 3).1
The alkaline-pH- and lipid-asymmetry-responsive Rim101 signaling pathway may commit yeast to liponecrotic PCD by reducing the level of PE in the extracellular leaflet of the PM
We found that WT cells exposed to various concentrations of exogenous POA cause medium alkalinisation in a POA concentration-dependent manner (data not shown). To examine if such medium alkalinisation is essential for the commitment of yeast to POA-induced liponecrosis, we compared the susceptibility of WT cells to this PCD mode in buffered and unbuffered culture media. The pH of buffered media supplemented with various concentrations of POA was maintained at approximately 6.5, close to the initial pH of unbuffered media containing the same POA concentrations (data not shown). We revealed that an exposure of WT yeast to POA commits these cells to liponecrotic PCD as efficiently in buffered media as it does in unbuffered media (data not shown). Thus, the POA-induced alkalinisation of culture medium by yeast cells is not essential for the ability of POA to trigger liponecrosis. Our unpublished data validate this conclusion; as we found, a hypomorphic allele of the gene encoding the PM ATPase Pma1p,19 a single-gene-deletion mutation eliminating the PM Na+ (K+)/H+ antiporter Nha1p,20 and a single-gene-deletion mutation removing the PM K+ transporter Trk1p21 exhibit considerable differential effects on the extent of culture medium alkalinisation by cells exposed to POA but do not alter the susceptibility of yeast to POA-induced liponecrosis (Feldman et al., in preparation).
Alkalinisation of culture medium can induce the alkaline-pH-responsive Rim101 signaling pathway in yeast.22,23 As we found, the POA-induced alkalinisation of medium by yeast does not elicit liponecrotic PCD despite the demonstrated ability of such alkalinisation to trigger this signaling pathway. The Rim101 pathway can also be induced in response to changes in the asymmetrical distribution of various PL species across the PM bilayer.22,23 It needs to be pointed out that: (1) an active maintenance of the proper PL asymmetry in the PM bilayer depends on the abundance and composition of different PL species in this bilayer;24-26 (2) POA-containing species of the PA, PS, PE, PC and PtdIns classes of PL undergo excessive accumulation within cellular membranes of yeast committed to POA-induced liponecrosis (Fig. 2); (3) all these PL classes are known to be delivered to the PM bilayer via PM-ER junctions after being synthesized in the ER or mitochondria (Fig. S3).7-18 It is conceivable therefore that: (1) POA-containing species of PA, PS, PE, PC and PI could amass within the PM bilayer of yeast treated with POA; and (2) such buildup of POA-containing species of PL within the PM could alter the proper PL asymmetry in the PM bilayer and, thus, could induce the alkaline-pH- and lipid-asymmetry-responsive Rim101 signaling pathway.
Upon stimulation of the Rim101 pathway, endocytic internalization of its pH sensing protein complex in the PM initiates a series of events leading to a proteolytic processing of the transcriptional factor Rim101p by the protease Rim13p on the surface of the endosome (Fig. 4A).23,27-33 Processed Rim101p is delivered to the nucleus, where it activates transcription of the RSB1 gene (Fig. 4A).23,31,34 A protein product of the RSB1 gene is a transporter within the PM.22,35 Rsb1p is known to: (1) stimulate the Lem3p-dependent transport of PE from the extracellular (outer) leaflet of the PM to its intracellular (inner) leaflet; and (2) suppress the Yor1p-dependent transport of PE in the opposite direction (Fig. 4A).22,35-37 Thus, an activation of the Rim101 pathway is likely to reduce the level of PE in the extracellular leaflet of the PM (Fig. 4A).22,23,31,34-37 We therefore sought to investigate if an exposure of yeast to POA alters the relative levels of PE in the 2 leaflets of the PM. To attain this objective, we used the tetracyclic peptide antibiotic cinnamycin whose specific binding to PE in the extracellular leaflet of the PM is known to cause cell lysis;36,38 hence, the level of PE in this leaflet of the PM bilayer positively correlates with the sensitivity of yeast to cinnamycin.22 We found that an exposure of WT cells to POA decreases the sensitivity of yeast to cinnamycin (Fig. 4B) and, thus, reduces the level of PE in the extracellular leaflet of the PM. Moreover, the yor1Δ mutation eliminating a protein required for PE transport from the intracellular leaflet of the PM to its extracellular leaflet22,37 further decreased the sensitivity of pre-treated with POA cells to cinnamycin (Fig. 4B). In contrast, the lem3Δ mutation eliminating a protein required for PE transport from the extracellular leaflet of the PM to its intracellular leaflet22,36 increased the sensitivity of pre-treated with POA cells to cinnamycin (Fig. 4B). In sum, these findings suggest that a depletion of PE in the extracellular leaflet of the PM, likely due to its enhanced translocation to the intracellular leaflet of the PM, is a hallmark event of POA-induced liponecrosis. It is conceivable that this characteristic trait of liponecrotic PCD is due to the ability of POA to trigger the Rim101 pathway, thereby increasing the level of Rsb1p in the PM. The increased abundance of Rsb1p may: (1) enhance its stimulating effect on the Lem3p-dependent transport of PE from the extracellular leaflet to the intracellular leaflet of the PM; and (2) amplify its inhibitory effect on the Yor1p-dependent transport of PE within the PM bilayer in the opposite direction. Of note, there could be other (i.e., Rsb1p-, Lem3p- and/or Yor1p-independent) ways of reducing the level of PE in the extracellular leaflet of the PM in yeast.
Figure 4.
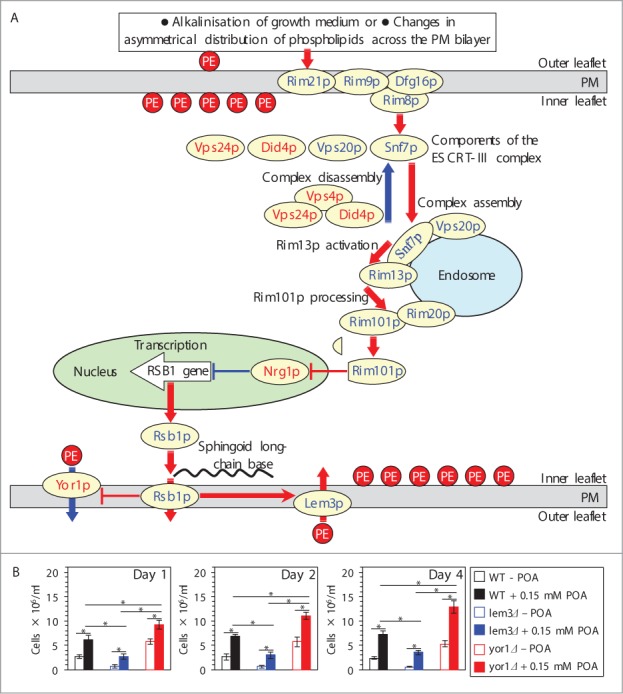
A depletion of PE in the extracellular leaflet of the PM is a characteristic trait of POA-induced liponecrotic PCD, perhaps due to the ability of POA to trigger the alkaline-pH- and lipid-asymmetry-responsive Rim101 signaling pathway. (A) Outline of the Rim101 signaling pathway operating in yeast cells. This pathway can be induced by alkalinisation of culture medium and/or in response to certain changes in asymmetrical distribution of some species of PL across the PM bilayer. The pathway includes the pH sensing protein complex on the PM and the proteolytic processing complex for the transcriptional factor Rim101p on the endosomal membrane. After Rim101p is proteolytically processed by the protease Rim13p on the surface of the endosome, its cleaved form is delivered to the nucleus. In the nucleus, cleaved Rim101p activates transcription of the RSB1 gene by suppressing its repressor, Nrg1p. A protein product of the RSB1 gene is a putative sphingoid long-chain base-specific translocase/transporter within the PM. This protein stimulates the Lem3p-dependent transport of PE from the outer (extracellular) leaflet of the PM to its inner (intracellular) leaflet and suppresses the Yor1p-dependent transport of PE across the PM bilayer in the opposite direction. See text for additional details. The names of proteins whose lack causes a decrease or increase of the susceptibility of yeast to POA-induced liponecrosis are displayed in blue or red color, respectively. Activation arrows and inhibition bars denote pro-death processes (displayed in red color) or pro-survival processes (displayed in blue color) for liponecrotic PCD. (B) WT, lem3Δ and yor1Δ cells were recovered at days 1, 2 and 4 of culturing in YP medium initially containing 0.2% glucose as carbon source. Each cell suspension was divided into 2 equal aliquots. Cells in one of these aliquots were treated for 2 h with 0.15 mM POA; cells in the other aliquot remained untreated. Cells in each aliquot were then diluted in YP medium containing 2% glucose and supplemented with 25 μM cinnamycin. Both aliquots were incubated for 8 h, and the total number of cells in each aliquot was determined using a hemacytometer. Data are presented as means ± SEM (n = 5–6). Abbreviations: ESCRT-III, endosomal sorting complex (type III) required for transport; PE, phosphatidylethanolamine; PL, phospholipids; PM, plasma membrane; POA, palmitoleic acid; PS, phosphatidylserine; TAG, triacylglycerols.
Based on the above findings, we hypothesized that the Rim101 signaling pathway operates as a pro-death process which may commit yeast to POA-induced liponecrotic PCD. To verify this hypothesis, we examined how single-gene-deletion mutations eliminating different protein components of this pathway influence the susceptibility of yeast to POA-induced liponecrosis. We found that the rim21Δ, rim9Δ, dfg16Δ, rim8Δ, snf7Δ, vps20Δ, rim13Δ, rim20Δ and rim101Δ mutations reduce the susceptibility of yeast to liponecrotic PCD (Figs. 5A–I). All these mutations are known to impair the ability of the transcriptional factor Rim101p to activate transcription of the RSB1 gene (Fig. 4A).23,31,34 Thus, all of them are expected to diminish the stimulating effect of Rsb1p on the Lem3p-dependent transport of PE from the extracellular leaflet of the PM to its intracellular leaflet (Fig. 4A).22,36,37 Moreover, all these mutations are also anticipated to lessen the inhibitory effect of Rsb1p on the Yor1p-dependent transport of PE across the PM bilayer in the opposite direction (Fig. 4A).22,36,37 In sum, the rim21Δ, rim9Δ, dfg16Δ, rim8Δ, snf7Δ, vps20Δ, rim13Δ, rim20Δ and rim101Δ mutations reducing the susceptibility of yeast to POA-induced liponecrosis are likely to decrease the extent of PE depletion in the extracellular leaflet of the PM, a hallmark event of this PCD subroutine. Furthermore, the rsb1Δ and lem3Δ mutations made yeast less susceptible to POA-induced liponecrosis (Figs. 5J and K). Because both these mutations are known to diminish the stimulating effect of Rsb1p on the Lem3p-dependent transport of PE from the extracellular leaflet of the PM to its intracellular leaflet (Fig. 4A),22,36,37 they are expected to lessen the degree of PE depletion in the extracellular leaflet of the PM. Moreover, we found that the vps4Δ, vps24Δ and did4Δ mutations increase the susceptibility of yeast to POA-induced liponecrosis (Figs. 6A–C). Because all these mutations are known to enhance the ability of Rim101p to activate transcription of the RSB1 gene (Fig. 4A),28,31 they are likely to increase the extent of PE depletion in the extracellular leaflet of the PM. In addition, we found that the nrg1Δ mutation eliminating a transcriptional repressor of the RSB1 gene (Fig. 4A)23,31,34 and the yor1Δ mutation impairing the transport of PE from the intracellular leaflet of the PM to its extracellular leaflet (Fig. 4A)22,36,37 make yeast more susceptible to POA-induced liponecrosis (Figs. 6D and E). Because the nrg1Δ mutation is known to attenuate the Yor1p-dependent transport of PE from the intracellular leaflet of the PM to its extracellular leaflet and the yor1Δ mutation has been shown to abolish it (Fig. 4A),22,36,37 both mutations are expected to increase the extent of PE depletion in the extracellular leaflet of the PM. Altogether, these data support the hypothesis that the Rim101 signaling pathway operates as a pro-death process which may commit yeast to liponecrosis by reducing the level of PE in the extracellular leaflet of the PM.
Figure 5.

Single-gene-deletion mutations eliminating different protein components of the Rim101 signaling pathway reduce the susceptibility of yeast to POA-induced liponecrotic PCD if they decrease the extent of PE depletion in the extracellular leaflet of the PM, a hallmark event of this PCD. WT and mutant cells were recovered at days 1, 2 and 4 of culturing in YP medium initially containing 0.2% glucose as carbon source. Cell survival was assessed by measuring the clonogenicity of cells after 2 h of treatment with various concentrations of exogenous POA. Data are presented as means ± SEM (n = 8–10).
Figure 6.

Single-gene-deletion mutations eliminating different protein components of the Rim101 signaling pathway make yeast more susceptible to POA-induced liponecrotic PCD if they increase the degree of PE depletion in the extracellular leaflet of the PM, a hallmark event of this PCD. WT and mutant cells were recovered at days 1, 2 and 4 of culturing in YP medium initially containing 0.2% glucose as carbon source. Cell survival was assessed by measuring the clonogenicity of cells after 2 h of treatment with various concentrations of exogenous POA. Data are presented as means ± SEM (n = 6–13).
The commitment of yeast to liponecrotic PCD alters mitochondrial processes indispensable for cell viability
As we demonstrated in this study, an exposure of yeast to exogenous POA elicits an excessive accumulation of the CL and PE classes of PL, including their POA-containing species, within cellular membranes (Fig. 2; Fig. S2). Mitochondria are known to be the only site within a yeast cell where CL resides; this signature PL of the mitochondrion is synthesized in its inner membrane from PA that is formed exclusively in the ER membrane and delivered to mitochondria via mitochondria-ER junctions (Fig. S3).9-13,15 Furthermore, although PE resides in all cellular membranes, it is known to be synthesized only in the IMM from PS that is formed exclusively in the ER membrane and delivered to mitochondria via mitochondria-ER junctions (Fig. S3).9-13,15 Moreover, the relative levels of other PL classes in both mitochondrial membranes are known to be defined by a bidirectional exchange of PL between the mitochondrial and ER membranes; such exchange integrates lipid dynamics within these organellar membranes into an intricate network (Fig. S3).9-13,15 Taken together, these findings suggest that in yeast exposed to exogenous POA: (1) POA-containing species of CL and PE (and, perhaps, of other PL classes) could amass within both mitochondrial membranes; and (2) such buildup of POA-containing species of PL within mitochondrial membranes could alter certain vital cellular processes that are confined to mitochondria and modulated by PL compositions of both mitochondrial membranes.11,39-41 Moreover, we recently reported that the cyc3Δ mutation, which impairs mitochondrial respiration, makes yeast more susceptible to POA-induced liponecrosis.1
All these considerations prompted us to investigate how the commitment of yeast to liponecrotic PCD influences several mitochondrial processes indispensable for cell viability. Furthermore, because findings reported here further support our recently proposed hypothesis1 on the essential pro-survival role of Atg32p-driven mitophagy in protecting yeast from liponecrotic PCD, we examined how the atg32Δ-dependent mutational block of mitophagy impacts several vital mitochondrial processes in yeast committed to this PCD subroutine. We found that the concentration of exogenous POA added to WT cells: (1) is inversely proportional to the rate of mitochondrial respiration (Fig. 7A), electrochemical potential across the IMM (Fig. 7B) and cellular level of ATP (Fig. 7C), which in yeast is produced mainly in mitochondria;42 and (2) positively correlates with the intracellular level of reactive oxygen species (ROS) (Fig. 7D) known to be generated primarily as by-products of mitochondrial respiration.43 Moreover, we also found that the atg32Δ-dependent mutational block of mitophagy: (1) elevates the degree to which an exposure of yeast to exogenous POA decreases the rate of mitochondrial respiration (Fig. 7A), electrochemical potential across the IMM (Fig. 7B) and cellular level of ATP (Fig. 7C); and (2) elevates the extent to which such exposure increases the intracellular level of ROS (Fig. 7D). Noteworthy, mitophagy has been shown to sustain a healthy population of functional mitochondria that actively respire, exhibit high membrane potential, efficiently synthesize ATP and generate low levels of ROS.13
Figure 7.

Yeast cells committed to liponecrotic PCD exhibit significant changes in the magnitude of 4 mitochondrial processes indispensable for their viability. WT and atg32Δ cells were recovered at days 1, 2 and 4 of culturing in YP medium initially containing 0.2% glucose as carbon source. The rate of mitochondrial respiration (A), electrochemical potential across the inner mitochondrial membrane (ΔΨ) (B), cellular level of ATP (C) and intracellular concentration of reactive oxygen species (ROS) (D) were measured as described in Materials and Methods. Data are presented as means ± SEM (n = 4–7; *P < 0.01).
In sum, these findings imply that the commitment of yeast to liponecrotic PCD: (1) impairs such vital mitochondrial processes as respiration, membrane potential maintenance and ATP synthesis; and (2) rises the intracellular level of ROS by stimulating their production in mitochondria. Because the inability of yeast cells deficient in mitophagy to maintain functional mitochondria increases their susceptibility to POA-induced liponecrosis, it is conceivable that the magnitude of mitochondrial functionality could define the extent of such susceptibility. Furthermore, one could envision that the excessive accumulation of POA-containing species of CL and PE (and, perhaps, of other PL classes) in mitochondrial membranes of yeast cells treated with POA could contribute to their commitment to liponecrotic PCD by altering mitochondrial functionality.
The commitment of yeast to liponecrotic PCD elevates the extent of oxidative damage to cellular proteins and lipids
Because yeast cells undergoing liponecrotic PCD exhibit elevated intracellular levels of ROS (Fig. 7D), we sought to investigate how the commitment of yeast to this PCD subroutine influences the magnitude of oxidative damage to cellular proteins and lipids. We found that the concentration of exogenous POA added to WT cells positively correlates with both the degree of oxidative carbonylation of cellular proteins (Figs. 8A and B) and the extent of oxidative damage to cellular lipids (Fig. 8C). Moreover, we also found that the atg32Δ-dependent mutational block of mitophagy elevates the magnitude of oxidative damage to cellular proteins (Figs. 8A and B) and lipids (Fig. 8C) in yeast exposed to POA. Because Atg32p-driven mitophagy plays an essential pro-survival role in protecting yeast from liponecrotic PCD (see above), one could envision that a rise in oxidative damage to cellular proteins and lipids above certain critical level could contribute to the commitment of yeast to this PCD subroutine.
Figure 8.

Yeast cells committed to liponecrotic PCD exhibit elevated oxidative damage to cellular proteins and lipids. WT and atg32Δ cells were recovered at days 1, 2 and 4 of culturing in YP medium initially containing 0.2% glucose as carbon source. Immunodetection of carbonyl groups of oxidatively damaged cellular proteins (A) and biochemical assays for measuring oxidatively damaged cellular proteins (B) and lipids (C) were performed as described in Materials and Methods. Data are presented as means ± SEM (n = 3–5; *P < 0.01).
The Nma111p-dependent activation of the metacaspase Yca1p is an essential pro-survival process which protects yeast from liponecrotic PCD by maintaining cellular proteostasis
The characteristic of liponecrotic PCD changes in such vital mitochondrial processes as membrane potential maintenance and ROS production are also typical of the caspase-dependent and caspase-independent pathways of apoptotic PCD orchestrated by yeast mitochondria.44-50 A key protein component of the caspase-dependent apoptotic pathway operating in yeast is the metacaspase Yca1p.51 It is involved in degrading various cytosolic proteins in about 40% of the numerous apoptotic PCD scenarios initiated in response to a variety of stimuli.52-54 Yca1p can be activated by: (1) ROS and/or cytochrome c released from mitochondria into the cytosol;49,53,55 and (2) Nma111p, a serine protease which in the nucleus degrades Bir1p, the only presently known inhibitor of Yca1p (Fig. 9A).49,56,57 The caspase-independent apoptotic pathway in yeast includes the “moonlighting“ endonucleases Aif1p and Nuc1p; in response to some pro-apoptotic stimuli, they can move from the mitochondrion to the nucleus where they promote chromatin condensation and DNA degradation (Fig. 9A).49,58-60
Figure 9 (See previous page).

Although the Yca1p- and Nma111p-driven pathway of protein degradation plays an essential pro-death role in caspase-dependent apoptotic PCD elicited by exogenous hydrogen peroxide, it is an indispensable pro-survival process which protects yeast exposed to POA from liponecrotic PCD. (A) Outline of the caspase-dependent and caspase-independent pathways of apoptotic PCD orchestrated by yeast mitochondria in response to hydrogen peroxide (H2O2). A key protein component of the caspase-dependent apoptotic pathway is the metacaspase Yca1p. Yca1p can be activated by reactive oxygen species (ROS) and/or cytochrome c following their release from mitochondria into the cytosol; Yca1p can also be activated by the serine protease Nma111p known to degrade Bir1p, an inhibitor of Yca1p. The caspase-independent apoptotic pathway includes Aif1p and Nuc1p; in response to H2O2 and some other pro-apoptotic stimuli, these 2 endonucleases can move from the mitochondrion to the nucleus where they cause chromatin condensation and/or DNA degradation. See text for additional details. Activation arrows and inhibition bars denote pro-death processes (displayed in red color) or pro-survival processes (displayed in blue color) for apoptotic PCD elicited by H2O2. (B–E) WT and mutant cells were recovered at days 1, 2 and 4 of culturing in YP medium initially containing 0.2% glucose as carbon source. Cell survival was assessed by measuring the clonogenicity of cells after 2 h of treatment with various concentrations of exogenous H2O2. Data are presented as means ± SEM (n = 5–9). (F–I) WT and mutant cells were recovered at days 1, 2 and 4 of culturing in YP medium initially containing 0.2% glucose as carbon source. Cell survival was assessed by measuring the clonogenicity of cells after 2 h of treatment with various concentrations of exogenous POA. Data are presented as means ± SEM (n = 8–13).
Although our recent study revealed that yeast cells committed to POA-induced liponecrotic PCD do not display such characteristic traits of apoptotic PCD as nuclear fragmentation and PS externalization,1 one could envision that the caspase-dependent and/or caspase-independent apoptotic pathways could be in some way integrated into a cellular network orchestrating liponecrosis. We therefore investigated how the single-gene-deletion mutations eliminating key protein components of these pathways influence the susceptibility of yeast to liponecrosis; in a series of positive control experiments, we examined how these mutations impact the susceptibility of yeast to apoptotic PCD elicited by exposure to low doses of hydrogen peroxide. We found that mutations impairing either caspase-dependent or caspase-independent apoptotic pathway make yeast less susceptible to apoptotic PCD (Figs. 9B–E). Moreover, while the aif1Δ and nuc1Δ mutations that impair the caspase-independent apoptotic pathway did not alter the susceptibility of yeast to POA-induced liponecrosis (Figs. 9F and G), the mutational blocks of caspase-dependent apoptotic pathway increased such susceptibility (Figs. 9H and I). Thus, although the Yca1p- and Nma111p-driven pathway of protein degradation plays an essential pro-death role in caspase-dependent apoptosis, it is an indispensable pro-survival process which protects yeast exposed to POA from liponecrotic PCD.
One could envision that the essential pro-survival roles of the metacaspase Yca1p and the serine protease Nma111p in POA-induced liponecrotic PCD consist in promoting a selective proteolytic degradation of oxidatively damaged cellular proteins; such proteins amass in yeast committed to this PCD (Figs. 8A and B). It is conceivable that an impairment of the ability of Yca1p and Nma111p to promote the selective proteolytic degradation of oxidatively damaged cellular proteins may lead to their unfolding and the ensuing aggregation, thereby causing a buildup of damaged, dysfunctional, unfolded and aggregated proteins in yeast committed to liponecrotic PCD. Our findings support this hypothesis. Indeed, the concentration of exogenous POA added to WT cells positively correlated with the extent of protein aggregation observed in these cells (Fig. 10A). Moreover, the yca1Δ and nma111Δ mutations substantially elevated the magnitude of protein aggregation in yeast cells committed to liponecrotic PCD (Fig. 10A).
Figure 10.

The metacaspase Yca1p and the serine protease Nma111p play essential pro-survival roles in POA-induced liponecrosis by promoting a proteolytic degradation of aggregated cellular proteins in yeast committed to this PCD. (A) WT, yca1Δ and nma111Δ cells were recovered at day 4 of culturing in YP medium initially containing 0.2% glucose as carbon source. Recovery of insoluble aggregates of denatured cellular proteins and monitoring the extent of protein aggregation were performed as described in Materials and Methods. (B and C) Outlines of the establishment of a pro-death (B) or pro-survival (C) cellular pattern of Yca1p- and Nma111p-dependent protein degradation in yeast committed to POA-induced liponecrosis. See text for additional details. Activation arrows and inhibition bars denote pro-death processes (displayed in red color) or pro-survival processes (displayed in blue color) for liponecrosis elicited by POA. The thickness of activation arrows and inhibition bars correlates with the rates and/or efficacies of the pro-death or pro-survival processes.
Collectively, these data suggest the following hypothetical scenario for the establishment of a pro-death or pro-survival cellular pattern of Yca1p- and Nma111p-dependent protein degradation in yeast committed to liponecrotic PCD. A pro-death cellular pattern is instituted if the efficacy with which Nma111p degrades Bir1p, an inhibitor of Yca1p, is lowered (Fig. 10B). The resulting rise in the abundance of Bir1p reduces the Yca1p-driven degradation of damaged, dysfunctional, unfolded and aggregated cytosolic proteins in yeast cells committed to liponecrosis, thereby causing an excessive accumulation of such proteins (Fig. 10B). The consequent inability of yeast to maintain cellular protein homeostasis (proteostasis) creates the extreme cellular stress, thus accelerating liponecrotic PCD (Fig. 10B). In contrast, a pro-survival cellular pattern is established if the Nma111p-dependent degradation of Bir1p is activated (Fig. 10C). The proteolysis of Bir1p promotes the Yca1p-driven degradation of damaged, dysfunctional, unfolded and aggregated cytosolic proteins in yeast cells committed to liponecrosis, thereby enabling these cells to maintain efficient proteostasis, avoid the excessive cellular stress and decelerate a progression of the liponecrotic PCD process (Fig. 10C).
Various pathways of autophagy differ in their roles in POA-induced liponecrotic PCD
We recently hypothesized that: (1) the cytosolic serine/threonine protein kinase Atg1p61-64 may orchestrate the execution of POA-induced liponecrotic PCD by activating all currently known pathways for non-selective and selective autophagic degradation of dysfunctional and damaged organelles and macromolecules;1 and (2) Atg32p, a mitochondrial receptor involved in the cargo-specific mitophagy pathway for selective autophagic degradation of aged, dysfunctional, damaged or excessive mitochondria,5,6 may protect yeast from this mode of PCD by enabling to maintain a population of functional mitochondria.1 In this study, we sought to investigate how various pathways of non-selective or selective autophagy contribute to liponecrotic PCD. The protein machinery orchestrating the diverse autophagic pathways in yeast is well known.61-63 We examined how various single-gene-deletion mutations eliminating different protein components of this machinery impact the susceptibility of yeast to POA-induced liponecrosis. The atg1Δ, atg11Δ and atg17Δ mutations impair all of the known pathways for non-selective or selective autophagy, including (1) the non-selective autophagy pathway for massive degradation of various cellular organelles and macromolecules;61-63 (2) the “biosynthetic” cytoplasm-to-vacuole (Cvt) pathway for the delivery of several resident hydrolases to the vacuole;61-63 (3) the cargo-specific pexophagy pathway for selective autophagic degradation of aged, dysfunctional, damaged or excessive peroxisomes;61-63 and (4) the cargo-specific mitophagy pathway.61-63 We found that the atg1Δ, atg11Δ and atg17Δ mutations make yeast less susceptible to POA-induced liponecrosis (Figs. 11A–C). In contrast, the atg19Δ mutation, which impairs only the “biosynthetic” Cvt pathway,61-63 did not alter the susceptibility of yeast to this mode of PCD (Fig. 11D). Furthermore, the atg36Δ mutation, which eliminates only the cargo-specific pexophagy pathway,65 did not exhibit an effect on the susceptibility of yeast to POA-induced liponecrosis (Fig. 11E). Moreover, we confirmed our recent finding1 that the atg32Δ mutation, known to impair only the cargo-specific mitophagy pathway,5,6 increases the susceptibility of yeast to this mode of PCD (Fig. 11F).
Figure 11.

Various pathways of autophagy play different roles in POA-induced liponecrotic PCD. WT and mutant cells were recovered at days 1, 2 and 4 of culturing in YP medium initially containing 0.2% glucose as carbon source. Cell survival was assessed by measuring the clonogenicity of cells after 2 h of treatment with various concentrations of exogenous POA. Data for the effects of the atg1Δ and atg32Δ mutations on the susceptibility of yeast to POA-induced liponecrosis are from a different series of experiments than the ones reported in our recent publication [1]. Data are presented as means ± SEM (n = 11–19).
Altogether, these findings and our recently published data1 imply that various pathways of autophagy differ in their roles in POA-induced liponecrosis. The “biosynthetic” Cvt pathway and the cargo-specific pexophagy pathway are not involved in committing yeast to this PCD subroutine, executing it or protecting from it. The non-selective autophagy pathway for massive degradation of various cellular organelles and macromolecules plays a pivotal role in executing the liponecrotic mode of PCD. In contrast, the cargo-specific mitophagy pathway is a crucial pro-survival process which protects yeast from liponecrosis - likely by eliminating damaged and dysfunctional mitochondria, thereby enabling to sustain a population of functional mitochondria.
To verify our conclusion that the non-selective autophagy pathway for degradation of various cellular organelles and macromolecules plays an essential role in executing the liponecrotic mode of PCD, we used the important recent finding on the ability of the natural polyamine spermidine to extend yeast longevity by inducing this autophagy pathway.66 Noteworthy, the spe1Δ mutation which eliminates an enzyme catalyzing the first step of polyamine biosynthesis67,68 (Fig. S4A) greatly reduces the intracellular level of spermidine and shortens yeast longevity.66 Thus, the efficiency of spermidine synthesis in yeast cells positively correlates with the efficiency of a systemic biological effect which spermidine causes by inducing the non-selective autophagy pathway. We first examined how the spe1Δ, spe2Δ and spe3Δ mutations influence the susceptibility of yeast cells to POA-induced liponecrosis. Each of these mutations eliminates a different enzyme of the pathway for polyamine biosynthesis in yeast cells67,69,70 (Fig. S4A) and, thus, is expected to impair the ability of spermidine to induce the non-selective autophagy pathway of autophagy. We found that (1) the spe1Δ, spe2Δ and spe3Δ mutations reduce the susceptibility of yeast to liponecrosis (Figs. S4B, D, and F, respectively); (2) if added exogenously at the low concentration of 0.1 mM, spermidine reduces the protecting effect of each of these mutations against liponecrosis and restores the level of susceptibility to liponecrotic PCD seen for WT cells (Figs. S4C, E, and G, respectively); and (3) if added exogenously at the high concentration of 0.3 mM or 1 mM, spermidine makes spe1Δ, spe2Δ and spe3Δ cells even more susceptible to liponecrosis than WT cells (Figs. S4C, E, and G, respectively). Moreover, we also found that while exogenous spermidine increases the susceptibility of WT cells to liponecrotic PCD in a concentration-dependent manner (Fig. S4H), it does not alter the reduced susceptibility to liponecrosis of atg1Δ cells deficient in the non-selective pathway of autophagy (Fig. S4I).
Collectively, these findings further validate our conclusion that the non-selective autophagy pathway for degradation of various cellular organelles and macromolecules plays a pivotal role in executing liponecrosis. In addition, these findings suggest that (1) the intracellular level of spermidine positively correlates with the extent of yeast susceptibility to liponecrotic PCD; and (2) spermidine may increase such susceptibility by activating the non-selective pathway of autophagy, thereby triggering the process which executes this mode of PCD.
Based on findings reported here and on our published data,1 we propose the following model for mechanisms that in yeast underlie the opposing roles of various autophagy pathways in POA-induced liponecrotic PCD (Fig. 12). In yeast cells treated with POA, some mitochondria become “stressed," damaged and dysfunctional due to the observed in these cells excessive production of ROS in mitochondria. In atg32Δ mutant cells, these “stressed," damaged and dysfunctional mitochondria cannot be selectively eliminated as efficiently as in WT cells, due to the atg32Δ-dependent mutational block of mitophagy (Fig. 12A). This intensifies processes that create an excessive level of oxidative cellular stress, thereby accelerating the onset of liponecrotic PCD in yeast impaired in Atg32p-driven mitophagy (Fig. 12A). In atg1Δ, atg11Δ, atg17Δ, spe1Δ, spe2Δ and spe3Δ cells exposed to POA (Fig. 12B), Atg32p-driven mitophagy eliminates the “stressed," damaged and dysfunctional mitochondria as selectively and efficiently as it does in WT, atg19Δ and atg36Δ cells treated with POA (Fig. 12C). Therefore, the dynamics of processes creating an excessive level of oxidative cellular stress in atg1Δ, atg11Δ, atg17Δ, spe1Δ, spe2Δ and spe3Δ mutants exposed to POA (Fig. 12B) is similar to the one in WT, atg19Δ and atg36Δ cells treated with POA (Fig. 12C). However, unlike WT, atg19Δ and atg36Δ cells undergoing liponecrotic PCD when the level of oxidative cellular stress exceeds a toxic threshold (Fig. 12C), atg1Δ, atg11Δ, atg17Δ, spe1Δ, spe2Δ and spe3Δ cells exhibit a decelerated rate of the execution of this PCD subroutine because they are impaired in the non-selective autophagy pathway for massive degradation of various cellular organelles and macromolecules (Fig. 12B).
Figure 12.
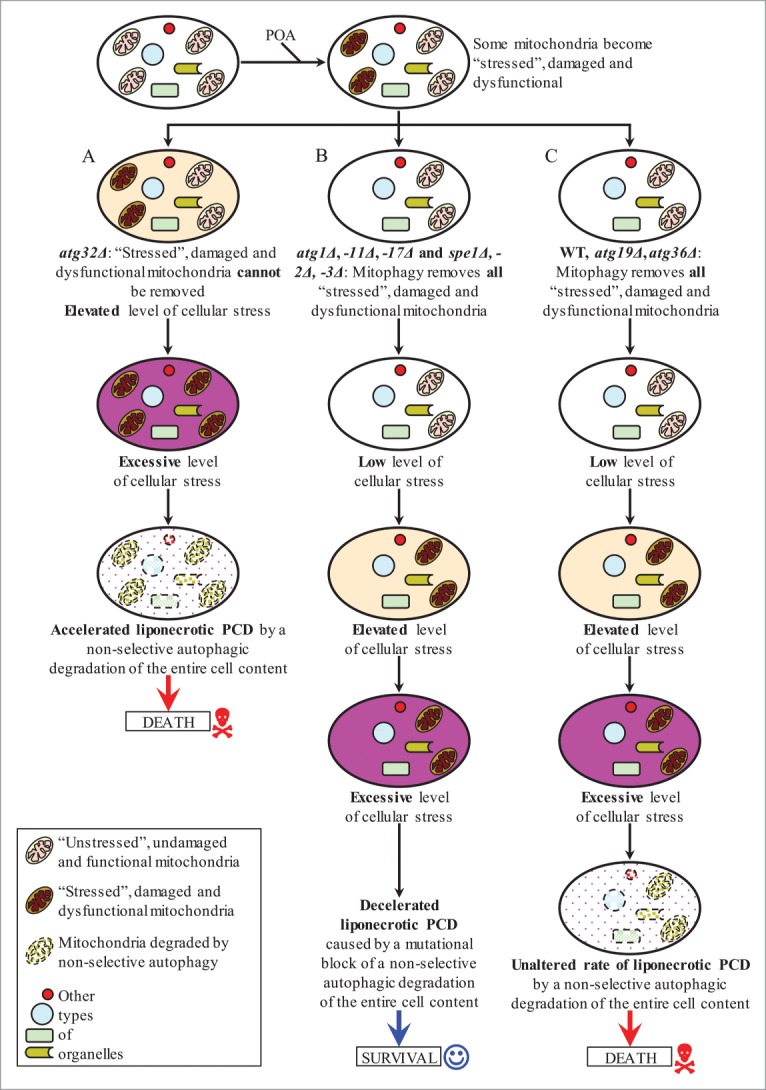
A model for mechanisms that in yeast underlie the opposing roles of various autophagy pathways in POA-induced liponecrotic PCD. See text for additional details. Activation arrows denote pro-death processes (displayed in red color) or pro-survival processes (displayed in blue color) for POA-induced liponecrotic PCD.
Discussion
This study and our recent published data1 suggest the following model for a mechanism underlying POA-induced liponecrotic PCD (Fig. 13). Exogenously added POA is incorporated into POA-containing PL. After being synthesized in the ER, excessive quantities of these POA-containing PL initially accumulate in the ER membrane and then amass in mitochondrial membranes as well as in the PM. The buildup of the POA-containing PL in the PM causes a reduction of PE level in its outer (extracellular) leaflet, thereby triggering the alkaline-pH- and lipid-asymmetry-responsive Rim101 signaling pathway (Fig. 13). This pathway stimulates transcription of the RSB1 gene; the resulting increased abundance of Rsb1p promotes further reduction of PE in the outer leaflet of the PM by (1) enhancing a stimulating effect of Rsb1p on the Lem3p-dependent transport of PE from the outer leaflet to the inner (intracellular) leaflet of the PM; and (2) amplifying an inhibitory effect of Rsb1p on the Yor1p-dependent transport of PE within the PM bilayer in the opposite direction. An ensuing depletion of PE in the outer leaflet of the PM elevates the permeability of this cellular membrane for small molecules (such as propidium iodide1,71), thereby contributing to the commitment of yeast to liponecrotic PCD.
Figure 13.

A model for a mechanism underlying POA-induced liponecrotic PCD in yeast. Exogenously added POA is incorporated into POA-containing phospholipids (PL) that amass in the endoplasmic reticulum (ER) membrane, mitochondrial membranes and the plasma membrane (PM). The buildup of the POA-containing PL in the PM reduces the level of phosphatidylethanolamine (PE) in its outer leaflet, thereby increasing PM permeability for small molecules and committing yeast to liponecrosis. The excessive accumulation of POA-containing PL in mitochondrial membranes impairs mitochondrial functionality and causes the excessive production of reactive oxygen species (ROS) in mitochondria. The resulting rise in cellular ROS above a critical level contributes to the commitment of yeast to liponecrosis by: (1) oxidatively damaging numerous cellular organelles, thereby triggering their autophagic degradation; and (2) oxidatively damaging various cytosolic proteins, thus impairing cellular proteostasis. Several processes in yeast exposed to POA can protect cells from liponecrosis. They include: (1) POA oxidation in peroxisomes, which reduces the flow of POA into PL synthesis pathways; (2) POA incorporation into triacylglycerols (TAG, a major form of neutral lipids), which prevents the excessive accumulation of POA-containing PL in cellular membranes; (3) mitophagy, a selective autophagic degradation of dysfunctional mitochondria, which sustains a population of functional mitochondria needed for POA incorporation into TAG; and (4) a degradation of damaged, dysfunctional and aggregated cytosolic proteins, which enables the maintenance of cellular proteostasis. Activation arrows and inhibition bars denote pro-death processes (displayed in red color) or pro-survival processes (displayed in blue color) for POA-induced liponecrotic PCD.
The excessive accumulation of POA-containing PL in both mitochondrial membranes of cells exposed to POA contributes to the commitment of yeast to liponecrotic PCD by impairing several mitochondrial processes that are indispensable for cell viability, such as respiration, membrane potential and ATP synthesis (Fig. 13). These mitochondrial processes are essential for the ability of functional mitochondria to provide energy needed for the incorporation of POA into TAG, a pro-survival process which prevents the buildup of POA-containing PL within the ER membrane, mitochondrial membranes and the PM by reducing the flow of POA into pathways for PL synthesis and interorganellar transport (Fig. 13).
Moreover, the buildup of POA-containing PL in both mitochondrial membranes of cells exposed to POA contributes to the commitment of yeast to liponecrotic PCD also by causing the excessive production of ROS in mitochondria. A significant rise in cellular ROS above a critical level contributes to such commitment by: (1) oxidatively damaging protein and lipid constituents of various cellular organelles, thereby triggering a non-selective autophagic degradation of numerous “stressed”, damaged and dysfunctional organelles in a pro-death process orchestrated by Atg1p, Atg11p and Atg17p; and (2) oxidatively damaging various cytosolic proteins, thereby impairing a pro-survival process of the maintenance of cellular proteostasis (Fig. 13).
Several cellular processes in yeast exposed to POA can protect cells from liponecrosis. These pro-survival cellular processes include: (1) POA oxidation within functional peroxisomes and POA incorporation into TAG deposited in LD, both of which attenuate the excessive accumulation of POA-containing PL in the ER membrane, mitochondrial membranes and the PM by lowering the flow of POA into pathways for PL synthesis and interorganellar transport (Fig. 13); (2) mitophagy, a selective autophagic degradation of “stressed”, damaged and dysfunctional mitochondria in an Atg32p-driven process which sustains a healthy population of functional mitochondria capable of providing energy for POA deposition within LD in the form of TAG (Fig. 13); and (3) a degradation of damaged, dysfunctional, unfolded and aggregated cytosolic proteins in a Yca1p (metacaspase)- and Nma111p-dependent process which enables the efficient maintenance of protein homeostasis in cells exposed to POA (Fig. 13).
In the future, it would be important to explore a mechanism through which the observed depletion of PE in the outer (extracellular) leaflet of the PM enclosing yeast cells exposed to POA elevates the permeability of this cellular membrane for small molecules, thereby committing yeast to liponecrotic PCD. Another challenge for the future will be to define mechanisms by which the observed buildup of POA-containing PL in both mitochondrial membranes of yeast cells exposed to POA alters such vital mitochondrial processes as respiration, membrane potential maintenance, ATP synthesis and ROS production – thus contributing to the commitment of yeast to liponecrotic PCD. Moreover, it would be interesting to establish the identities of numerous oxidatively damaged and aggregated proteins that accumulate in yeast cells exposed to POA and to explore a pro-survival mechanism underlying the degradation of these proteins in a Yca1p (metacaspase)- and Nma111p-dependent manner.
Materials and Methods
Yeast strains and growth conditions
The WT strain BY4742 (MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0) and single-gene-deletion mutant strains in the BY4742 genetic background (all from Thermo Scientific/Open Biosystems) were grown in YP medium (1% yeast extract, 2% peptone) initially containing 0.2% glucose as carbon source. Cells were cultured at 30°C with rotational shaking at 200 rpm in Erlenmeyer flasks at a “flask volume/medium volume” ratio of 5:1.
Cell viability assay for monitoring the susceptibility of yeast to liponecrotic PCD
A sample of cells was taken from a culture at days 1, 2 and 4 of culturing. A fraction of the sample was diluted in order to determine the total number of cells using a hemacytometer. Eight × 107 cells were harvested by centrifugation for 1 min at 21,000 × g at room temperature and resuspended in 8 ml of YP medium containing 0.2% glucose as carbon source. Each cell suspension was divided into 8 equal aliquots. Three pairs of aliquots were supplemented with POA from a 50 mM stock solution (in 10% chloroform, 45% hexane and 45% ethanol). The final concentration of POA was 0.05 mM, 0.1 mM or 0.15 mM for each pair of aliquots; in all these aliquots, the final concentrations of chloroform, hexane and ethanol were 0.03%, 0.135% and 0.135%, respectively. One pair of aliquots was supplemented only with chloroform, hexane and ethanol added to the final concentrations of 0.03%, 0.135% and 0.135%, respectively. All aliquots were then incubated for 2 h at 30°C on a Labquake rotator set for 360° rotation. Serial dilutions of cells were plated in duplicate onto plates containing YP medium with 2% glucose as carbon source. After 2 d of incubation at 30°C, the number of colony forming units (CFU) per plate was counted. The number of CFU was defined as the number of viable cells in a sample. For each aliquot of cells exposed to POA, the% of viable cells was calculated as follows: (number of viable cells per ml in the aliquot exposed to POA/number of viable cells per ml in the control aliquot that was not exposed to POA) × 100.
Mass spectrometric identification and quantitation of cellular lipids
Yeast cells were resuspended in 155 mM ammonium bicarbonate (pH 8.0) and disrupted by glass beads. Cell lysates were diluted to 50 μg of protein equivalent per 1 ml, and mixed with 20 μl of internal lipid standard mixture prepared in chloroform. The mixture contained 100 μg/μl of PA 14:0-14:0, 100 μg/μl of PE 14:0-14:0, 20 μg/μl of PG 14:0-14:0, 100 μg/μl of PS 14:0-14:0, 100 μg/μl of PC 13:0-13:0, 100 μg/μl of PtdIns 17:0-17:0, 50 μg/μl of DAG 14:0-14:0, 100 μg/μl of TAG 13:0-13:0-13:0, 100 μg/μl of CL 14:0-14:0-14:0-14:0 and 20 μg/μl of nonadecylic FFA 19:0. Samples were extracted with 3 ml of chloroform/methanol (17:1) for 2 h. The lower organic 17:1 phase lipid extract was collected. The remaining aqueous sample material was re-extracted with 1.5 ml of chloroform/methanol (2:1) for 2 h. The lower organic 2:1 phase lipid extract was collected and combined with the lower organic 17:1 phase lipid extract. The lipid extracts were evaporated under nitrogen flow. The lipid film was dissolved in 100 μl of chloroform/methanol (1:2). Mass spectrometric analyses of lipid extracts were performed with a Thermo OrbitrapVelos mass spectrometer equipped with a HESI-II ion source. Lipid species were separated by Fourier transform tandem mass spectrometry. Mass spectra were converted to an open format using the ProteoWizard MSConvert software (http://proteowizard.sourceforge.net/). The raw data were then imported into the open source software LipidXplorer (https://wiki.mpi-cbg.de/wiki/lipidx/index.php/Main_Page) for the automated detection and quantitation of lipid species.
Miscellaneous procedures
Measurement of changes in the pH of culture medium,72 measurement of PE level in the extracellular leaflet of the PM,22 cellular respiration assay,72 monitoring of the mitochondrial membrane potential,72 ROS measurement,73 preparation of cellular extracts and a microanalytic assay for measuring ATP,74 recovery of insoluble aggregates of denatured proteins and an assay for monitoring the extent of protein aggregation,73 immunodetection of carbonyl groups of oxidatively damaged cellular proteins,73 SDS-PAGE,75 and an assay for measuring oxidatively damaged cellular lipids13 were performed as previously described.
Statistical analysis
Statistical analysis was performed using Microsoft Excel's Analysis ToolPack-VBA. All data are presented as mean ± SEM. The p values were calculated using an unpaired 2-tailed t test.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We acknowledge the Center for Biological Applications of Mass Spectrometry at Concordia University for outstanding services.
Funding
This study was supported by grants from the NSERC of Canada and Concordia University Chair Fund. AB and VRR were supported by Frederick Banting and Charles Best Doctoral Scholarship Awards from the Canadian Institutes of Health Research. MTB was supported by a Doctoral Research Fellowship Award from the Fonds Québécois de la Recherche sur la Nature et les Technologies. PK was supported by Doctoral Research Fellowship Awards from the Fonds de recherché en santé du Quebec and from the Fonds québécois de la recherche sur la nature et les technologies. VIT is a Concordia University Research Chair in Genomics, Cell Biology and Aging.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- 1. Sheibani S, Richard VR, Beach A, Leonov A, Feldman R, Mattie S, Khelghatybana L, Piano A, Greenwood M, Vali H, et al. Macromitophagy, neutral lipids synthesis, and peroxisomal fatty acid oxidation protect yeast from "liponecrosis," a previously unknown form of programmed cell death. Cell Cycle 2014; 13:138-47; PMID:24196447; http://dx.doi.org/ 10.4161/cc.26885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kroemer G, El-Deiry WS, Golstein P, Peter ME, Vaux D, Vandenabeele P, Zhivotovsky B, Blagosklonny MV, Malorni W, Knight RA, et al. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death. Cell Death Differ 2005; 12 (Suppl 2):1463-7; PMID:16247491; http://dx.doi.org/ 10.1038/sj.cdd.4401724 [DOI] [PubMed] [Google Scholar]
- 3. Kroemer G, Galluzzi L, Vandenabeele P, Abrams J, Alnemri ES, Baehrecke EH, Blagosklonny MV, El-Deiry WS, Golstein P, Green DR, et al. Classification of cell death: recommendations of the nomenclature committee on cell death 2009. Cell Death Differ 2009; 16:3-11; PMID:18846107; http://dx.doi.org/ 10.1038/cdd.2008.150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Galluzzi L, Vitale I, Abrams JM, Alnemri ES, Baehrecke EH, Blagosklonny MV, Dawson TM, Dawson VL, El-Deiry WS, Fulda S, et al. Molecular definitions of cell death subroutines: recommendations of the nomenclature committee on cell death 2012. Cell Death Differ 2012; 19:107-20; PMID:21760595; http://dx.doi.org/ 10.1038/cdd.2011.96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kanki T, Wang K, Cao Y, Baba M, Klionsky DJ. Atg32 is a mitochondrial protein that confers selectivity during mitophagy. Dev Cell 2009; 17:98-109; PMID: 19619495; http://dx.doi.org/ 10.1016/j.devcel.2009.06.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Okamoto K, Kondo-Okamoto N, Ohsumi Y. Mitochondria-anchored receptor Atg32 mediates degradation of mitochondria via selective autophagy. Dev Cell 2009; 17:87-97; PMID:19619494; http://dx.doi.org/ 10.1016/j.devcel.2009.06.013 [DOI] [PubMed] [Google Scholar]
- 7. Carrasco S, Meyer T. STIM proteins and the endoplasmic reticulum-plasma membrane junctions. Annu Rev Biochem 2011; 80:973-1000; PMID:21548779; http://dx.doi.org/ 10.1146/annurev-biochem-061609-165311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Friedman JR, Voeltz GK. The ER in 3D: a multifunctional dynamic membrane network. Trends Cell Biol 2011; 21:709-17; PMID:21900009; http://dx.doi.org/ 10.1016/j.tcb.2011.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Henry SA, Kohlwein SD, Carman GM. Metabolism and regulation of glycerolipids in the yeast Saccharomyces cerevisiae. Genetics 2012; 190:317-49; PMID:22345606; http://dx.doi.org/ 10.1534/genetics.111.130286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rowland AA, Voeltz GK. Endoplasmic reticulum-mitochondria contacts: function of the junction. Nat Rev Mol Cell Biol 2012; 13:607-25; PMID:22992592; http://dx.doi.org/ 10.1038/nrm3440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Beach A, Richard VR, Leonov A, Burstein MT, Bourque SD, Koupaki O, Juneau M, Feldman R, Iouk T, Titorenko VI. Mitochondrial membrane lipidome defines yeast longevity. Aging (Albany NY) 2013; 5:551-74; PMID:23924582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Horvath SE, Daum G. Lipids of mitochondria. Prog Lipid Res 2013; 52:590-614; PMID:24007978; http://dx.doi.org/ 10.1016/j.plipres.2013.07.002 [DOI] [PubMed] [Google Scholar]
- 13. Richard VR, Leonov A, Beach A, Burstein MT, Koupaki O, Gomez-Perez A, Levy S, Pluska L, Mattie S, Rafesh R, et al. Macromitophagy is a longevity assurance process that in chronologically aging yeast limited in calorie supply sustains functional mitochondria and maintains cellular lipid homeostasis. Aging (Albany NY) 2013; 5:234-69; PMID:23553280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tavassoli S, Chao JT, Young BP, Cox RC, Prinz WA, de Kroon AI, Loewen CJ. Plasma membrane-endoplasmic reticulum contact sites regulate phosphatidylcholine synthesis. EMBO Rep 2013; 14:434-40; PMID: 23519169; http://dx.doi.org/ 10.1038/embor.2013.36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tatsuta T, Scharwey M, Langer T. Mitochondrial lipid trafficking. Trends Cell Biol 2014; 24:44-52; PMID: 24001776; http://dx.doi.org/ 10.1016/j.tcb.2013.07.011 [DOI] [PubMed] [Google Scholar]
- 16. Kohlwein SD, Veenhuis M, van der Klei IJ. Lipid droplets and peroxisomes: key players in cellular lipid homeostasis or a matter of fat - store 'em up or burn 'em down. Genetics 2013; 193:1-50; PMID:23275493; http://dx.doi.org/ 10.1534/genetics.112.143362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Markgraf DF, Klemm RW, Junker M, Hannibal-Bach HK, Ejsing CS, Rapoport TA. An ER protein functionally couples neutral lipid metabolism on lipid droplets to membrane lipid synthesis in the ER. Cell Reports 2014; 6:44-55; PMID:24373967; http://dx.doi.org/ 10.1016/j.celrep.2013.11.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Faergeman NJ, DiRusso CC, Elberger A, Knudsen J, Black PN. Disruption of the Saccharomyces cerevisiae homologue to the murine fatty acid transport protein impairs uptake and growth on long-chain fatty acids. J Biol Chem 1997; 272:8531-8; PMID:9079682; http://dx.doi.org/ 10.1074/jbc.272.13.8531 [DOI] [PubMed] [Google Scholar]
- 19. Porat Z, Wender N, Erez O, Kahana C. Mechanism of polyamine tolerance in yeast: novel regulators and insights. Cell Mol Life Sci 2005; 62:3106-16; PMID:16374585; http://dx.doi.org/ 10.1007/s00018-005-5341-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sychrová H, Ramírez J, Peña A. Involvement of Nha1 antiporter in regulation of intracellular pH in Saccharomyces cerevisiae. FEMS Microbiol Lett 1999; 171:167-72; http://dx.doi.org/ 10.1016/S0378-1097(98)00597-7 [DOI] [PubMed] [Google Scholar]
- 21. Orij R, Brul S, Smits GJ. Intracellular pH is a tightly controlled signal in yeast. Biochim Biophys Acta 2011; 1810:933-44; PMID:21421024; http://dx.doi.org/ 10.1016/j.bbagen.2011.03.011 [DOI] [PubMed] [Google Scholar]
- 22. Ikeda M, Kihara A, Denpoh A, Igarashi Y. The Rim101 pathway is involved in Rsb1 expression induced by altered lipid asymmetry. Mol Biol Cell 2008; 19:1922-31; PMID:18287536; http://dx.doi.org/ 10.1091/mbc.E07-08-0806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Maeda T. The signaling mechanism of ambient pH sensing and adaptation in yeast and fungi. FEBS J 2012; 279:1407-13; PMID:22360598; http://dx.doi.org/ 10.1111/j.1742-4658.2012.08548.x [DOI] [PubMed] [Google Scholar]
- 24. Fadeel B, Xue D. The ins and outs of phospholipid asymmetry in the plasma membrane: roles in health and disease. Crit Rev Biochem Mol Biol 2009; 44:264-77; PMID:19780638; http://dx.doi.org/ 10.1080/10409230903193307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mattiazzi M, Jambhekar A, Kaferle P, Derisi JL, Krizaj I, Petrovic U. Genetic interactions between a phospholipase A2 and the Rim101 pathway components in S. cerevisiae reveal a role for this pathway in response to changes in membrane composition and shape. Mol Genet Genomics 2010; 283:519-30; PMID:20379744; http://dx.doi.org/ 10.1007/s00438-010-0533-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. van Meer G. Dynamic transbilayer lipid asymmetry. Cold Spring Harb Perspect Biol 2011; 3:a004671; http://dx.doi.org/ 10.1101/cshperspect.a004671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Barwell KJ, Boysen JH, Xu W, Mitchell AP. Relationship of DFG16 to the Rim101p pH response pathway in Saccharomyces cerevisiae and Candida albicans. Eukaryot Cell 2005; 4:890-9; PMID:15879523; http://dx.doi.org/ 10.1128/EC.4.5.890-899.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Obara K, Yamamoto H, Kihara A. Membrane protein Rim21 plays a central role in sensing ambient pH in Saccharomyces cerevisiae. J Biol Chem 2012; 287:38473-81; PMID:23019326; http://dx.doi.org/ 10.1074/jbc.M112.394205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shukla AK, Xiao K, Lefkowitz RJ. Emerging paradigms of β-arrestin-dependent seven transmembrane receptor signaling. Trends Biochem Sci 2011; 36:457-69; PMID: 21764321; http://dx.doi.org/ 10.1016/j.tibs.2011.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Xu W, Smith FJ, Jr, Subaran R, Mitchell AP. Multivesicular body-ESCRT components function in pH response regulation in Saccharomyces cerevisiae and Candida albicans. Mol Biol Cell 2004; 15:5528-37; PMID:15371534; http://dx.doi.org/ 10.1091/mbc.E04-08-0666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hayashi M, Fukuzawa T, Sorimachi H, Maeda T. Constitutive activation of the pH-responsive Rim101 pathway in yeast mutants defective in late steps of the MVB/ESCRT pathway. Mol Cell Biol 2005; 25:9478-90; PMID:16227598; http://dx.doi.org/ 10.1128/MCB.25.21.9478-9490.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Babst M, Katzmann DJ, Estepa-Sabal EJ, Meerloo T, Emr SD. Escrt-III: an endosome-associated heterooligomeric protein complex required for MVB sorting. Dev Cell 2002; 3:271-82; PMID:12194857; http://dx.doi.org/ 10.1016/S1534-5807(02)00220-4 [DOI] [PubMed] [Google Scholar]
- 33. Weiss P, Huppert S, Kölling R. Analysis of the dual function of the ESCRT-III protein Snf7 in endocytic trafficking and in gene expression. Biochem J 2009; 424:89-97; PMID:19725809; http://dx.doi.org/ 10.1042/BJ20090957 [DOI] [PubMed] [Google Scholar]
- 34. Lamb TM, Mitchell AP. The transcription factor Rim101p governs ion tolerance and cell differentiation by direct repression of the regulatory genes NRG1 and SMP1 in Saccharomyces cerevisiae. Mol Cell Biol 2003; 23:677-86; PMID: 12509465; http://dx.doi.org/ 10.1128/MCB.23.2.677-686.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kihara A, Igarashi Y. Identification and characterization of a Saccharomyces cerevisiae gene, RSB1, involved in sphingoid long-chain base release. J Biol Chem 2002; 277:30048-54; PMID:12034738; http://dx.doi.org/ 10.1074/jbc.M203385200 [DOI] [PubMed] [Google Scholar]
- 36. Kato U, Emoto K, Fredriksson C, Nakamura H, Ohta A, Kobayashi T, Murakami-Murofushi K, Kobayashi T, Umeda M. A novel membrane protein, Ros3p, is required for phospholipid translocation across the plasma membrane in Saccharomyces cerevisiae. J Biol Chem 2002; 277:37855-62; PMID:12133835; http://dx.doi.org/ 10.1074/jbc.M205564200 [DOI] [PubMed] [Google Scholar]
- 37. Kihara A, Igarashi Y. Cross talk between sphingolipids and glycerophospholipids in the establishment of plasma membrane asymmetry. Mol Biol Cell 2004; 15:4949-59; PMID:15342785; http://dx.doi.org/ 10.1091/mbc.E04-06-0458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Aoki Y, Uenaka T, Aoki J, Umeda M, Inoue K. A novel peptide probe for studying the transbilayer movement of phosphatidylethanolamine. J Biochem 1994; 116:291-7; PMID:7822246 [DOI] [PubMed] [Google Scholar]
- 39. Böttinger L, Horvath SE, Kleinschroth T, Hunte C, Daum G, Pfanner N, Becker T. Phosphatidylethanolamine and cardiolipin differentially affect the stability of mitochondrial respiratory chain supercomplexes. J Mol Biol 2012; 423:677-86; PMID:22971339; http://dx.doi.org/ 10.1016/j.jmb.2012.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Claypool SM, Koehler CM. The complexity of cardiolipin in health and disease. Trends Biochem Sci 2012; 37:32-41; PMID:22014644; http://dx.doi.org/ 10.1016/j.tibs.2011.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Joshi AS, Thompson MN, Fei N, Hüttemann M, Greenberg ML. Cardiolipin and mitochondrial phosphatidylethanolamine have overlapping functions in mitochondrial fusion in Saccharomyces cerevisiae. J Biol Chem 2012; 287:17589-97; PMID:22433850; http://dx.doi.org/ 10.1074/jbc.M111.330167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Fraenkel DG. Respiration. In: Fraenkel DG. Yeast Intermediary Metabolism. Cold Spring Harbor, USA: Cold Spring Harbor Laboratory Press, 2011:135-71. [Google Scholar]
- 43. Giorgio M, Trinei M, Migliaccio E, Pelicci PG. Hydrogen peroxide: a metabolic by-product or a common mediator of ageing signals? Nat Rev Mol Cell Biol 2007; 8:722-8; PMID:17700625; http://dx.doi.org/ 10.1038/nrm2240 [DOI] [PubMed] [Google Scholar]
- 44. Madeo F, Fröhlich E, Ligr M, Grey M, Sigrist SJ, Wolf DH, Fröhlich KU. Oxygen stress: a regulator of apoptosis in yeast. J Cell Biol 1999; 145:757-67; PMID:10330404; http://dx.doi.org/ 10.1083/jcb.145.4.757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Pozniakovsky AI, Knorre DA, Markova OV, Hyman AA, Skulachev VP, Severin FF. Role of mitochondria in the pheromone- and amiodarone-induced programmed death of yeast. J Cell Biol 2005; 168:257-69; PMID:15657396; http://dx.doi.org/ 10.1083/jcb.200408145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Skulachev VP. Bioenergetic aspects of apoptosis, necrosis and mitoptosis. Apoptosis 2006; 11:473-85; PMID:16532373; http://dx.doi.org/ 10.1007/s10495-006-5881-9 [DOI] [PubMed] [Google Scholar]
- 47. Eisenberg T, Büttner S, Kroemer G, Madeo F. The mitochondrial pathway in yeast apoptosis. Apoptosis 2007; 12:1011-23; PMID:17453165; http://dx.doi.org/ 10.1007/s10495-007-0758-0 [DOI] [PubMed] [Google Scholar]
- 48. Pereira C, Silva RD, Saraiva L, Johansson B, Sousa MJ, Côrte-Real M. Mitochondria-dependent apoptosis in yeast. Biochim Biophys Acta 2008; 1783:1286-302; PMID:18406358; http://dx.doi.org/ 10.1016/j.bbamcr.2008.03.010 [DOI] [PubMed] [Google Scholar]
- 49. Carmona-Gutierrez D, Eisenberg T, Büttner S, Meisinger C, Kroemer G, Madeo F. Apoptosis in yeast: triggers, pathways, subroutines. Cell Death Differ 2010; 17:763-73; PMID:20075938; http://dx.doi.org/ 10.1038/cdd.2009.219 [DOI] [PubMed] [Google Scholar]
- 50. Guaragnella N, Zdralević M, Antonacci L, Passarella S, Marra E, Giannattasio S. The role of mitochondria in yeast programmed cell death. Front Oncol 2012; 2:70; PMID:22783546; http://dx.doi.org/ 10.3389/fonc.2012.00070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Madeo F, Herker E, Maldener C, Wissing S, Lächelt S, Herlan M, Fehr M, Lauber K, Sigrist SJ, Wesselborg S, Fröhlich KU. A caspase-related protease regulates apoptosis in yeast. Mol Cell 2002; 9:911-7; PMID:11983181; http://dx.doi.org/ 10.1016/S1097-2765(02)00501-4 [DOI] [PubMed] [Google Scholar]
- 52. Mazzoni C, Falcone C. Caspase-dependent apoptosis in yeast. Biochim Biophys Acta 2008; 1783:1320-7; PMID:18355456; http://dx.doi.org/ 10.1016/j.bbamcr.2008.02.015 [DOI] [PubMed] [Google Scholar]
- 53. Madeo F, Carmona-Gutierrez D, Ring J, Büttner S, Eisenberg T, Kroemer G. Caspase-dependent and caspase-independent cell death pathways in yeast. Biochem Biophys Res Commun 2009; 382:227-31; PMID: 19250922; http://dx.doi.org/ 10.1016/j.bbrc.2009.02.117 [DOI] [PubMed] [Google Scholar]
- 54. Wilkinson D, Ramsdale M. Proteases and caspase-like activity in the yeast Saccharomyces cerevisiae. Biochem Soc Trans 2011; 39:1502-8; PMID:21936842; http://dx.doi.org/ 10.1042/BST0391502 [DOI] [PubMed] [Google Scholar]
- 55. Guaragnella N, Antonacci L, Passarella S, Marra E, Giannattasio S. Achievements and perspectives in yeast acetic acid-induced programmed cell death pathways. Biochem Soc Trans 2011; 39:1538-43; PMID: 21936848; http://dx.doi.org/ 10.1042/BST0391538 [DOI] [PubMed] [Google Scholar]
- 56. Fahrenkrog B, Sauder U, Aebi U. The S. cerevisiae HtrA-like protein Nma111p is a nuclear serine protease that mediates yeast apoptosis. J Cell Sci 2004; 117:115-26; PMID:14657274; http://dx.doi.org/ 10.1242/jcs.00848 [DOI] [PubMed] [Google Scholar]
- 57. Walter D, Wissing S, Madeo F, Fahrenkrog B. The inhibitor-of-apoptosis protein Bir1p protects against apoptosis in S. cerevisiae and is a substrate for the yeast homologue of Omi/HtrA2. J Cell Sci 2006; 119:1843-51; PMID:16608876; http://dx.doi.org/ 10.1242/jcs.02902 [DOI] [PubMed] [Google Scholar]
- 58. Wissing S, Ludovico P, Herker E, Büttner S, Engelhardt SM, Decker T, Link A, Proksch A, Rodrigues F, Corte-Real M, et al. An AIF orthologue regulates apoptosis in yeast. J Cell Biol 2004; 166:969-74; PMID: 15381687; http://dx.doi.org/ 10.1083/jcb.200404138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Liang Q, Li W, Zhou B. Caspase-independent apoptosis in yeast. Biochim Biophys Acta 2008; 1783:1311-9; PMID:18358844; http://dx.doi.org/ 10.1016/j.bbamcr.2008.02.018 [DOI] [PubMed] [Google Scholar]
- 60. Büttner S, Eisenberg T, Carmona-Gutierrez D, Ruli D, Knauer H, Ruckenstuhl C, Sigrist C, Wissing S, Kollroser M, Fröhlich KU, et al. Endonuclease G regulates budding yeast life and death. Mol Cell 2007; 25:233-46; PMID:17244531; http://dx.doi.org/ 10.1016/j.molcel.2006.12.021 [DOI] [PubMed] [Google Scholar]
- 61. Nakatogawa H, Suzuki K, Kamada Y, Ohsumi Y. Dynamics and diversity in autophagy mechanisms: lessons from yeast. Nat Rev Mol Cell Biol 2009; 10:458-67; PMID:19491929; http://dx.doi.org/ 10.1038/nrm2708 [DOI] [PubMed] [Google Scholar]
- 62. Inoue Y, Klionsky DJ. Regulation of macroautophagy in Saccharomyces cerevisiae. Semin Cell Dev Biol 2010; 21:664-70; PMID:20359542; http://dx.doi.org/ 10.1016/j.semcdb.2010.03.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Reggiori F, Klionsky DJ. Autophagic processes in yeast: mechanism, machinery and regulation. Genetics 2013; 194:341-61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Matsuura A, Tsukada M, Wada Y, Ohsumi Y. Apg1p, a novel protein kinase required for the autophagic process in Saccharomyces cerevisiae. Gene 1997; 192:245-50; PMID:9224897; http://dx.doi.org/ 10.1016/S0378-1119(97)00084-X [DOI] [PubMed] [Google Scholar]
- 65. Motley AM, Nuttall JM, Hettema EH. Pex3-anchored Atg36 tags peroxisomes for degradation in Saccharomyces cerevisiae. EMBO J 2012; 31:2852-68; PMID:22643220; http://dx.doi.org/ 10.1038/emboj.2012.151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Eisenberg T, Knauer H, Schauer A, Büttner S, Ruckenstuhl C, Carmona-Gutierrez D, Ring J, Schroeder S, Magnes C, Antonacci L, et al. Induction of autophagy by spermidine promotes longevity. Nat Cell Biol 2009; 11:1305-14; PMID:19801973; http://dx.doi.org/ 10.1038/ncb1975 [DOI] [PubMed] [Google Scholar]
- 67. Fonzi WA, Sypherd PS. The gene and the primary structure of ornithine decarboxylase from Saccharomyces cerevisiae. J Biol Chem 1987; 262:10127-33; PMID:3038869 [PubMed] [Google Scholar]
- 68. Minois N, Carmona-Gutierrez D, Madeo F. Polyamines in aging and disease. Aging (Albany NY) 2011; 3:716-32; PMID:21869457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Kashiwagi K, Taneja SK, Liu TY, Tabor CW, Tabor H. Spermidine biosynthesis in Saccharomyces cerevisiae. Biosynthesis and processing of a pro-enzyme form of S-adenosylmethionine decarboxylase. J Biol Chem 1990; 265:22321-8; PMID:2266128 [PubMed] [Google Scholar]
- 70. Hamasaki-Katagiri N, Tabor CW, Tabor H. Spermidine biosynthesis in Saccharomyces cerevisiae: polyamine requirement of a null mutant of the SPE3 gene (spermidine synthase). Gene 1997; 187:35-43; PMID:9073064; http://dx.doi.org/ 10.1016/S0378-1119(96)00660-9 [DOI] [PubMed] [Google Scholar]
- 71. Goldberg AA, Richard VR, Kyryakov P, Bourque SD, Beach A, Burstein MT, Glebov A, Koupaki O, Boukh-Viner T, Gregg C, et al. Chemical genetic screen identifies lithocholic acid as an anti-aging compound that extends yeast chronological life span in a TOR-independent manner, by modulating housekeeping longevity assurance processes. Aging (Albany NY) 2010; 2:393-414; PMID:20622262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Goldberg AA, Bourque SD, Kyryakov P, Gregg C, Boukh-Viner T, Beach A, Burstein MT, Machkalyan G, Richard V, Rampersad S, et al. Effect of calorie restriction on the metabolic history of chronologically aging yeast. Exp Gerontol 2009; 44:555-71; PMID:19539741; http://dx.doi.org/ 10.1016/j.exger.2009.06.001 [DOI] [PubMed] [Google Scholar]
- 73. Kyryakov P, Beach A, Richard VR, Burstein MT, Leonov A, Levy S, Titorenko VI. Caloric restriction extends yeast chronological lifespan by altering a pattern of age-related changes in trehalose concentration. Front Physiol 2012; 3:256; PMID:22783207; http://dx.doi.org/ 10.3389/fphys.2012.00256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Lin SS, Manchester JK, Gordon JI. Enhanced gluconeogenesis and increased energy storage as hallmarks of aging in Saccharomyces cerevisiae. J Biol Chem 2001; 276:36000-7; PMID:11461906; http://dx.doi.org/ 10.1074/jbc.M103509200 [DOI] [PubMed] [Google Scholar]
- 75. Titorenko VI, Smith JJ, Szilard RK, Rachubinski RA. Pex20p of the yeast Yarrowia lipolytica is required for the oligomerization of thiolase in the cytosol and for its targeting to the peroxisome. J Cell Biol 1998; 142:403-20; PMID:9679140; http://dx.doi.org/ 10.1083/jcb.142.2.403 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.