

Cellular senescence is a stress response that reveals itself as a multifaceted player in tumor suppression, tissue repair, aging, and cancer therapy.1 By definition, cellular senescence is considered a form of proliferative cell death, but unlike truly dying apoptotic or necrotic cells, senescent cells persist in vivo and their accumulation can perturb tissue functions. This is particularly relevant in tissues affected by age-associated pathologies or damaged by cancer therapies like radiation and chemotherapy. Unfortunately, our knowledge about the precise role of residing senescent cells in tissues remains scarce, which reduces our ability to develop potential therapeutic interventions targeting the detrimental effects associated with senescence. This problem arises from 2 limitations. First, traditional strategies involving the genetic abrogation of key senescence genes in mice trigger cancer, limiting their potential to recapitulate long-term senescence-associated phenotypes such as aging. For example, successful attempts at preventing cellular senescence in animal models via the genetic inactivation of essential senescence/tumor suppressor genes like p53 or p14ARF/p16INK4A (CDKN2A loci) have led to rapid secondary increases in cancer rates.2 Second, and most importantly, it is still extremely difficult to detect relatively rare senescent cells in tissues. This last limitation is now rapidly being overcome as recent powerful tools have emerged to facilitate real-time visualization3-5 and spatio-temporal targeted elimination of senescent cells in tissues.5,6
Cellular senescence was originally defined as the replicative exhaustion of normal cells in culture leading to a permanent growth arrest in the G0-G1 phase of the cell cycle. Replicative senescence is caused either via telomere shortening or occurs prematurely in response to stresses such as oxidation and excessive mitogenic signals. Similarly, strong pro-mitogenic oncogenes like Ras trigger OIS (oncogene-induced senescence), which has been observed both in vitro and in vivo in pre-neoplastic lesions, and act as a barrier to cancer progression.1 Overall, it is now clear that senescence supress neoplastic transformation. Besides the prototypical senescence-associated proliferation arrest, senescent cells develop a paracrine senescence-associated secretory phenotype (SASP), which includes the secretion of extracellular matrix altering proteases, cytokines and growth factors. The SASP is a tool that senescent cells use to communicate with, and manipulate, their immediate microenvironment. Importantly, the SASP can have beneficial or detrimental effects that are context dependent. For example, during liver injury repair, the genes p53 and p14ARF/p16INK4A promote the senescence of activated hepatic stellate cells, which then produce a SASP that coordinates immune-mediated resolution of the liver fibrosis.2 Alternatively, detrimental effects of the SASP are well described, such that it can create microenvironments that promote cancer progression.1
In light of this evidence, is there a need for new senescence-centric models? The classic strategies used earlier have not enabled a direct and definitive link between senescent cells themselves and observed tissue phenotypes. In other words, in the absence of dedicated models, the direct impact of tissue resident senescent cells and their SASP could remain mostly coincidental or correlative.
The new tissue senescence-tracking mouse models are simply based on the coupling of reporter-killer genes to the only known senescence-specific promoter, that of the p16INK4A gene product, an alternatively spliced transcript produced from the CDKN2A loci. The first 2 models are similar, having coupled luciferase reporter genes to the p16INK4A promoter, allowing for real-time visualization of senescent cells in tissues (Fig. 1A). These senescence-associated (SA)-reporters have elegantly confirmed the accumulation of senescent cells during aging and in damaged tissues.3,4 They also revealed that senescence is systematically induced in tissues harbouring an emerging neoplastic lesion.3 The consequences of this activation remain to be explored but the observation of this phenomena would have been difficult using conventional models.. The next model is conceptually different. It allows visualization via GFP fluorescence and targeted elimination of tissue resident senescent cells in a controlled manner (Fig. 1B). In this model, an inducible caspase-like killer transgene is activated via dimerization following the addition of a small molecule ligand (AP20187). Activation of the SA-killer triggers apoptosis only in senescent cells essentially resulting in their clearance. Incredibly, elimination of senescent cells was sufficient to delay age-associated phenotypes in a mouse model of premature aging, demonstrating without doubt and for the first time that resident senescent cells in tissues can have detrimental effects.6 Whether the accumulation of senescent cells is also responsible for tissue phenotypes associated with natural healthy aging remains to be shown in future studies.
Figure 1.
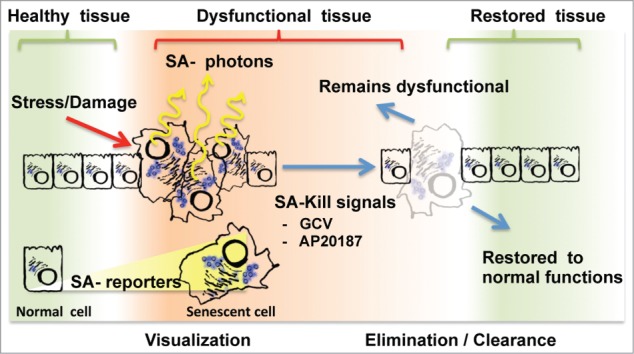
Current strategies for senescence visualization and manipulation in tissues. The accumulation of senescent cells in healthy tissues following chronic stress or acute damage is pictured from left to right. Alternatively, dysfunctional tissues can be restored to health following senescent cells elimination/clearance (pictured on the far right). To visualize senescent cells (left), the new models use senescence-associated (SA) luminescent reporters that will emit SA-photons suitable for tissue imaging. To eliminate/clear senescent cells (right), SA-Kill signals like the small molecules GCV and AP20187 specifically induce apoptosis in tissue resident senescent cells. Following senescent cell elimination, it is expected that some, but not all, tissues will revert to health.
Similarly, the last and most recent model targets a trimodality reporter (3MR) to senescent cells. The 3MR combines both red fluorescence and luciferase activity with the herpes simplex virus thymidine kinase (HSKtk), allowing multi-modal SA-reporter imaging via fluorescence, luminescence, or nuclear medicine tracers. Exploiting the 3MR, mitochondrial DNA damage-induced apoptosis can be specifically triggered in senescent cells via exposure to the synthetic nucleoside analog ganciclovir (GCV), which becomes toxic in the presence of the SA-killer HSVtk5,7 (Fig. 1B). Using this system, the authors demonstrate that senescent cells and their SASP are essential to coordinate myofibroblast activities during skin wound repair.5 These findings define for the first time that senescent cells themselves have beneficial context-dependent effects.
In summary, the spatio-temporal detection and the targeted elimination of senescent cells is now a real and useful tool to investigate the impact and functions of these cells in the context of complex organs and pathologies. Obviously, this will be invaluable in the future if we want to selectively target beneficial and detrimental effects of senescence using pharmaceutical approaches.
References
- 1.Rodier F, et al.. J Cell Biol 2011; 192:547-56; PMID:21321098; http://dx.doi.org/ 10.1083/jcb.201009094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Krizhanovsky V, et al.. Cell 2008; 134:657-67; PMID:18724938; http://dx.doi.org/ 10.1016/j.cell.2008.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Burd CE, et al.. Cell 2013; 152:340-51; PMID:23332765; http://dx.doi.org/ 10.1016/j.cell.2012.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yamakoshi K, et al.. J Cell Biol 2009; 186:393-407; PMID:19667129; http://dx.doi.org/ 10.1083/jcb.200904105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Demaria M, et al.. Dev Cell 2014; 31:722-33; PMID:25499914; http://dx.doi.org/ 10.1016/j.devcel.2014.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baker DJ, et al.. Nature 2011; 479:232-6; PMID:22048312; http://dx.doi.org/ 10.1038/nature10600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Laberge RM, et al.. Cell Death Dis 2013; 4:e727; PMID:23868060; http://dx.doi.org/ 10.1038/cddis.2013.199. [DOI] [PMC free article] [PubMed] [Google Scholar]