

Abstract
Tumor suppressor p53 is one of the most frequently mutated genes in cancer, with almost 50% of all types of cancer expressing a mutant form of p53. p53 transactivates the expression of its primary negative regulator, HDM2. HDM2 is a ubiquitin ligase, which initiates the proteasomal degradation of p53 following ubiquitination. Proteasome inhibitors, by targeting the ubiquitin proteasome pathway inhibit the degradation of the majority of cellular proteins including wild-type p53. In contrast, in this study we found that the protein expression of mutant p53 was suppressed following treatment with established or novel proteasome inhibitors. Furthermore, for the first time we demonstrated that Arsenic trioxide, which was previously shown to suppress mutant p53 protein level, exhibits proteasome inhibitory activity. Proteasome inhibitor-mediated suppression of mutant p53 was partially rescued by the knockdown of HDM2, suggesting that the stabilization of HDM2 by proteasome inhibitors might be responsible for mutant p53 suppression to some extent. This study suggests that suppression of mutant p53 is a general property of proteasome inhibitors and it provides additional rationale to use proteasome inhibitors for the treatment of tumors with mutant p53.
Keywords: Arsenic trioxide, HDM2, p53, proteasome inhibitors, RNAi
Abbreviations
- HDM2
human double minute 2
- PUMA
p53 upregulated modulator of apoptosis
- Bcl-2
B-cell lymphoma 2
- Bax
Bcl-2-associated X
- FOXM1
Forkhead Box M1, Mcl-1, Myeloid cell leukemia sequence 1
Introduction
The tumor suppressor gene p53 is a transcription factor that regulates many critical cellular processes such as maintenance of genomic stability, senescence, cell cycle arrest and apoptosis (reviewed in).1,2 The main function of p53 as a tumor suppressor is carried out by its role as a sequence specific transcription factor that regulates the expression of genes. p53 accumulates in the nucleus following stress induction and binds to its receptive genes and promotes their activation.3,4 Some of the genes that are activated following p53 transactivation include p21, PUMA, Gadd45, Bcl-2 family genes such as Bax, which can cause cell cycle arrest or apoptosis.5-7 In addition to direct activation of its target genes, p53 is also involved in the repression of certain genes. The genes that get suppressed by p53 include Bcl-2, Bcl-xL, survivin, cyclin B1, FOXM1 etc.8-10 It has been demonstrated that nearly 80% of p53 responsive genes are suppressed by this transcription factor.11 p53 also activates the expression of HDM2, (MDM2 in mouse), which is the main negative regulator of p53. HDM2 is an E3 ubiquitin ligase that ubiquitinates the C terminus of p53 and targets p53 for proteasomal degradation,12 (reviewed in).13 Therefore, a negative feedback loop between HDM2 and p53 results in low level of wild-type p53 in normal cells (reviewed in).14
The significance of p53 is underscored by the observation that p53 is the most commonly mutated gene in human malignancies, with more that 50% of all human cancers expressing mutated form of p53.15,16 The mutations in p53 result in the loss of transcriptional activity of p53 causing p53 to lose its tumor suppressor function. Furthermore, in addition to the loss of transcriptional activity, gain of function is observed in the mutant form of p53 resulting in oncogenic functionality.17 The mutant form of p53 is overexpressed in many types of human cancers because of its longer half-life as opposed to wild-type p53, which has a half-life of 10–30 minutes.18 As a result, the oncogenic function of the mutant form of p53 has been identified as a target for development of novel anti-cancer therapeutics (reviewed in).19,20 The rationale for these potential drugs is to suppress the activity of mutant p53 by degradation or by reverting the mutant p53 back to its wild-type conformation.21
Proteasome inhibitors are a novel class of anti-cancer therapeutics targeting the activity of the proteasome, which is involved in targeted degradation of proteins.22,23 Inhibition of the proteasome complex results in the stabilization of proteins that induce cell cycle arrest and apoptosis, including wild-type p53.24 In this study, however, we observed that in cells carrying the mutant form of p53 the level of mutant p53 is suppressed after treatment with proteasome inhibitors. Knockdown of HDM2 by siRNA rescues in part the suppression of mutant p53 following treatment with proteasome inhibitors, suggesting that HDM2 stabilization by proteasome inhibitors leads to the degradation of mutant p53. Therefore, proteasome inhibitors could be used to target the oncogenic activity of mutant p53.
Results
Proteasome inhibitors suppress mutant p53 protein level
Based on our initial observations that in contrast to wild-type p53 proteasome inhibitors suppress mutant p53, we decided to compare the effect of different proteasome inhibitors on wild-type and mutant p53 protein. We utilized MCF7 and U2OS cells expressing wild-type p53, and MDA-MB-231 (R280K),25 MIA-PaCa-2 (R248W)26 and DU145 (P223L, V274F)27 cells carrying mutant p53. The cells were treated with known proteasome inhibitors MG132, bortezomib, carfilzomib28 and thiostrepton.29 As expected, a concentration dependent induction of wild-type 53 level was found following treatment with the proteasome inhibitors in the wild-type p53 expressing cells U2OS and MCF7 (Fig. 1A and B). However, we observed a concentration dependent inhibition of p53 level following treatment with the proteasome inhibitors in cells expressing the mutant form of p53, such as MDA-MB-231, MIA-PaCa-2 and DU145 (Fig. 2A-C). Taken together, though treatment with proteasome inhibitors leads to an increase in the level of p21, wild-type p53 and HDM2, mutant p53 is suppressed by proteasome inhibitors.
Figure 1.
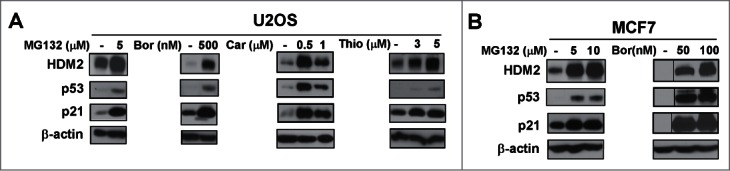
Wild-type p53 is stabilized by proteasome inhibitors. (A and B) U2OS osteosarcoma cells and MCF7 breast cancer cells were treated with the indicated concentrations of proteasome inhibitors MG132, bortezomib (Bor), carfilzomib (Car) or thiostrepton (Thio). Immunoblotting was performed for HDM2, p53, p21. β-actin was used as the loading control.
Figure 2.
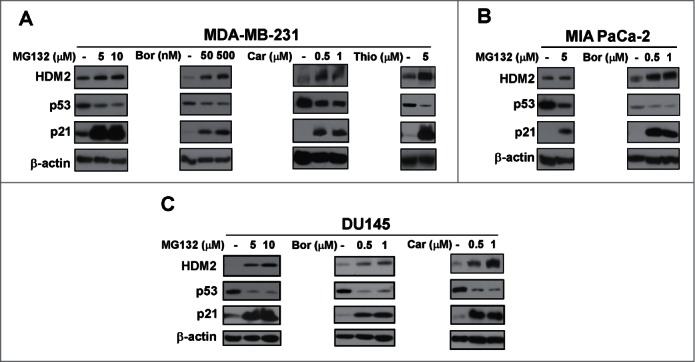
Mutant p53 is suppressed by proteasome inhibitors. (A–C) MDA-MB-231 breast, MIA PaCa-2 pancreatic and DU145 prostate cancer cells were treated with the indicated concentrations of proteasome inhibitors MG132, bortezomib (Bor), carfilzomib (Car) or thiostrepton (Thio). Immunoblot analysis of HDM2, p53, p21 and β-actin as the loading control was carried out after treatment.
Arsenic trioxide is a proteasome inhibitor
Arsenic trioxide is an anti-cancer drug, which has been shown to suppress the expression of mutant p53 in various cancer cells.30 Because several proteasome inhibitors down-regulate mutant p53 levels according to our current data, we hypothesized that Arsenic trioxide might exhibit proteasome inhibitory activity. First, we evaluated the effect of Arsenic trioxide on FOXM1 transcriptional activity because proteasome inhibitors universally inhibit the transcriptional activity of FOXM1.29 We observed a significant inhibition of FOXM1 transcriptional activity and protein expression in different human cancer cell lines following Arsenic trioxide treatment (Fig. 3). We further observed that Arsenic trioxide stabilized the expression of a number of proteins, which are targets of proteasome-dependent degradation including HDM2, Mcl-1 and p21 (Fig. 3B-D). The proteasome inhibitory activity of Arsenic trioxide was also demonstrated by the formation of ubiquitin conjugates following treatment in MIA PaCa-2 cells (Fig. 3B). In addition, we found that Arsenic trioxide suppressed mutant p53 expression (Fig. 3C) in agreement with previously published data.30 Altogether, these data suggest that Arsenic trioxide acts as a typical proteasome inhibitor.
Figure 3.
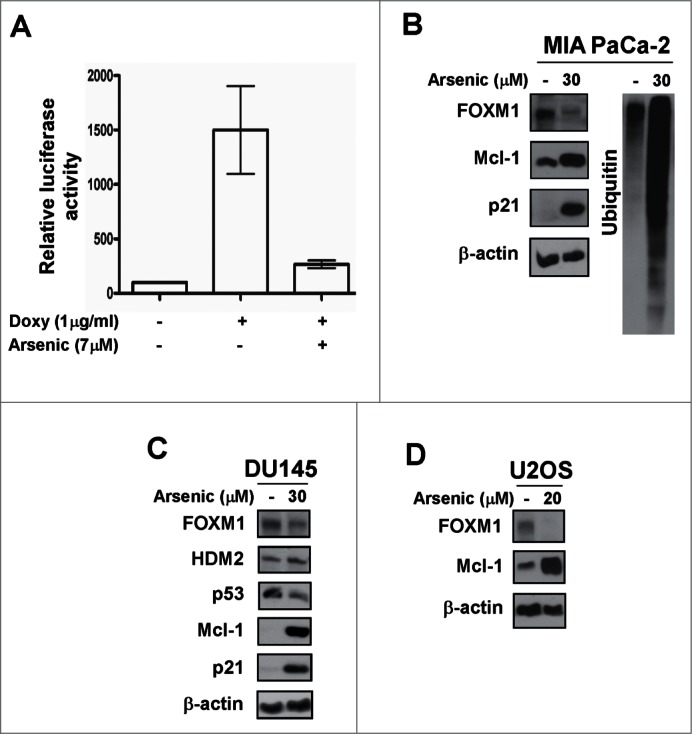
Arsenic trioxide acts as a proteasome inhibitor. (A) C3-luc cells were treated as indicated for overnight and luciferase activity was measured using the Luciferase Assay System kit from Promega. Graph shows mean ± SEM of 2 independent experiments. (B–D) MIA PaCa-2, DU145 and U2OS cells were treated as indicated. Immunoblotting was performed with antibodies specific for FOXM1, HDM2, p53, Mcl-1, p21 and ubiquitin. β-actin was used as the loading control.
RNAi-mediated knockdown of HDM2 partially rescues the suppression of mutant p53 following proteasome inhibitor treatment
In cells with mutant p53 HDM2 level is low because its transcription is not up-regulated by p5331 and consequently mutant p53 level is high. To test the hypothesis that the strong up-regulation of HDM2 by proteasome inhibitors is responsible for targeting mutant p53 for degradation, MIA PaCa-2 pancreatic and MDA-MB-231 breast cancer cells were transfected with anti-HDM2 siRNA. The transfection of anti-HDM2 siRNA was followed by treatment with proteasome inhibitors MG132 and bortezomib. In cells that were transfected with control-siRNA (non specific to HDM2), the level of p53 decreased on treatment with the proteasome inhibitors (Fig. 4A-C). However, in cells transfected with anti-HDM2 siRNA, suppression of mutant p53 after treatment with the proteasome inhibitors was alleviated (Fig. 4A-C). These data suggest that following treatment with proteasome inhibitors, the stabilization of HDM2 is partially responsible for the suppression of mutant p53.
Figure 4.

Proteasome inhibitor-associated suppression of mutant p53 is partially rescued by HDM2 knockdown. (A–C) MIA PaCa-2 and MDA-MB-231 cells were transfected with either control or HDM2 specific siRNA. Following a 72-hour transfection cells were treated as indicated. Cell lysates were immunoblotted for HDM2, p53, p21 and β-actin as the loading control.
Discussion
In this paper, in agreement with previous observations32,33 we showed that proteasome inhibitors suppress mutant, but not wild-type p53. More importantly, we demonstrated by RNA interference that mutant p53 suppression by proteasome inhibitors was modulated by HDM2 (Fig. 4). In addition, for the first time we showed that Arsenic trioxide exhibits proteasome inhibitory activity. It is a significant finding because this drug has been used against acute promyelocytic leukemia (APL)34 and it is important to understand the mechanism of its action. Furthermore, we showed previously that proteasome inhibitors suppress FOXM129 potentially via the stabilization of a negative regulator of FOXM1 (NRFM) that inhibits the transcriptional activity of FOXM1 on its own promoter,35 because of the FOXM1 auto-regulation loop.36 Similarly, in this paper we demonstrated that suppression of mutant p53 is also a general feature of proteasome inhibitors. However, the mechanisms of suppression of mutant p53 or FOXM1 are unrelated. Suppression of FOXM1 by proteasome inhibitors is based on the FOXM1 autoregulatory loop.35,36 On the other hand, suppression of mutant p53 is linked to the stabilization of the low cellular level of HDM2 by proteasome inhibitors, leading to the degradation of mutant p53 (Fig. 5). Mutations occur mainly in the DNA-binding domain of p53 resulting in a transcriptionally inactive protein, which cannot up-regulate HDM2, its negative regulator. Consequently, we can assume that in cells with p53 mutations following treatment with proteasome inhibitors mutant p53 protein level will decrease because HDM2 level will significantly increase and will target mutant p53 for proteasomal degradation.
Figure 5.

Model of the HDM2-mediated suppression of mutant p53 after treatment with proteasome inhibitors. (A) The basal level of wild-type p53 is low because HDM2, its transcriptional target and negative regulator marks it for proteasomal degradation. After treatment with proteasome inhibitors both wild-type p53 and HDM2 are stabilized. Though HDM2 continues to degrade wild-type p53, but its overall level increases because its degradation by HDM2 is overridden by its stabilization by proteasome inhibitors. (B) The basal level of mutant p53 is high because it cannot transactivate its negative regulator HDM2. Following proteasome inhibitor treatment both mutant p53 and HDM2 are stabilized, but the overall level of mutant p53 decreases because the increased amount of HDM2 efficiently targets it for degradation, thus its stabilization by proteasome inhibitors is overridden by its HDM2-mediated degradation.
Mutant p53 is known to contribute to malignant function by acquisition of activities that include increased ability of proliferation, invasion and anti-cancer therapy resistance of tumor cells.17 Consequently, our findings greatly support the use of proteasome inhibitors in the treatment of tumors harboring mutant p53. Additional experiments are needed to determine whether mutant p53 is one of the critical targets of proteasome inhibitors in cancer.
Materials and Methods
Cell culture and chemical compounds
MIA PaCa-2 pancreatic, DU145 prostate cancer cell lines (ATCC), U2OS osteosarcoma and osteosarcoma-derived C3-luc cells37 were grown in DMEM medium (10–017-CV; Cellgro). MDA-MB-231 and MCF7 (ATCC) breast cancer cell lines were grown in RPMI medium (10–040-CV; Cellgro). The media were supplemented with 10% fetal bovine serum (S11550; Atlanta Biologicals) and 1% penicillin-streptomycin (15140; GIBCO). All the cells were maintained at 37°C in 5% CO2. Bortezomib (Velcade; Millenium Pharmaceuticals), MG132 (474791; Calbiochem), thiostrepton (T8902; Sigma) and Carfilzomib (A1098; Active Biochemicals) were dissolved in dimethyl sulfoxide (DMSO) (BP231; Fisher Scientific), Arsenic trioxide (202673; Sigma) in NaOH and Doxycycline (D5897; LKT Laboratories) in phosphate buffered saline (PBS).
Immunoblot analysis
Treated cells were harvested and lysed by using IP buffer (20mM HEPES, 1% Triton X-100, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 100mM NaF, 10 mM Na4P2O7, 1 mM sodium orthovanadate, 0.2 mM PMSF supplemented with protease inhibitor tablet (11836153001; Roche Applied Sciences)). Protein concentration was determined by the Bio-Rad Protein Assay reagent (500–0006; BIO-RAD). Isolated proteins were separated on SDS-PAGE and transferred to PVDF membrane (Millipore). Immunoblotting was carried out with antibodies specific for HDM2 (sc-813; Santa Cruz), p53 (sc-126; Santa Cruz), Mcl-1 (MS-683-P0; Lab Vision), ubiquitin (sc-271289; Santa Cruz), FOXM1 (the rabbit polyclonal antibody against FOXM1 was described previously),38 p21 (556431; BD-PharMingen) and β-actin (A5441; Sigma).
Luciferase assay
Cells were treated with the combination of 1μg/ml Doxycycline and the indicated concentrations of the drugs for overnight. The luciferase activity was determined by the Luciferase Assay System (E1500; Promega) according to the recommendations of the manufacturer. The data were normalized on the amount of protein in the samples.
Transfection and siRNA
Control (universal negative control #1) small interfering RNA (siRNA) and siRNA specific to HDM2 (AGGCAAAUGUGCAAUACCA) were synthesized by Sigma. 50nM of siRNA duplexes were transfected into cells using Lipofectamine 2000 (11668–019; Invitrogen) according to the manufacturer's recommendation. Cells were treated 72 hours after transfection.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Dr. Ming Wang for technical assistance. We also thank Dr. Pradip Raychaudhuri for the kind gift of ubiquitin antibody.
References
- 1. Levine AJ, Oren M. The first 30 years of p53: growing ever more complex. Nat Rev Cancer 2009; 9:749-58; PMID:19776744; http://dx.doi.org/ 10.1038/nrc2723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ko LJ, Prives C. p53: puzzle and paradigm. Genes Dev 1996; 10:1054-72; PMID:8654922; http://dx.doi.org/ 10.1101/gad.10.9.1054 [DOI] [PubMed] [Google Scholar]
- 3. Bargonetti J, Friedman PN, Kern SE, Vogelstein B, Prives C. Wild-type but not mutant p53 immunopurified proteins bind to sequences adjacent to the SV40 origin of replication. Cell 1991; 65:1083-91; PMID:1646078; http://dx.doi.org/ 10.1016/0092-8674(91)90560-L [DOI] [PubMed] [Google Scholar]
- 4. Funk WD, Pak DT, Karas RH, Wright WE, Shay JW. A transcriptionally active DNA-binding site for human p53 protein complexes. Mol Cell Biol 1992; 12:2866-71; PMID:1588974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Miyashita T, Reed JC. Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell 1995; 80:293-9; PMID:7834749; http://dx.doi.org/ 10.1016/0092-8674(95)90412-3 [DOI] [PubMed] [Google Scholar]
- 6. Nakano K, Vousden KH. PUMA, a novel proapoptotic gene, is induced by p53. Mol Cell 2001; 7:683-94; PMID:11463392; http://dx.doi.org/ 10.1016/S1097-2765(01)00214-3 [DOI] [PubMed] [Google Scholar]
- 7. Chao C, Saito S, Kang J, Anderson CW, Appella E, Xu Y. p53 transcriptional activity is essential for p53-dependent apoptosis following DNA damage. Embo J 2000; 19:4967-75; PMID:10990460; http://dx.doi.org/ 10.1093/emboj/19.18.4967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Haldar S, Negrini M, Monne M, Sabbioni S, Croce CM. Down-regulation of bcl-2 by p53 in breast cancer cells. Cancer Res 1994; 54:2095-7; PMID:8174112 [PubMed] [Google Scholar]
- 9. Innocente SA, Abrahamson JL, Cogswell JP, Lee JM. p53 regulates a G2 checkpoint through cyclin B1. Proc Natl Acad Sci U S A 1999; 96:2147-52; PMID:10051609; http://dx.doi.org/ 10.1073/pnas.96.5.2147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pandit B, Halasi M, Gartel AL. p53 negatively regulates expression of FoxM1. Cell Cycle 2009; 8:3425-7; PMID:19806025; http://dx.doi.org/ 10.4161/cc.8.20.9628 [DOI] [PubMed] [Google Scholar]
- 11. Mirza A, Wu Q, Wang L, McClanahan T, Bishop WR, Gheyas F, Ding W, Hutchins B, Hockenberry T, Kirschmeier P, et al. . Global transcriptional program of p53 target genes during the process of apoptosis and cell cycle progression. Oncogene 2003; 22:3645-54; PMID:12789273; http://dx.doi.org/ 10.1038/sj.onc.1206477 [DOI] [PubMed] [Google Scholar]
- 12. Blagosklonny MV, Demidenko ZN, Fojo T. Inhibition of transcription results in accumulation of Wt p53 followed by delayed outburst of p53-inducible proteins: p53 as a sensor of transcriptional integrity. Cell Cycle 2002; 1:67-74; PMID:12429911 [PubMed] [Google Scholar]
- 13. Michael D, Oren M. The p53-Mdm2 module and the ubiquitin system. Semin Cancer Biol 2003; 13:49-58; PMID:12507556; http://dx.doi.org/ 10.1016/S1044-579X(02)00099-8 [DOI] [PubMed] [Google Scholar]
- 14. Mihara M, Erster S, Zaika A, Petrenko O, Chittenden T, Pancoska P, Moll UM. p53 has a direct apoptogenic role at the mitochondria. Mol Cell 2003; 11:577-90; PMID:12667443; http://dx.doi.org/ 10.1016/S1097-2765(03)00050-9 [DOI] [PubMed] [Google Scholar]
- 15. Hollstein M, Sidransky D, Vogelstein B, Harris CC. p53 mutations in human cancers. Science 1991; 253:49-53; PMID:1905840; http://dx.doi.org/ 10.1126/science.1905840 [DOI] [PubMed] [Google Scholar]
- 16. Hollstein M, Rice K, Greenblatt MS, Soussi T, Fuchs R, Sorlie T, Hovig E, Smith-Sørensen B, Montesano R, Harris CC. Database of p53 gene somatic mutations in human tumors and cell lines. Nucleic Acids Res 1994; 22:3551-5; PMID:7937055 [PMC free article] [PubMed] [Google Scholar]
- 17. Cooks T, Pateras IS, Tarcic O, Solomon H, Schetter AJ, Wilder S, Lozano G, Pikarsky E, Forshew T, Rosenfeld N, et al. . Mutant p53 prolongs NF-kappaB activation and promotes chronic inflammation and inflammation-associated colorectal cancer. Cancer Cell 2013; 23:634-46; PMID:23680148; http://dx.doi.org/ 10.1016/j.ccr.2013.03.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Blagosklonny MV. Loss of function and p53 protein stabilization. Oncogene 1997; 15:1889-93; PMID:9365234; http://dx.doi.org/ 10.1038/sj.onc.1201374 [DOI] [PubMed] [Google Scholar]
- 19. Farnebo M, Bykov VJ, Wiman KG. The p53 tumor suppressor: a master regulator of diverse cellular processes and therapeutic target in cancer. Biochem Biophys Res Commun 2010; 396:85-9; PMID:20494116; http://dx.doi.org/ 10.1016/j.bbrc.2010.02.152 [DOI] [PubMed] [Google Scholar]
- 20. Bullock AN, Fersht AR. Rescuing the function of mutant p53. Nat Rev Cancer 2001; 1:68-76; PMID:11900253; http://dx.doi.org/ 10.1038/35094077 [DOI] [PubMed] [Google Scholar]
- 21. Bykov VJ, Issaeva N, Shilov A, Hultcrantz M, Pugacheva E, Chumakov P, Bergman J, Wiman KG, Selivanova G. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat Med 2002; 8:282-8; PMID:11875500; http://dx.doi.org/ 10.1038/nm0302-282 [DOI] [PubMed] [Google Scholar]
- 22. Adams J, Palombella VJ, Sausville EA, Johnson J, Destree A, Lazarus DD, Maas J, Pien CS, Prakash S, Elliott PJ. Proteasome inhibitors: a novel class of potent and effective antitumor agents. Cancer Res 1999; 59:2615-22; PMID:10363983 [PubMed] [Google Scholar]
- 23. Almond JB, Cohen GM. The proteasome: a novel target for cancer chemotherapy. Leukemia 2002; 16:433-43; PMID:11960320; http://dx.doi.org/ 10.1038/sj.leu.2402417 [DOI] [PubMed] [Google Scholar]
- 24. An WG, Hwang SG, Trepel JB, Blagosklonny MV. Protease inhibitor-induced apoptosis: accumulation of wt p53, p21WAF1/CIP1, and induction of apoptosis are independent markers of proteasome inhibition. Leukemia 2000; 14:1276-83; PMID:10914553 [DOI] [PubMed] [Google Scholar]
- 25. Girardini JE, Napoli M, Piazza S, Rustighi A, Marotta C, Radaelli E, Capaci V, Jordan L, Quinlan P, Thompson A, et al. . A Pin1/mutant p53 axis promotes aggressiveness in breast cancer. Cancer Cell 2011; 20:79-91; PMID:21741598; http://dx.doi.org/ 10.1016/j.ccr.2011.06.004 [DOI] [PubMed] [Google Scholar]
- 26. Yoshikawa H, Nagashima M, Khan MA, McMenamin MG, Hagiwara K, Harris CC. Mutational analysis of p73 and p53 in human cancer cell lines. Oncogene 1999; 18:3415-21; PMID:10362363; http://dx.doi.org/ 10.1038/sj.onc.1202677 [DOI] [PubMed] [Google Scholar]
- 27. Bajgelman MC, Strauss BE. The DU145 human prostate carcinoma cell line harbors a temperature-sensitive allele of p53. Prostate 2006; 66:1455-62; PMID:16741917; http://dx.doi.org/ 10.1002/pros.20462 [DOI] [PubMed] [Google Scholar]
- 28. Herndon TM, Deisseroth A, Kaminskas E, Kane RC, Koti KM, Rothmann MD, Habtemariam B, Bullock J, Bray JD, Hawes J, et al. . U S Food and Drug Administration approval: carfilzomib for the treatment of multiple myeloma. Clin Cancer Res 2013; 19:4559-63; PMID:23775332; http://dx.doi.org/ 10.1158/1078-0432.CCR-13-0755 [DOI] [PubMed] [Google Scholar]
- 29. Bhat UG, Halasi M, Gartel AL. FoxM1 is a general target for proteasome inhibitors. PLoS One 2009; 4:e6593; PMID:19672316; http://dx.doi.org/ 10.1371/journal.pone.0006593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yan W, Zhang Y, Zhang J, Liu S, Cho SJ, Chen X. Mutant p53 protein is targeted by arsenic for degradation and plays a role in arsenic-mediated growth suppression. J Biol Chem 2011; 286:17478-86; PMID:21454520; http://dx.doi.org/ 10.1074/jbc.M111.231639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Radhakrishnan SK, Gierut J, Gartel AL. Multiple alternate p21 transcripts are regulated by p53 in human cells. Oncogene 2006; 25:1812-5; PMID:16261158; http://dx.doi.org/ 10.1038/sj.onc.1209195 [DOI] [PubMed] [Google Scholar]
- 32. An WG, Chuman Y., Fojo T., Blagosklonny MV. Inhibitors of transcription, proteasome inhibitors, and DNA-damaging drugs differentially affect feedback of p53 degradation. Exp Cell Res 1998; 244:54-60; PMID:9770348; http://dx.doi.org/ 10.1006/excr.1998.4193 [DOI] [PubMed] [Google Scholar]
- 33. Blagosklonny MV. p53 from complexity to simplicity: mutant p53 stabilization, gain-of-function, and dominant-negative effect. FASEB J 2000; 14:1901-7; PMID:11023974; http://dx.doi.org/ 10.1096/fj.99-1078rev [DOI] [PubMed] [Google Scholar]
- 34. Aldoss I, Mark L, Vrona J, Ramezani L, Weitz I, Mohrbacher AM, Douer D. Adding ascorbic acid to arsenic trioxide produces limited benefit in patients with acute myeloid leukemia excluding acute promyelocytic leukemia. Ann Hematol 2014; PMID:24906216 [DOI] [PubMed] [Google Scholar]
- 35. Gartel AL. Thiostrepton, proteasome inhibitors and FOXM1. Cell Cycle 2011; 10:4341-2; PMID:22134246; http://dx.doi.org/ 10.4161/cc.10.24.18544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Halasi M, Gartel AL. A novel mode of FoxM1 regulation: positive auto-regulatory loop. Cell Cycle 2009; 8:1966-7; PMID:19411834; http://dx.doi.org/ 10.4161/cc.8.12.8708 [DOI] [PubMed] [Google Scholar]
- 37. Radhakrishnan SK, Bhat UG, Hughes DE, Wang IC, Costa RH, Gartel AL. Identification of a Chemical Inhibitor of the Oncogenic Transcription Factor Forkhead Box M1. Cancer Res 2006; 66:9731-35; PMID:17018632; http://dx.doi.org/ 10.1158/0008-5472.CAN-06-1576 [DOI] [PubMed] [Google Scholar]
- 38. Major ML, Lepe R, Costa RH. Forkhead box M1B transcriptional activity requires binding of Cdk-cyclin complexes for phosphorylation-dependent recruitment of p300/CBP coactivators. Mol Cell Biol 2004; 24:2649-61; PMID:15024056; http://dx.doi.org/ 10.1128/MCB.24.7.2649-2661.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]