

Abstract
The transcription mechanism(s) of renal cell matrix accumulation in diabetes does not explored. Phosphorylation of the transcription factor cAMP-responsive element binding protein (CREB) significantly increased in cells treated with high glucose (HG) compared to cell grown in normal glucose (NG). Cells pretreated with rapamycin before exposure to HG showed significant decrease phosphorylation of CREB, increase in AMPK activity and decrease protein/mRNA and promoter activity of fibronectin. In addition, cells transfected with siRNA against CREB showed significant increase in AMPK activity, decrease in protein/mRNA and promoter activity of fibronectin. Cells treated with HG showed nuclear localization of p-CREB while pretreated cells with rapamycin reversed HG effect. Moreover, gel shift analysis shows increase binding of CREB to fibronectin promoter in cells treated with HG while cells pretreated with rapamycin reversed the effect of HG. Furthermore, db/db mice treated with rapamycin showed significant increase in AMPK activity, decrease in expression of p-CREB and protein/mRNA of fibronectin. Strong staining of fibronectin and p-CREB was detected in kidney cortex of db/db mice while treated mice with rapamycin reversed hyperglycemia effect. In summary, our data provide a novel mechanism of transcriptional regulation of fibronectin through CREB that may be used as therapeutic approach to prevent diabetes complications.
Keywords: AMPK, cell matrix, CREB, fibronectin, renal cells, transcription
Introduction
Renal hypertrophy, matrix protein accumulation and tubulointerstitial fibrosis are major pathological features of diabetic nephropathy (DN) and contribute to the eventual decline in glomerular filtration rate in humans and experimental models of DN.1-3 Proximal tubular epithelial cells exposed to high glucose concentrations undergo hypertrophy4 and increased matrix proteins accumulation.5,6 In vitro studies in cultured tubular epithelial cells and in vivo studies in diabetic kidney cortex have demonstrated that PI3K/Akt pathway is activated while the AMPK pathway is inactivated by high glucose concentration and by hyperglycemia.7
AMPK is the primary energy sensor in cells. Upon depletion of intracellular ATP, AMP levels rise and bind to an AMPK regulatory subunit. AMP binding places AMPK in a conformation that makes it accessible to its upstream activating kinase LKB1, which activates AMPK via phosphorylation of its catalytic subunit at Thr.1728,9 AMPK is inactivated in diabetes and this inactivation contributes to tuberin inactivation and impaired ability to act as Rheb-GAP.10 One critical downstream target of mTOR is pS6 kinase. Activation of S6K and its subsequent phosphorylation of ribosomal protein S6 are required for biosynthesis of the cellular translational apparatus, a critical component of cell growth. S6K phosphorylates downstream substrates such as ribosomal protein S6 and eIF4B to promote mRNA translation.11-14 The central role of S6K and ribosomal protein S6 has been further demonstrated by the use of rapamycin, a macrolide that specifically and directly inhibits the mammalian target of rapamycin (mTOR), an obligated upstream activator of pS6K15,16 which results in 4E-BP1 phosphorylation and increased expression of eIF4E. While mTOR signaling is best characterized for its role in regulation of translation, recent evidence suggests that the mTOR pathway also controls the transcription of several genes.17 Microarray analyses using rapamycin-treated mammalian cells demonstrate that mTOR signaling regulates transcription of numerous genes, particularly those involved in metabolic and biosynthetic pathways.18 In the steady state, the vast majority of cellular mTOR is found in the cytoplasm but a cytoplasmic-nuclear shuttling behavior for mTOR has been identified19,20 indicating a nuclear function for mTOR. Several lines of evidence have implicated the direct involvement of mTOR and its downstream S6K in the regulation of transcription.
The mechanism(s) of transcription regulation of cell matrix proteins in diabetes does not characterized. While strict metabolic control prevents many of the complications of diabetes, normoglycemia is often difficult to achieve and is fraught with complications. Therefore, understanding the mechanism by which glucose exerts its deleterious effects through accumulation of renal cell matrix proteins will help design adjunct therapy to prevent/treat diabetic complications.
Results
Inhibition of AMPK activity by HG is associated with increase phosphorylation of CREB and promoter activity/protein expression of fibronectin
To determine the effect of AMPK on regulation of CREB and fibronectin expression, MCT cells were treated with HG for different time points (4,8,12 and 24hrs) as well as cells pretreated with rapamycin (20nM) before expose to HG at all time points. Protein were extracted from all cells at all time points and subjected to Western blot analysis. Cells treated with HG (25mM) showed significant decrease in AMPK activity, increase phosphorylation of CREB and increase in protein expression of fibronectin at all time points (Fig. 1A–L) Cells pretreated with rapamycin before expose to HG showed significant increase in AMPK activity, decrease phosphorylation of CREB and decrease protein expression of fibronectin (Fig. 1A–L) Maximum effect of rapamycin on upregulation of AMPK activity, decrease p-CREB and decrease protein expression of fibronectin was detected at 24hrs of treatment (Fig. 1H, J, L) To test the effect of HG and rapamycin on regulation of promoter activity of fibronectin, 293 cells were transfected with luciferase reporter plasmid containing the fibronectin promoter. Luciferase activity of fibronectin was measured in cells treated with HG or pretreated with rapamycin at all time points. No significant differences was detected in promoter activity of fibronectin in cells treated with HG or pretreated with rapamycin compared to cells grown in NG at 4h and 12h (Fig. 1M) While cells treated with HG at 8h only showed significant increase compared to cells grown in NG. The data also showed significant increase in promoter activity of fibronectin in cell treated with HG for 24h (Fig. 1M) while sharp decrease in promoter activity of fibronectin was detected in cells pretreated with rapamycin for 24h (Fig. 1M) These data suggests that activation of AMPK by rapamycin is sufficient to decrease CREB phosphorylation and to decrease the promoter activity and protein expression of fibronectin in cells under exposure to high glucose to the maximum at 24 hr.
Figure 1.
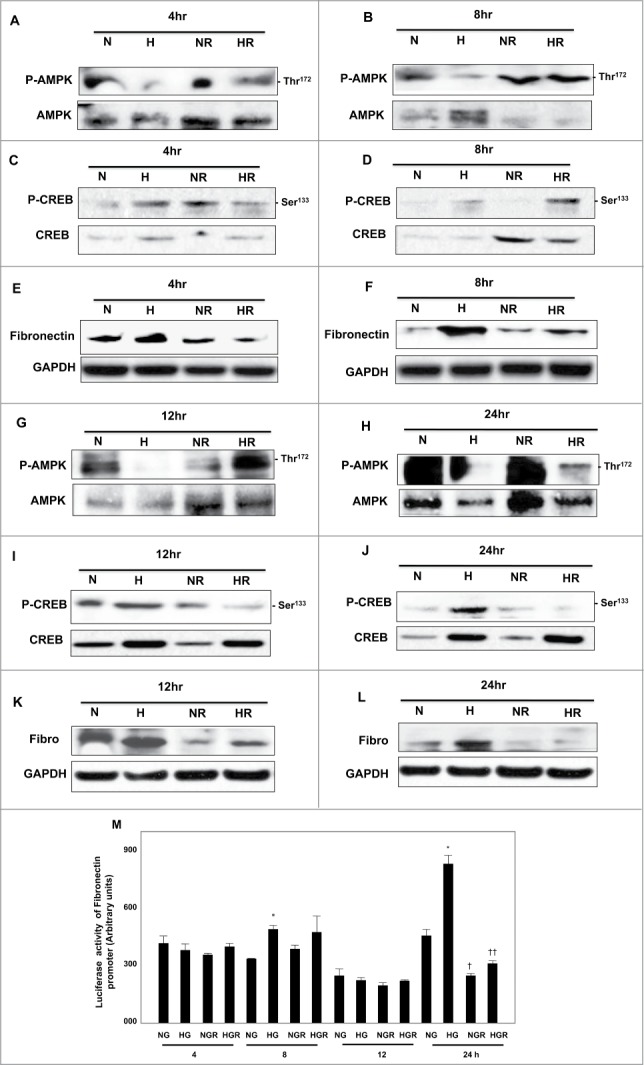
Rapamycin activates AMPK and decreases phosphorylation of CREB at Ser133 resulting in decreased protein expression and promoter activity of fibronectin in HG-treated renal proximal tubular cells. High glucose significantly decreased AMPK phosphorylation (A, B, G, H), increased phosphorylation of CREB at Ser133 (C, D, I, J) and protein expression of fibronectin (E, F, K, L) at all time points (4–24hrs). (M) A reporter plasmid containing the fibronectin promoter driving expression of the luciferase and a control Renilla reporter gene were co-transfected into the cells using LipofectAMINE Plus Reagent™. Rapamycin pretreatment reversed the increase effect of HG on fibronectin promoter activity was detected only on 24 hr. Experiment represent means±SE (n=6). Significant difference from cells grown in normal glucose is indicated by *P<0 .01, cells grown in NG and pretreated with rapamycin by †P<0 .01 and cells exposed to HG and treated with rapamycin compared to cells grown in HG by ††P<0 .01.
AMPK is a key kinase in regulating of fibronectin
To explore the effect of AMPK in regulating fibronectin, cells were transfected with DN-AMPK or treated with compound C (AMPK inhibitor) then treated with HG for 24 h. Data in Figure 2A, B showed that downregulation of AMPK using DN-AMPK in cells grown in NG or treated with HG resulted in significant increase in phosphorylation of CREB at Ser133 and significant accumulation of fibronectin protein expression under both NG and HG conditions. In addition, cells transfected with DN-AMPK then transfected with luciferase reporter plasmid containing the fibronectin promoter showed significant increase in fibronectin promoter activity compared to cells without DN-AMPK (Fig. 2E) Moreover, cells grown in NG or HG and treated with compound C for 24h showed also increase in p-CREB and fibronectin protein expression (Fig. 2C, D) as well as significant increase in in fibronectin promoter activity (Fig. 2F) These data suggests that AMPK plays a major role in regulating fibronectin activity through CREB.
Figure 2.
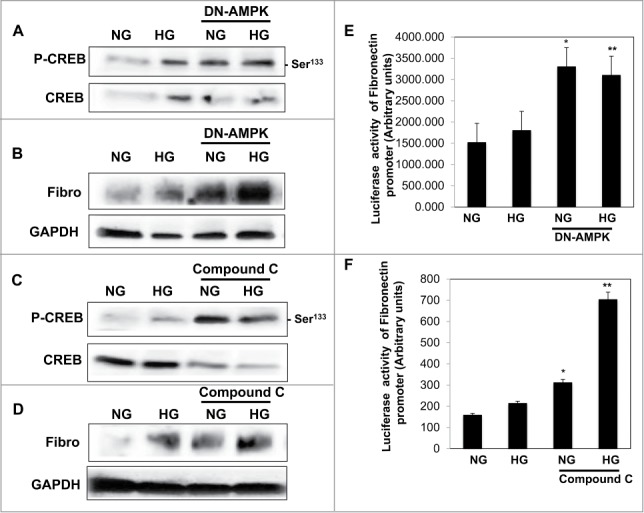
Downregulation or inhibition of AMPK resulted in increase phosphorylation of CREB at Ser133 and led to a significant increase in protein expression and promoter activity of fibronectin in renal proximal tubular cells treated with HG. (A) Cells grown in NG or exposed to HG then transfected with DN-AMPK for 24 h resulted in significant increase in (A) phosphorylation of CREB at Ser133 and protein expression of fibronectin. In addition, cells grown in NG or exposed to HG then treated with compound C for 24 h resulted in significant increase in (C) phosphorylation of CREB at Ser133 and (D) protein expression of fibronectin. (B) Cells treated with compound C or co-transfected with DN-AMPK, a reporter plasmid construct carrying the fibronectin promoter and a control Renilla reporter gene. Significant increased in fibronectin promoter activity detected in cells co-transfected with DN-AMPK (E) or treated with (F) compound C under low and high concentrations of glucose indicating the role of AMPK as a major kinase in regulating fibronectin through CREB. Experiment represent means±SE (n=6). Significant difference from cells grown in NG and transfected with DN-AMPK or treated with compound C is indicated by *P<0 .01, cells grown in HG and transfected with DN-AMPK or treated with compound C by **P<0 .01.
Downregulation of CREB significantly decreased mRNA/protein and promoter activity of fibronectin
To determine whether decreases mRNA and protein expression will be reflected on changes in fibronectin promoter activity through CREB, MCT cells were transfected with siRNA of CREB. Cells transfected with siRNA of CREB and pre-treated with rapamycin showed significant decrease in fibronectin expression compared to cells transfected with siRNA of CREB alone under NG and HG conditions (Fig. 3A–D) In addition, transcription regulation of fibronectin in cells transfected with siRNA of CREB and pre-treated with rapamycin showed abolish of mRNA of fibronectin expression compared to cells pretreated only with rapamycin (Fig. 3C, D) Moreover, cells transfected with siRNA of CREB and/or pretreated with rapamycin then transfected with luciferase reporter plasmid containing the fibronectin promoter showed sharp decrease in fibronectin promoter activity compared to cells pretreated only with rapamycin under NG and HG conditions (Fig. 3E) These data suggest that CREB is a major transcription factor involve in regulating fibronectin expression.
Figure 3.
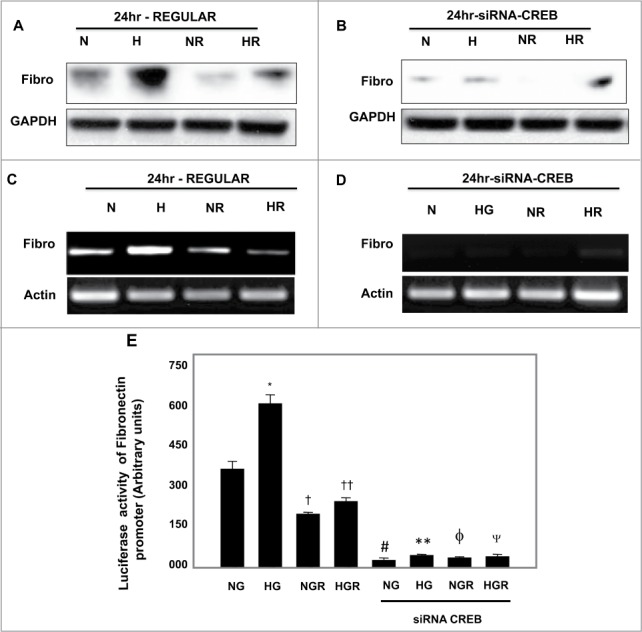
Downregulation of CREB by siRNA showed stronger effect on decrease protein, mRNA and promoter activity of fibronectin compared to rapamycin treatment in renal proximal tubular cells treated with HG. Cells pretreated with rapamycin showed significant decrease in (A) protein expression and (C) mRNA expression of fibronectin compared to cells exposed to HG. Transfected cells with siRNA against CREB resulted in (B) significant decrease in protein expression and (D) mRNA expression of fibronectin compared to cells pretreated with rapamycin. Sharp decrease in (B) protein and (D) mRNA of fibronectin was detected in cells transfected with siRNA of CREB and pretreated with rapamycin compared to cells pretreated with rapamycin under NG and HG conditions. (E) Cells co-transfected with siRNA against CREB, a reporter plasmid construct carrying the fibronectin promoter and a control Renilla reporter gene showed strong significant decrease in fibronectin promoter activity in cells grown under low and high concentrations compared to cells treated only with rapamycin. Experiment represent means±SE (n=6). Significant difference from cells grown in NG compared to cells in HG is indicated by *P<0 .01, cells grown in NG and pretreated with rapamycin by †P<0 .01 compared to cells grown in NG, cells exposed to HG and pretreated with rapamycin compared to cells grown in HG by ††P<0 .01. Cells transfected with siRNA of CREB and grown in NG by #P<0 .01 and in HG by **P<0 .0. Cells transfected with siRNA of CREB and pretreated with rapamycin grown in NG by ϕP<0 .01 and in HG by ΨP<0 .01.
siRNA of eiF4E and rapamycin significantly decreased protein/mRNA and promoter activity of fibronectin
In another set of experiments, we tested the effect of siRNA of eiF4E on protein, mRNA and promoter activity of fibronectin in cells grown in NG or treated with HG as well as cells pretreated with rapamycin. Cell lysates were collected for Western blot analysis. Cells transfected with siRNA of eiF4E and pretreated with rapamycin showed significant decrease in eiF4E and fibronectin expression compared to cells transfected with siRNA of eiF4E alone under NG and HG conditions (Fig. 4A–D) In addition, transcription regulation of fibronectin in cells transfected with siRNA of eiF4E and pretreated with rapamycin showed abolish of mRNA of fibronectin expression compared to cells transfected only with siRNA of eiF4E (Fig. 4E, F) Moreover, cells transfected with siRNA of eiF4E or pretreated with rapamycin then transfected with luciferase reporter plasmid containing the fibronectin promoter showed sharp decrease in fibronectin promoter activity compared to cells transfected only with siRNA of eiF4E under NG and HG conditions (Fig. 3G)
Figure 4.
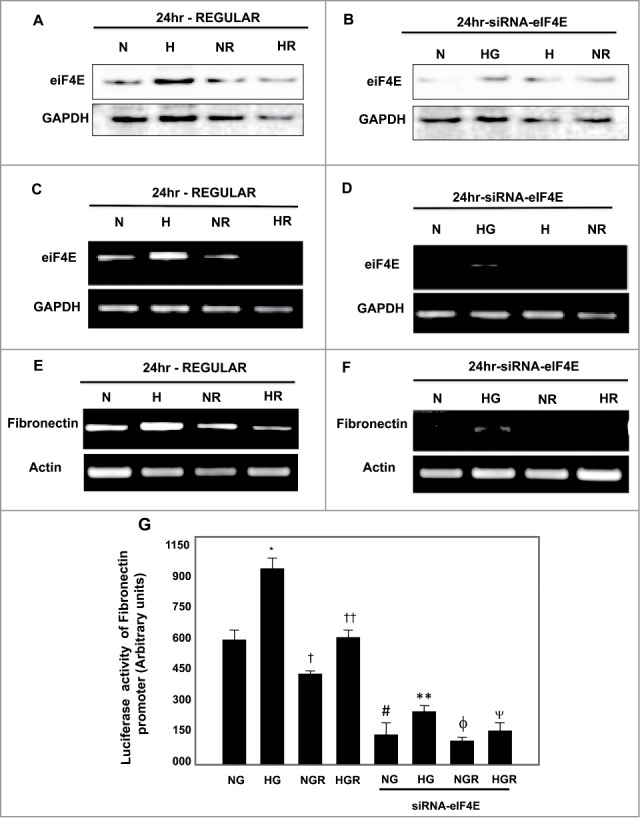
Downregulation of eIF4E by siRNA showed stronger effect on decrease protein, mRNA and promoter activity of fibronectin compared to rapamycin treatment in renal proximal tubular cells treated with HG. (A) Cells transfected with siRNA against eIF4E resulted in (B) decrease protein expression and sharp decrease in (D) mRNA of eIF4E compared to (A, C) cells treated only with rapamycin. In addition, downregulation of eIF4E resulted in strong decrease in mRNA of fibronectin compared to cells pretreated only with rapamycin under both conditions of NG and HG (E, F). Cells transfected with siRNA and pretreated with rapamycin hardly show any mRNA expression of fibronectin under both conditions of NG and HG. (G) Cells co-transfected with siRNA against eIF4E, a reporter plasmid construct carrying the fibronectin promoter and a control Renilla reporter gene showed significant decrease in fibronectin promoter activity in cells grown under low and high concentrations compared to cells treated only with rapamycin. Experiment represent means±SE (n=6). Significant difference from cells grown in NG compared to cells in HG is indicated by *P<0 .01, cells grown in NG and pretreated with rapamycin by †P<0 .01 compared to cells grown in NG, cells exposed to HG and pretreated with rapamycin compared to cells grown in HG by ††P<0 .01. Cells transfected with siRNA of eIF4E and grown in NG by #P<0 .01 and in HG by **P<0 .0. Cells transfected with siRNA of CREB and pretreated with rapamycin grown in NG by ϕP<0 .01 and in HG by ΨP<0 .01.
Rapamycin treatment results in redistribution of p-CREB from the nucleus to cytoplasm in MCT cells
Immunofluorescene staining was used to detect the localization of p-CREB in pretreated MCT cells with rapamycin (20 nM) for 24 hours under NG and HG conditions. Overlay staining of p-CREB and DNA staining, demonstrating nuclear localization of p-CREB in HG treated MCT cells while cytoplasmic staining of p-CREB was detected in cells grown under normal glucose condition (Fig. 5A, B) On the other hand, cells pre-treated with rapamycin before expose to HG showed majority of p-CREB staining within the cytoplasm (Fig. 5C, D) These data were further confirmed by Western blot analysis using cytoplasmic and nuclear fractions of MCT cells pre-treated with 20nm rapamycin (Fig. 5E, F) before exposure to HG. The redistribution of p-CREB localization by rapamycin indicating that activation of the mTOR pathway plays an important role in the activation of fibrosis signaling in MCT cells.
Figure 5.
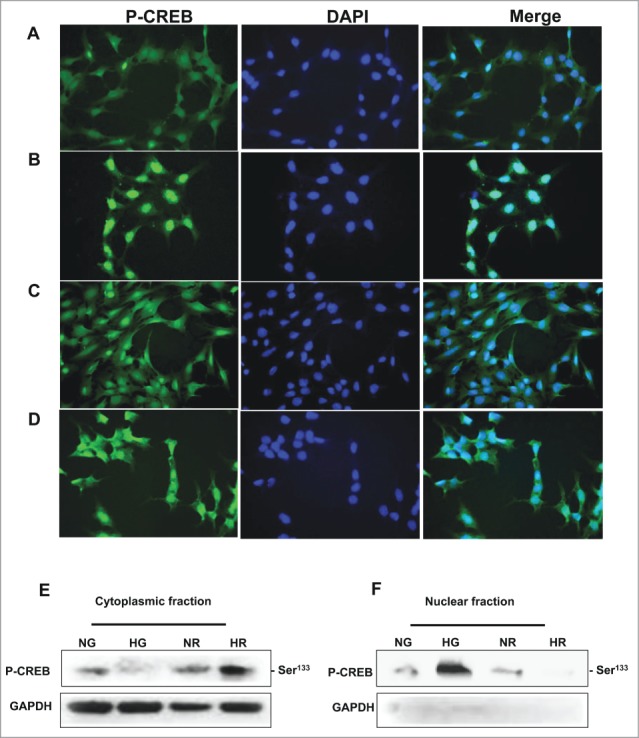
High glucose exposure redistributes p-CREB from cytoplasm into nucleus and rapamycin treatment reverses HG effects. Immunostaining and western blot analysis were used to detect the localization of p-CREB in cells grown in NG or exposed HG and treated with rapamycin. (A) p-CREB was mainly detected in cytoplasmic region of the cells grown in NG, while cells exposed to (B) HG showed a nuclear staining localization of p-CREB. Cells pretreated with rapamycin and grown in (C) NG or (D) HG showed the majority of p-CREB within the cytoplasmic area. FITC green signals for p-CREB was detected using a filter with an excitation range of 535 nm and DAPI blue signals for nuclear DNA using a filter with excitation at 488 nm. Merge of the p-CREB and DAPI confirm the localization of p-CREB within the cytoplasm or nuclear compartment. Cytoplasmic and nuclear fractionation confirmed the localization of p-CREB within (E, F) cytoplasmic fraction in cells grown in NG and in nuclear fraction in cells exposed to HG by Western blot. Cells pretreated with rapamycin showed the reverse of the localization of p-CREB indicating the role of mTOR in regulation CREB localization in renal cells.
CREB is a major transcription factor binds to the fibronectin promoter element
To further investigate the mechanism by which inhibition of mTOR affect the binding of CREB to DNA-binding site of fibronectin promoter, EMSA was performed using nuclear extracts of MCT cells grown in NG, exposed to HG, and pretreated with rapamycin. Intact double-strand end-labeled oligonucleotides covering the regions: −639 to −609 of the CREB that binds to the fibronectin promoter were used as a DNA probe (Fig. 6A) Labeled probe was incubated with nuclear extracts isolated from non-treated and treated MCT cells. Increase binding of CREB to fibronectin promoter element was detected in the nuclear extracts treated with HG compared to cells grown in NG (Fig. 6B) Pretreatment of MCT cells with 20nM rapamycin significantly reduced CREB to binds to fibronectin promoter element in the nuclear extracts compared to HG treated cells (Fig. 6B) To confirm the specificity of the protein-DNA interaction, the cell extracts were also pre-incubated with an antibody recognizing CREB (Active Motif, Carlsbad, CA). The DNA-protein complexes were significantly shifted in the presence of the CREB antibody indicating that CREB is indeed a component of these complexes (Fig. 6C) These data suggest that inhibition of mTOR results in regulation the binding of CREB to fibronectin promoter to block cell fibrosis in MCT cells.
Figure 6.
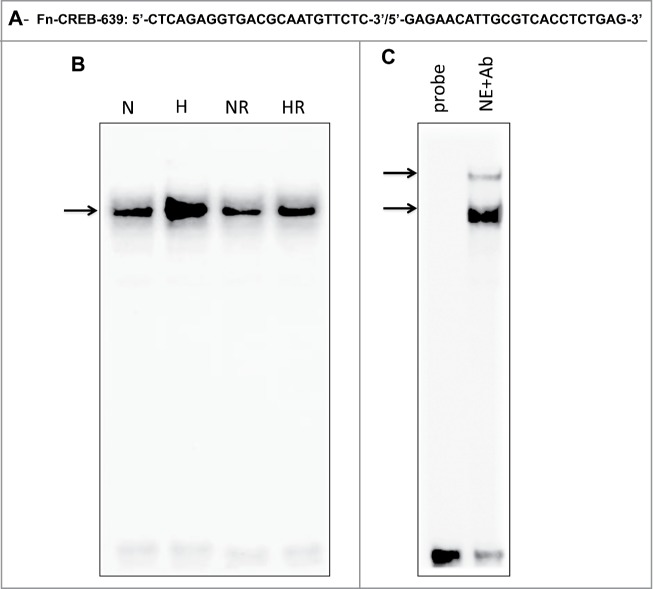
High glucose treatment significantly induces binding of CREB to the fibronectin promoter element and rapamycin treatment reversed the effect of HG in renal proximal tubular cells. (A) EMSA analysis of a DNA probe corresponding to the putative CREB binding site in the fibronectin promoter. Labeled probes were incubated with nuclear extracts isolated from MCT cells grown in NG or HG and treated or non-treated with rapamycin (20 nM). (B) Treatment of MCT cells with HG significantly induced binding of CREB to fibronectin promoter compared to cells grown in NG. While rapamycin pretreatment showed significant decreased in binding of CREB to fibronectin promoter in cells exposed to HG. (C) The specificity of binding of the DNA/protein complex to CREB was demonstrated by adding a CREB antibody (1ug) to the reaction mixture. Including the CREB antibody in the reaction results in partial shift of the DNA/protein complex and indicate the CREB is the component of the complex.
Activation of AMPK by rapamycin significantly reduced phosphorylation of CREB in kidney of diabetic mice
AMPK characterized as a protein activated by nutrient and bioenergetic stress that raises intracellular AMP and lowers ATP. Therefore, we demonstrated the role of CREB in regulation of AMPK, which lead to accumulate of cell matrix protein in diabetic mice. Diabetic mice treated with rapamycin showed a significant increase in AMPK activity compared to non-treated mice (Fig. 7A, B) To determine the role of rapamycin in regulation of p-CREB, Western blot and immunofluorescence staining were performed in kidney homogenates and kidney sections of non-treated and treated diabetic mice. Data in Figure 7C, D showed that treatment of diabetic mice with rapamycin significantly reduced the phosphorylation of CREB at Ser133 compared to non-treated mice. In addition, majority of staining of p-CREB showed within the nuclear compartment in kidney sections of diabetic mice (Fig. 7E) while treatment diabetic mice with rapamycin redistribute p-CREB from the nucleus to cytoplasm (Fig. 7F) These data were further confirmed by Western blot analysis using cytoplasmic and nuclear fractions of kidney homogenates from non-treated and treated mice (data not shown).
Figure 7.

Activation of AMPK by rapamycin significantly reduced phosphorylation of CREB and protein/mRNA of fibronectin expression in kidney of diabetic mice. (A, B) Representative Immunoblot analysis shows a significant increase in AMPK phosphorylation at Thr172 in kidney cortex of db/db mice treated with rapamycin compared to non-treated mice. Activation of AMPK resulted in significant decrease of (C&D) p-CREB in mice treated with rapamycin compared to non-treated mice. Actin was used as a loading control. (B&D) Histograms represent means±SE and significant difference from non-treated mice is indicated by **P<0 .01. FITC green signals for p-CREB and propidium iodide (PI) red signals for nuclear DNA showed the majority of staining of p-CREB within the nuclear compartment of kidney sections of diabetic mice (E), while treatment diabetic mice with rapamycin redistributed p-CREB from the nucleus to cytoplasm (F). Merge image showed the overlap of P-CREB with the cytoplasm and nuclear areas. (G, H) Diabetic showed significant increase in protein expression of fibronectin, while animals treated with rapamycin showed strong decrease in fibronectin expression by Western blot. These data further confirmed by immunoperoxidase staining of fibronectin in kidney sections from (I) non-treated and (J) rapamycin treated mice. (K, L) RT-PCR product of fibronectin showed that rapamycin treatment significantly decreased mRNA of fibronectin in kidney homogenate of diabetic mice compared to non-treated mice.
Downregulation of p-CREB is associated with significant decrease of mRNA and protein expression of fibronectin in kidney of diabetic mice
Protein and mRNA expression of fibronectin were measured in non-treated wand rapamycin-treated diabetic mice. Data in Fig. 7G, H showed significant decrease in protein expression of fibronectin in kidney homogenates from mice treated with rapmycin compared to non-treated mice by Western blot analysis. In addition, kidney sections of rapamycin-treated mice showed reduced in fibronectin immunoperoxidase staining compared to non-treated mice. Moreover, significant decrease in mRNA of fibronectin was detected by RT-PCR in kidney homogenate of treated mice compared to non-treated mice (Fig. 7K, L) These data suggests that CREB is a major downstream target of AMPK that involve in regulation of fibronectin. In addition, transcription regulation of fibronectin in mouse renal cells and kidney of mice showed through redistribution of p-CREB between nucleus and cytoplasmic compartment.
Discussion
This study provides the first evidence of transcriptional regulation of cell matrix protein in diabetes through regulation of CREB. Our data showed several lines of evidence that have implicated the AMPK/mTOR pathway on the transcriptional regulation of fibronectin through CREB. First, cells pretreated with rapamycin before exposure to HG showed significant increase in AMPK activity, decrease in phosphorylation of CREB and decrease in protein/mRNA/promoter activity of fibronectin. Second, cells transfected with siRNA against CREB showed significant increase in AMPK activity, decrease in protein/mRNA/promoter activity of fibronectin. Third, cells pretreated with rapamycin before exposure to HG showed redistribution of nuclear p-CREB to cytoplasm. Fourth, increase binding of CREB to fibronectin promoter element in cells treated with HG while cells pretreated with rapamycin reversed the effect of HG. In addition, in vivo data showed significant increase in AMPK activity, decrease in expression of p-CREB and decrease in protein/mRNA expression of fibronectin in db/db mice treated with rapamycin. In addition, redistribution of p-CREB from nuclear to cytoplasm was strongly associated with decrease in protein/mRNA of fibronectin expression in kidney cortex of db/db mice treated with rapamycin. Taken together, these data provide a novel transcriptional mechanism of regulation of fibronectin through CREB and added a novel role of AMPK/mTOR pathway in controlling cell matrix protein transcription in diabetes.
Microarray analyses using rapamycin-treated mammalian cells demonstrated that mTOR signaling regulates transcription of numerous genes, particularly those involved in metabolic and biosynthetic pathways 18 In the steady-state, the vast majority of cellular mTOR is found in the cytoplasm but a cytoplasmic-nuclear shuttling behavior for mTOR has been identified20 indicating a nuclear function for mTOR. Activation of S6K phosphorylates CREB and resulting in enhancement of their transcriptional activity. Of interest, these effects are blocked by rapamycin, implicating mTOR, the upstream activator of S6K. Immunofluorescence staining showed that treatment of diabetic mice with rapamycin significantly reduced the phosphorylation of CREB at Ser133 compared to non-treated mice. In addition, the majority of staining of p-CREB showed within the nuclear compartment in kidney sections of diabetic mice, while treatment diabetic mice with rapamycin redistribute p-CREB from the nucleus to cytoplasm confirming cell culture immunostaining and immunoblot of cytoplasmic and nuclear fractionation data. In addition, the decrease in CREB phosphorylation in cells pretreated with rapamycin before HG exposure was associated with significant decrease of mRNA and protein expression of fibronectin. The in vitro and in vivo data suggesting that CREB is a major transcription factor involve in regulation of fibronectin.
AMPK activity significantly reduced in kidneys from both diabetic mice and humans. Inhibition of AMPK activity by compound C or DN-AMPK resulted in significant increase in phosphorylation of CREB and promoter activity/protein expression of fibronectin in MCT cells. In addition, renal cells pretreated with rapamycin and diabetic mice treated with rapamycin showed a significant increase in AMPK activity and reduced the phosphorylation of CREB at Ser133 compared to non-treated cells and mice. Decrease CREB phosphorylation in cell culture and in db/db mice pretreated with rapamycin was associated with significant decrease in mRNA and protein expression of fibronectin suggesting that CREB is a major downstream target of AMPK that involve in regulation of fibronectin.
The decrease in fibronectin mRNA in MCT cells suggests that decreased transcription is one potential mechanism responsible for downregulation of fibronectin protein. Our data provided strong role AMPK/mTOR pathway in the transcriptional regulation of fibronectin through CREB. Cells pretreated with rapamycin showed significant decrease in promoter activity of fibronectin. In addition, cells pretreated compound C or transfected with DN-AMPK before exposure to HG (upstream kinase of mTOR) showed significant increase in promoter activity of fibronectin. Moreover, cells transfected with siRNA against CREB showed significant increase in promoter activity of fibronectin. Data from previous report showed that serum-stimulated 293 cells to activate the upstream target of mTOR/Akt/PKB, which potently induced phosphorylation of CREB at Ser133 while inhibition of PI-3K by LY 294002 decreased CREB phosphorylation suggesting that CREB may contribute importantly to cell survival in response to growth factor stimulation.21 In addition, pharmacologic inhibition of protein kinase C (PKC) and protein kinase A (PKA) that involved in hexosamine-mediated matrix production, blocked fibronectin synthesis under high glucose and glucosamine conditions in mesangial cells. The hexosamine-induced fibronectin protein synthesis in mesangial cells was associated with increases in CREB phosphorylation through activation protein kinases A and C.22
Two oligos that covered 2 regions of the CREB that binds to fibronectin promoter was tested by EMSA. First oligo covered a region from −352 to −322 (data not shown) and second oligo from −639 to −609 of CREB that binds to the fibronectin promoter. The second oligo only showed a strong binding to DNA-protein complex under HG condition while rapamycin treatment significantly reduced its binding to fibronectin promoter element in the nuclear extracts. The DNA-protein complexes were significantly shifted in the presence of the CREB antibody indicating that CREB is indeed a component of these complexes and providing a novel role of CREB in regulating fibronectin promoter to block cell fibrosis in renal cells and confirm the role of mTOR in decrease the binding CREB to DNA–protein complex of fibronectin.
In summary, these data showed a novel mechanistic role of CREB in the regulation of fibronectin under hyperglycemia condition in kidney of dbdb mice and in renal cells treated with HG. These data also support the role of AMPK activation and mTOR inhibitor in decreasing the phosphorylation of CREB and reduced the transcriptional activity and subsequently the translational activity of fibronectin (Fig. 8) The data provided a strong evidence of the role CREB as a major transcription factor controls cell matrix protein accumulation in diabetic kidney that may be used as therapeutic approach to prevent development and progression of early and advanced manifestations of diabetic nephropathy.
Figure 8.
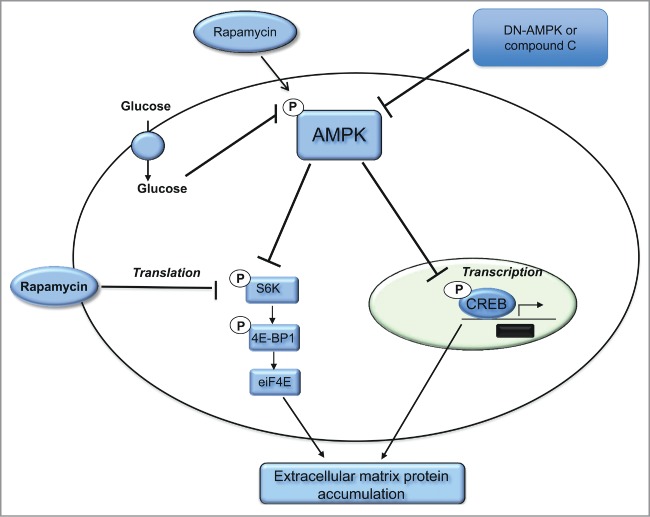
Proposed model of role of CREB in regulating the transcriptional activity of fibronectin in diabetic kidney. Activation of AMPK and inhibition of mTOR by rapamycin resulted in decrease phosphorylation of CREB at Ser133 and prevent the shuttling of the p-CREB to the nucleus to bind to fibronectin and activate its function under HG condition. In addition, inactivation of AMPK by DN-AMPK or compound C enhances mTOR activation to increase the translation of fibronectin through increase eIF4E as well as increase the transcriptional activity of fibronectin through enhances CREB phosphorylation. The consequences steps of translation and transcription of fibronectin resulted in significant accumulation of cell matrix protein in diabetic kidney, which is the major pathological feature of diabetic nephropathy.
Materials and Methods
Cell culture
The murine proximal tubular epithelial (MCT) cells were grown in DMEM containing 10% fetal bovine serum, 5-mmol/l glucose, 100-units/ml penicillin, 100μg/ml streptomycin, and 2mmol/l glutamine. Confluent cells were growth-arrested overnight in serum-free DMEM before experiments.
Cells treatment
MCT cells were grown to 80–90% confluency in 60 mm petri dish in normal glucose (NG) (5mM) or Hg (25mM). Cells were treated with rapamycin (20nM) for 4, 8, 12 and 24h or compound C (10μM) before exposed to HG for 24 h. Rapmaycin was obtained from Cayman Chemical (Ann Arbor, MI) and compound C from Abcam (Cambridge, MA). The cells were lysed in a lysis buffer as described previously.23 Cell lyastes were used for Western blot or promoter activity analysis.
Downregulation of AMPK
HEK 293 cells grown to 60–70% confluency in 6-well plates were transfected with a recombinant plasmid expressing DN-AMPK. The plasmid containing AMPK carrying K45R mutation of the α1-subunit (pCAGGS) was transfected into the cells using lipofectamine and Lipo-plus reagent (Invitrogen) as described previously.24 Cells were treated with HG (25mM) 24h before harvesting for Western blot or promoter activity analysis.
Protein extraction and immunoblot analysis
Protein concentration of the cell lysates or kidney cortex homogenates was determined with the Bradford reagent using bovine serum albumin as a standard.25 Western blot analysis was performed as previously described.23 P-AMPK, AMPK, p-CREB, CREB, eIF4E and fibronectin antibodies were purchased from Cell Signaling Technology. GADPH antibody was obtained from Santa Cruz Biotechnology. Expression of each protein was quantified by densitometry using NIH Image 1.62 software.
Transcriptional activity of fibronectin promoter
A luciferase reporter plasmid containing the fibronectin promoter was used to determine fibronectin gene transcription activity.12 MCT cells were grown in 12-well plates to 60–70% confluence, then co-transfected with both renilla and fibronectin plasmid covering the region of CREB that binds to fibronectin promoter using oligofectamine and Plus Reagent as described previously.12 Cells were treated with HG (25mM) and/or rapamycin (20nM) for 4, 8, 12 and 24 hours or compound C for 24h before harvesting the cells for luciferase assay. In addition, siRNA of CREB and siRNA of eIF4E as well as DN-AMPK were transfected into the cells for 24h before treatment with HG. Luciferase activity of fibronectin was determined using Dual-Luciferase Reporter Assay System in a luminometer (Promega, WI) and normalized to Renilla luciferase.
Downregulation of CREB and eIF4E by siRNA
MCT cells were grown in 6-well plates in NG medium. Selected siRNA duplexes against CREB and eIF4E and control siRNA were obtained as a kit from Santa Cruz Biotechnology (Santa Cruz, CA). Cells were transfected with specific siRNA of CREB or eIF4E or nonspecific siRNA duplexes (control) as described previously.24 Cells were treated with HG for 24 hours before harvesting for Western blot analysis.
Quantitation of mRNA by RT-PCR
RNA was extracted from MCT cells using RNA Isolation Solvent (Tel-Test) and from kidney homogenates using RNeasy Mini Kit (Qiagen, CA). RNA was quantified by spectrophotometer at 260 nm, and its integrity was tested by formaldehyde/agarose gel electrophoresis. RTPCR was performed as previously described24 using the primers (5′-GGAGAGCAAGCCTGAACCTGAAGAACC-3′/5′CCTGGTGTCCTGATCATTGCAC) for fibronectin and 5′-CCTTCCCTGAAGGTTCCTCC-3′/5′-TGCTATCTTATCACCTTTAG-3′ for eIF4E. The amplified product was 356-bp long for fibronectin and 425-bp for eIF4E.. For GAPDH, an internal control of amplification, upstream/reverse primers were 5′- GCCACCCAGAAGACTGTGGAT/5′- GAAGGCCATGCCAGTGAGCT synthesising a 528-bp product. The PCR products were analyzed by electrophoresis on agarose gels and ethidium bromide staining.
Immunofluorescence staining of p-CREB in MCT cells
A double fluorescence labeling method was used as described previously with minor modifications.26 MCT cells plated in chamber slides were serum-starved for overnight then treated with 25 mM glucose (HG) or HG and rapamycin for 24 hours. The cells were washed with PBS, fixed, and incubated with rabbit antibody against p-CREB antibody, followed by incubated with anti-rabbit IgG (1:200) conjugated with FITC. The cells were reacted with Vectashield Mounting Medium with 4′,6-diamidino-2-phenylindole (DAPI) for DNA staining. p-CREB was identified by the primary monoclonal antibody and FITC conjugated secondary antibody. FITC green signals for p-CREB was detected using a filter with an excitation range of 535 nm and DAPI blue signals for nuclear DNA using a filter with excitation at 488 nm. FITC and DAPI were detected using Nikon Research microscope equipped for epifluorescence with excitations and band pass filters. To show staining specificity, control cells were stained without primary antibody.
Cells fractionation
Cytoplasmic and nuclear fractions were extracted from MCT cell lysates using a nuclear and cytoplasmic fractionation kit (Pierce). The protein concentration of the nuclear and cytoplasmic extracts was determined using the Bradford method.25 Nuclear and cytoplasmic extracts were used for Western blot analysis while nuclear extracts used also for EMSA.
Electrophoretic mobility shift assay
Nuclear proteins were extracted from MCT cells as described previously.24 The protein concentration of the nuclear extracts was determined using Bradford method.25 Electrophoretic mobility shift assay (EMSA)-binding reactions were incubated in a 20 μl final volume for 20 min at room temperature containing 5 μg of the nuclear extract, 20–30 fmol of the 5′ end-labeled double-stranded oligonucleotide: Fn-CREB-639: 5′-CTCAGAGGTGACGCAATGTTCTC-3′/5′-GAGAACATTGCGTCACCTCTGAG-3′ covering a region of the CREB that binds to fibronectin promoter, and 1 μl of poly (dI-dC). The super shift assays were performed by pre-incubating nuclear extracts with 2 μg of CREB antibody (Cell Signaling Technology, MA) into the reaction. The reaction was carried out at room temperature for 30 min prior to adding the radiolabelled probe. The complexes were resolved using a 5% non-denaturing polyacrylamide gel. The gels were dried and exposed overnight at −70°C.
Animals
Two-month-old male db/db mice (Strain BKC.Cg.m +/+Leprdb/J) were purchased from Jackson Laboratory. The animals were allowed food and water ad libitum prior to and during the experiments. The mice were divided into 2 groups. Mice in group 1 (controls) were injected with an equal amount of DMSO. Mice in group 2 were injected i.p. with 2mg/kg body weight rapamycin 5d/week for 4weeks. Injections were carried out under isofluorane inhalation anesthesia (Abbott, Abbott Park, IL). Animals were euthanized at 4weeks of treatment and the kidneys were removed rapidly for dissection and biochemical analysis. Kidney sections and homogenates of kidney cortex were prepared as described previously.27
Immunoperoxidase staining of fibronectin
Detection of fibronectin was performed on paraffin kidney sections by immunoperoxidase histochemical staining.28 Kidney sections were incubated with rabbit anti-fibronectin antibody (Abcam, Cambridge, MA) for 30 min then washed twice with PBS. Sections were then incubated with horseradish peroxidase labeled anti-rabbit antibody for 30 min. The horseradish peroxidase was developed with diaminobenzidine tetrahydrochloride and hydrogen peroxide in PBS. To demonstrate staining specificity, control kidney sections were stained without primary antibody. Kidney sections were viewed and photographed using a Nikon Research microscope equipped for epifluorescence with excitation and band pass filters.
Immunofluorescence staining of p-CREB
A fluorescent labeling method of p-CREB immunostaining was performed in kidney sections of db/db mice as described previously.27 FITC green signals for p-CREB were detected using a filter with excitation range 450–490 nm and propidium iodide (PtdIns) red signals for nuclear DNA using a filter with excitation at 535 nm. Kidney sections were viewed and photographed using a Nikon Research microscope equipped for epifluorescence with excitation and band pass filters. To demonstrate staining specificity, control kidney sections were stained without primary antibody.
Statistics
Data are presented as mean ± standard error. Statistical differences were determined using ANOVA followed by Student Dunnett's (Exp. versus Control) test using one trial analysis. P-values less than 0.01 and 0.05 were considered statistically significant.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This work was supported in part by grants from the American Heart Association and Merit Review Award from South Texas Veterans Healthcare System (to SLH).
References
- 1.Gilbert RE, Cooper ME. The tubulointerstitium in progressive diabetic kidney disease: more than an aftermath of glomerular injury? Kidney Int 1999; 56:1627-37; PMID:10571771; http://dx.doi.org/ 10.1046/j.1523-1755.1999.00721.x [DOI] [PubMed] [Google Scholar]
- 2.Jones SC, Saunders HC, Qi W, Pollock CA. Intermittent high glucose enhances cell growth and collagen synthesis in cultured human tubulointerstitial cells. Diabetologia 1999; 42:1113-9; PMID:10447524; http://dx.doi.org/ 10.1007/s001250051279 [DOI] [PubMed] [Google Scholar]
- 3.Jones SC, Saunders HJ, Pollock CA. High glucose increases cell growth and collagen synthesis in cultured human tubulointerstitial cells. Diabet Med 1999; 16:932-8; PMID:10588523; http://dx.doi.org/ 10.1046/j.1464-5491.1999.00174.x [DOI] [PubMed] [Google Scholar]
- 4.Ziyadeh FN, Snipes ER, Watanabe M, Alvarez RJ, Goldfarb S, Haverty TP. High glucose induces cell hypertrophy and stimulates collagen gene transcription in proximal tubule. Am J Physiol Renal Fluid Electrolyte Physiol 1990; 259: F704-14; PMID:2221106 [DOI] [PubMed] [Google Scholar]
- 5.Park SH, Choi HJ, Lee JH, Woo CH, Kim JH, Han HJ: High glucose inhibits renal proximal tubule cell proliferation and involves PKC, oxidative stress and TGF-1. Kidney Int 2001; 59:1695-705; PMID:11318940; http://dx.doi.org/ 10.1046/j.1523-1755.2001.0590051695.x [DOI] [PubMed] [Google Scholar]
- 6.Tsao T, Fawcett J, Fervenza FC, Hsu FW, Huie P, Sibley RK, Rabkin R. Expression of insulin-like growth factor-I and transforming growth factor-β in hypokalemic nephropathy in the rat. Kidney Int 2001; 59:96-105; PMID:11135062; http://dx.doi.org/ 10.1046/j.1523-1755.2001.00470.x [DOI] [PubMed] [Google Scholar]
- 7.Lee MJ, Feliers D, Mariappan MM, Sataranatarajan K, Mahimainathan L, Musi N, Foretz M, Viollet B, Weinberg JM, Choudhury GG, Kasinath BS. AMP-activated protein kinase in diabetes-induced renal hypertrophy. Am J Physiol Renal Physiol. 2007; 292:F617-27; PMID:17018841; http://dx.doi.org/ 10.1152/ajprenal.00278.2006 [DOI] [PubMed] [Google Scholar]
- 8.Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell 2003; 115:577-90; PMID:14651849; http://dx.doi.org/ 10.1016/S0092-8674(03)00929-2 [DOI] [PubMed] [Google Scholar]
- 9.Corradetti MN, Inoki K, Bardeesy N, DePinho RA, Guan KL. Regulation of the TSC pathway by LKB1: evidence of a molecular link between tuberous sclerosis complex and Peutz-Jeghers syndrome. Genes Dev 2004; 18:1533-8; PMID:15231735; http://dx.doi.org/ 10.1101/gad.1199104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Inoki K, Li Y, Xu T, Guan KL. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev 2003; 17:1829-34; PMID:12869586; http://dx.doi.org/ 10.1101/gad.1110003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.White ES, Sagana RL, Booth AJ, Yan M, Cornett AM, Bloomheart CA, Tsui JL, Wilke CA, Moore BB, Ritzenthaler JD, Roman J, Muro AF. Control of fibroblast fibronectin expression and alternative splicing via the PI3K/Akt/mTOR pathway. Exp Cell Res 2010; 316:2644-53; PMID:20615404; http://dx.doi.org/ 10.1016/j.yexcr.2010.06.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Habib SL, Yadav M, Tizani S, Bhandari B, Valente AJ. Tuberin Inhibits Production of the Matrix Protein Fibronectin in Diabetes. J Am Soc Nephrology 2012; 23:1652-62; PMID:22904348; http://dx.doi.org/ 10.1681/ASN.2012030285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lieberthal W, Levine JS. The role of the mammalian target of rapamycin (mTOR) in renal disease. J Am Soc Nephrology 2009; 20:2493-502; ; http://dx.doi.org/ 10.1681/ASN.2008111186 [DOI] [PubMed] [Google Scholar]
- 14.Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell 2012; 149:274-93; PMID:22500797; http://dx.doi.org/ 10.1016/j.cell.2012.03.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev 2004; 18:1926-45; PMID:15314020; http://dx.doi.org/ 10.1101/gad.1212704 [DOI] [PubMed] [Google Scholar]
- 16.Choo AY, Blenis J. Not all substrates are treated equally: implications for mTOR, rapamycin-resistance and cancer therapy. Cell Cycle 2009; 8:567-72; PMID:19197153; http://dx.doi.org/ 10.4161/cc.8.4.7659 [DOI] [PubMed] [Google Scholar]
- 17.Mori S, Nada S, Kimura H, Tajima S, Takahashi Y, Kitamura A, Oneyama C, Okada M. The mTOR pathway controls cell proliferation by regulating the FoxO3a transcription factor via SGK1 kinase. PLoS One 2014; 9:e88891; PMID:24558442; http://dx.doi.org/ 10.1371/journal.pone.0088891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ortells MC, Morancho B, Drews-Elger K, Viollet B, Laderoute KR, López-Rodríguez C, Aramburu J. Transcriptional regulation of gene expression during osmotic stress responses by the mammalian target of rapamycin. Nucleic Acids Res 2012; 40:4368-84; PMID:22287635; http://dx.doi.org/ 10.1093/nar/gks038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim JE, Chen J. Regulation of peroxisome proliferator-activated receptor-gamma activity by mammalian target of rapamycin and amino acids in adipogenesis. Diabetes 2004; 53:2748-56; PMID:15504954; http://dx.doi.org/ 10.2337/diabetes.53.11.2748 [DOI] [PubMed] [Google Scholar]
- 20.Kim JE, Chen J. Cytoplasmic-nuclear shuttling of FKBP12-rapamycin-associated protein is involved in rapamycin-sensitive signaling and translation initiation. Proc Natl Acad Sci USA 2000; 97:14340-5; PMID:11114166; http://dx.doi.org/ 10.1073/pnas.011511898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Du K, Montminy M. CREB Is a Regulatory Target for the Protein Kinase Akt/PKB. JBC 1998; 273:32377-9; PMID:9829964; http://dx.doi.org/ 10.1074/jbc.273.49.32377 [DOI] [PubMed] [Google Scholar]
- 22.Singh LP, Andy J, Anyamale V, Greene K, Alexander M, Crook ED. Hexosamine-induced fibronectin protein synthesis in mesangial cells is associated with increases in cAMP responsive element binding (CREB) phosphorylation and nuclear CREB: the involvement of protein kinases A and C. Diabetes 2001; 50:2355-62; PMID:11574420; http://dx.doi.org/ 10.2337/diabetes.50.10.2355 [DOI] [PubMed] [Google Scholar]
- 23.Simone S, Gorin Y, Velagapudi C, Abboud HE, Habib SL. Mechanism of oxidative DNA damage in diabetes: tuberin inactivation and downregulation of DNA repair enzyme 8-oxo-7,8-dihydro-2′-deoxyguanosine-DNA glycosylase. Diabetes 2008; 57:2626-36; PMID:18599524; http://dx.doi.org/ 10.2337/db07-1579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Habib SL, Bhandari BK, Sadek N, Abboud-Werner SL, Abboud HE. Novel mechanism of regulation of the DNA repair enzyme OGG1 in tuberin-deficient cells. Carcinogenesis 2010; 31:2022-30; PMID:20837600; http://dx.doi.org/ 10.1093/carcin/bgq189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 1976; 72:248-54; PMID:942051; http://dx.doi.org/ 10.1016/0003-2697(76)90527-3 [DOI] [PubMed] [Google Scholar]
- 26.Liang S, Cuevas G, Tizani S, Salas T, Liu H, Li B, Habib SL. Novel mechanism of regulation of fibrosis in kidney tumor with tuberous sclerosis. Mol Cancer 2013; 12:1-11; PMID:23286373; http://dx.doi.org/ 10.1186/1476-4598-12-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Habib SL, Liang S. Hyperactivation of Akt/mTOR and deficiency in tuberin increased the oxidative DNA damage in kidney cancer patients with diabetes. Oncotarget 2014; 5:2542-50; PMID:24797175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Habib SL. Alterations in tubular epithelial cells in diabetic nephropathy. J Nephrol 2013; 26:865-9; PMID:24052469; http://dx.doi.org/ 10.5301/jn.5000287 [DOI] [PubMed] [Google Scholar]