

Recognition of mRNA transcripts by RNA binding proteins plays essential roles at various steps of gene expression, including splicing, translation, stability and general mRNA metabolism. Also the biogenesis and activity of non-coding RNAs (miRNAs, siRNAs, lncRNAs) and other regulatory RNA molecules depends on protein interactions. Cis elements, i.e. short RNA sequence motifs, in the mRNA are often degenerate and of low sequence complexity, for example, poly-(U) motifs that consist of a stretch of uridines.1 Recent genome-wide studies start to provide a catalog of cis regulatory elements to which RNA binding proteins (RBPs) are bound.2 In parallel, the repertoire of trans-acting factors, i.e., RNA binding proteins is being established using high-throughput approaches.3 Although it has been found that many so far unknown RNA binding proteins (RBPs) may exist, most RNA interactions rely on a limited number of RNA binding domains (RBDs).3 It is still poorly understood how this limited set of RBDs can achieve sufficient specificity to unambiguously identify and bind to specific cis elements within the RNA and thereby regulate distinct processes (Fig. 1).
Figure 1.
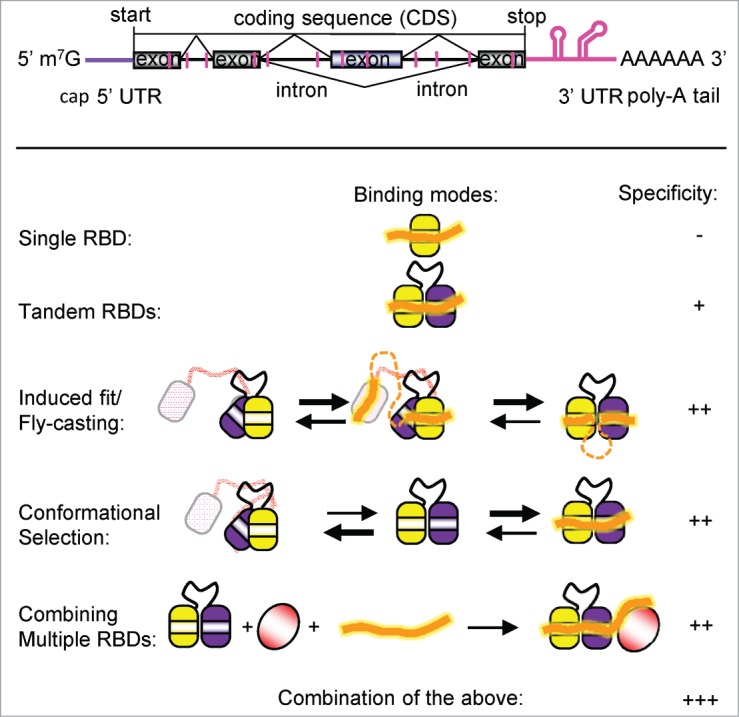
Recognition of mRNA by RNA binding proteins (RBPs). Top: A schematic view of a pre-mRNA indicating introns, exons, the poly(A)-tail, the untranslated regions (UTRs), and cis elements (pink). Bottom: Different binding modes how regulatory RBPs can bind to poly(U) cis elements. A combination of these modes yields the highest binding specificity.
Structural biology has provided unique and detailed insight into protein-RNA interactions. However, much of our structural knowledge about the recognition of cis RNA elements is based on structures of single domains bound to short oligonucleotides comprising 3–7 nucleotides (Fig. 1). These structures are useful to confirm and identify the cis element consensus motifs, but cannot explain how a given cis element is discriminated from others. Structural studies of tandem RNA binding domains bound to longer RNA sequences have shown how the code of RNA recognition can be expanded by combining multiple RNA binding domains (RBD). By recognizing longer (6–10 nucleotides) RNA sequences, tandem domains can achieve enhanced binding specificity and selectivity4 (Fig. 1). When combining 2 or more RBDs, cooperative effects and structural arrangements can be exploited for RNA recognition. For example, tandem domains may interact and present an extended RNA binding surface to induce a specific conformation in the single-stranded RNA ligand, as has been suggested for the looping-out of exons after RNA binding by PTB5 (Fig. 1). Alternatively, tandem domains may adopt an inactive closed arrangement where binding to a cognate RNA ligand induces an active, open domain arrangement6 (Fig. 1). The rearrangement can thus function as a rheostat that measures RNA binding affinity and avoid non-productive binding to non-cognate RNA motifs.6
Different arrangements of tandem RBDs (e.g. RNA recognition motifs – RRMs, or KH domains) have been found to bind to similar target sequences. For instance, trans-acting RNA binding proteins such as U2AF65, TIA-1, SXL, CPEB, and hnRNP C, are all able to recognize poly-(U) sequences of similar length and composition.1 In order to achieve specific recognition and functional readouts more complex and extended RNA sequence motifs need to be recognized. One possibility is to combine multiple RNA binding motifs to recognize an extended RNA sequence and form a specific cooperative complex (Fig. 1). We recently reported an example where extraordinary RNA binding cooperativity is achieved by combination of multiple RBDs. This complex plays a crucial role in translational regulation of dosage compensation in Drosophila. Two RRM domains of Sex-lethal (SXL), and the first of 5 cold shock domains (CSD1) of Upstream-of-N-Ras (UNR) sandwich the msl2 mRNA and form a specific complex to prevent translation of the transcript.7 Altogether 16 nucleotides of the mRNA are involved in ternary complex formation, and the affinity of both proteins for the RNA dramatically increases in the complex: fold10- for Sxl and ∼1000-fold for CSD1. Interestingly, Sxl alone has been shown previously to bind to a poly-(U) stretch in the transformer pre-mRNA where this interaction is involved in regulation of pre-mRNA splicing. Thus, depending on the presence of an additional component (UNR), recognition of poly-(U) stretches by SXL has distinct biological outcomes. The extended 16 nt RNA sequence recognized by SXL and UNR is present only twice within the complete 5′ and 3′ UTRs of msl2 mRNA, compared to the abundant presence of poly-(U) motifs in this transcript. In fact, the UNR CSD1 by itself is rather promiscuous and binds various short RNA sequences with similar affinities. Rather, SXL confers RNA binding specificity to CSD1. The cooperative RNA recognition by SXL-UNR also depends on unique protein-protein contacts within the ternary complex, which involve residues that are not present in all RRM and CSD domains. These residues are highly conserved throughout different phyla and are likely to mediate similar interactions in proteins of other species, even though these other species may not share the dosage compensation system of fruit flies.
The combination and cooperative RNA recognition by multiple RBDs represents a paradigm for how general and abundant RNA binding proteins may specifically identify their cis elements in extended single-stranded RNAs and thus regulate specific processes. Structural biology will continue to uncover the molecular principles underlying the code of protein-RNA recognition, an essential undertaking to decipher the regulation of gene expression.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1. Ray D, et al, Nature 2013; 499:172; PMID:23846655; http://dx.doi.org/ 10.1038/nature12311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Huppertz I, et al, Methods 2014; 65:274; PMID:24184352; http://dx.doi.org/ 10.1016/j.ymeth.2013.10.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Castello A.et al, Cell 2012; 149:1393; PMID:22658674; http://dx.doi.org/ 10.1016/j.cell.2012.04.031 [DOI] [PubMed] [Google Scholar]
- 4. Daubner GM. et al, Curr Opin Struct Biol 2013; 23, 100; PMID:23253355; http://dx.doi.org/ 10.1016/j.sbi.2012.11.006 [DOI] [PubMed] [Google Scholar]
- 5. Oberstrass FC, et al, Science 2005; 309:2054; PMID:16179478; http://dx.doi.org/ 10.1126/science.1114066 [DOI] [PubMed] [Google Scholar]
- 6. Mackereth CD, et al., Nature 2011; 475:408; PMID:21753750; http://dx.doi.org/ 10.1038/nature10171 [DOI] [PubMed] [Google Scholar]
- 7. Hennig J, et al, Nature 2014; 515:287-90 epub ahead of print; PMID:25209665; http://dx.doi.org/ 10.1038/nature13693 [DOI] [PubMed] [Google Scholar]