

Abstract
The product of the ecotropic virus integration site 1 (EVI1) gene, whose overexpression is associated with a poor prognosis in myeloid leukemias and some epithelial tumors, regulates gene transcription both through direct DNA binding and through modulation of the activity of other sequence specific transcription factors. Previous results from our laboratory have shown that EVI1 influenced transcription regulation in response to the myeloid differentiation inducing agent, all-trans retinoic acid (ATRA), in a dual manner: it enhanced ATRA induced transcription of the RARβ gene, but repressed the ATRA induction of the EVI1 gene itself. In the present study, we asked whether EVI1 would modulate the ATRA regulation of a larger number of genes, as well as biological responses to this agent, in human myeloid cells. U937 and HL-60 cells ectopically expressing EVI1 through retroviral transduction were subjected to microarray based gene expression analysis, and to assays measuring cellular proliferation, differentiation, and apoptosis. These experiments showed that EVI1 modulated the ATRA response of several dozens of genes, and in fact reinforced it in the vast majority of cases. A particularly strong synergy between EVI1 and ATRA was observed for GDF15, which codes for a member of the TGF-β superfamily of cytokines. In line with the gene expression results, EVI1 enhanced cell cycle arrest, differentiation, and apoptosis in response to ATRA, and knockdown of GDF15 counteracted some of these effects. The potential clinical implications of these findings are discussed.
Keywords: apoptosis, cell cycle, EVI1, all-trans retinoic acid, GDF15, gene expression profiling, myeloid differentiation
Abbreviations
- AML
acute myeloid leukemia
- APL
acute promyelocytic leukemia
- Ar
ATRA regulation
- ATRA
all-trans retinoic acid
- DMSO
dimethyl sulfoxide
- Em
EVI1 modulation
- Er
EVI1 regulation
- FBS
fetal bovine serum
- FC
fold change
- FDR
false discovery rate
- GFP
green fluorescent protein
- mcoEvi1
murine codon optimized Evi1
- MDS
myelodysplastic syndrome
- PSG
penicillin streptomycin glutamine
- RAR
retinoic acid receptor
- RARE
retinoic acid response element
- SE
standard error
Introduction
The ecotropic virus integration site 1 (EVI1) gene, originally identified because its activation through retroviral integration caused leukemia in a mouse model system,1 is aberrantly expressed in approximately 10% of patients with acute myeloid leukemia (AML).2-5 EVI1 overexpression has also been observed in variable proportions of patients with myelodysplastic syndromes (MDS),6-8 chronic myeloid leukemia (CML),9-11 acute lymphoblastic leukemia (ALL),12 and certain epithelial tumors.13-16 An association between elevated EVI1 mRNA levels and poor prognosis is particularly well documented for AML,2-5,17,18 but has also been reported for other malignancies.8,10,13,14 Corroborating its role as an oncogene capable of initiating malignant transformation, in a human gene therapy trial for chronic granulomatous disease clones with activating integrations of the therapeutic vector into the EVI1 locus expanded preferentially, followed by evolution into MDS and, ultimately, AML.19 Analogously, experimental expression of Evi1 effected development of an MDS like disease,20 and coexpression with other oncogenes such as HoxA9 + Meis1 or AML1D171N caused AML21,22 in murine bone marrow transplantation models. In vitro, EVI1 stimulated cell proliferation and inhibited differentiation and apoptosis in some experimental models,12,16,20,23-31 but elicited the opposite effects in others.20,31-39 This suggests that the fate of EVI1 overexpressing cells is influenced by lineage, maturation stage, cooperating molecular events, and/or environmental stimuli, and raises the possibility that it may be amenable to pharmacological modulation.
EVI1 is a nuclear zinc finger protein that is assumed to exert its biological effects predominantly by regulating gene transcription. Indeed, a number of direct EVI1 target genes have been reported.26,39-43 In addition, EVI1 interacted with other transcription factors, e.g. GATA1,44 RUNX1/AML1,45 PU.1,46 and SMAD3,47-49 to modulate their effects. Our own previous studies have shown that EVI1 enhanced or decreased transcriptional regulation by all-trans retinoic acid (ATRA) in a promoter specific manner.50
Retinoic acid is the biologically active metabolite of vitamin A and plays a crucial role during many developmental processes.51,52 It operates by binding to and regulating the activity of a nuclear receptor that is composed of a retinoic acid receptor (RAR) and a retinoid X receptor (RXR) subunit, each of which is encoded by 3 paralogous genes, α, β, and γ.53,54 The RAR/RXR heterodimer binds to its canonical DNA response elements both in the absence and presence of ligand, but changes its conformation from a corepressor binding to a coactivator binding, and therefore from a transcription repressing to an activating, state upon interaction with retinoic acid.53,54 Interestingly, the RARβ gene itself is strongly induced by ATRA through direct binding of RAR/RXR to a retinoic acid response element (RARE) in its promoter.55
In the context of hematopoiesis, ATRA promoted the formation of haematopoietic stem cells (HSCs) from the hemogenic endothelium.56 It enhanced the proliferation and in vivo long term repopulation ability of primitive haematopoietic precursor cells, but inhibited the proliferation and advanced the differentiation of more committed progenitor cells.57-60 The well documented differentiation promoting properties of ATRA have been put to therapeutic use in the context of acute promyelocytic leukemia (APL), a subtype of AML characterized by the presence of RARα fusion proteins with reduced sensitivity to ATRA.60 Combined treatment with superphysiological doses of ATRA and conventional chemotherapeutic drugs or arsenic trioxide has greatly improved the outcome of patients with this disease.60 In contrast, in non-APL AML no clear value for the addition of ATRA has so far been demonstrated.61 Nevertheless, because dosing and scheduling may require adaptation, a potential beneficial effect of ATRA in non-APL AML is still under active investigation.
Our own previous studies have shown that EVI1 was not only regulated by ATRA through direct binding of the retinoic acid receptor heterodimer to a canonical RARE located in the most 5' one of several alternative first exons,50,62 but that EVI1 in turn modulated ATRA regulated transcription: it counteracted its own induction by ATRA in a negative feedback loop, but augmented the ATRA induction of the RARβ gene.50 Based on these findings, we now asked whether EVI1 would modulate the regulation by ATRA of a larger number of genes, and whether it would also impact on biological responses to ATRA.
Results
EVI1 modulates transcriptional regulation by ATRA of a substantial number of genes in human myeloid cells
We have previously established a human myeloid cell line, U937_EVI1, which constitutively expresses ectopic EVI1 at levels comparable to those effected by a rearrangement of its gene locus in chromosome band 3q26.30 To address the question whether EVI1 would modulate the ATRA responses not only of its own and the RARβ, but of a larger number of genes, RNA was extracted from 3 independent replicate cultures of U937_EVI1 and control U937_vec cells treated with 1 μM ATRA or an equivalent amount of its solvent DMSO for 24 h. Gene expression microarray analysis revealed that 453 genes were induced and 240 repressed by ATRA, and 44 genes were up- and 67 down-regulated by EVI1 at a false discovery rate (FDR)63 of 0.05 (Fig 1A, B; Tables S1A, B). Genes were considered to exhibit an EVI1 modulated ATRA response if they fulfilled the following stringent criteria: the factor by which EVI1 significantly altered their expression in the presence of ATRA was equal to or greater than 2, and equal to or greater than the square of the factor by which EVI1 changed their mRNA levels in the absence of ATRA (see Materials and Methods for details). Dependent on whether genes were induced or repressed by ATRA, and whether EVI1 enhanced or counteracted this regulation, 4 categories were defined: category 1 comprised genes whose induction by ATRA was enhanced by EVI1; category 2, genes whose repression by ATRA was counteracted by EVI1; category 3, genes whose induction by ATRA was counteracted by EVI1; and category 4, genes whose repression by ATRA was enhanced by EVI1. 113, 3, 2, and 6 genes, respectively, fulfilled the definitions for inclusion into these categories (Fig. 1C), indicating i) that the ATRA response of a substantial number of genes was modulated by EVI1, and ii) that enhancement of the ATRA response by EVI1 was far more common than its inhibition (119 versus 5 genes, respectively).
Figure 1.
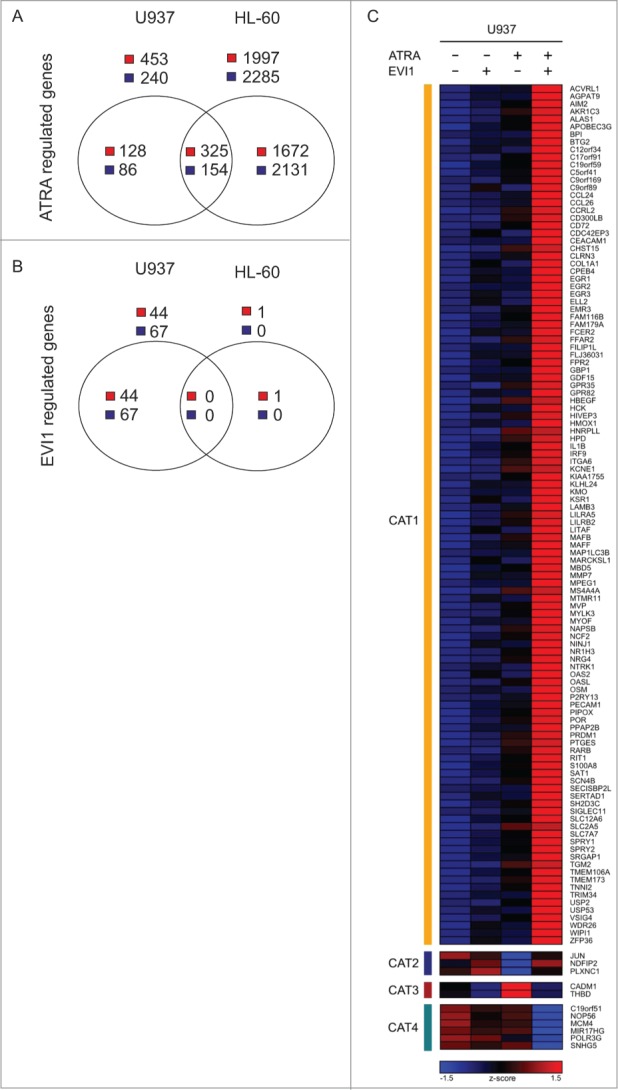
EVI1 and ATRA regulate gene expression both in an independent and a cooperative manner in human myeloid cells. (A) Numbers of genes significantly up- or down-regulated in response to ATRA in U937_vec cells, HL-60_vec cells, or both. (B) Numbers of genes significantly up- or downregulated in U937_EVI1 vs. U937_vec cells and in HL-60_Evi1 versus HL-60_vec cells. (C) Heatmap representing the expression (z-score) of genes whose induction by ATRA was enhanced by, or observed only in the presence of, EVI1 (category 1, CAT 1), whose repression by ATRA was counteracted by EVI1 (category 2, CAT 2), whose induction by ATRA was counteracted by EVI1 (category 3, CAT 3), and whose repression by ATRA was enhanced by, or observed only in the presence of, EVI1 (category 4, CAT 4) in U937 cells.
Figure 2.
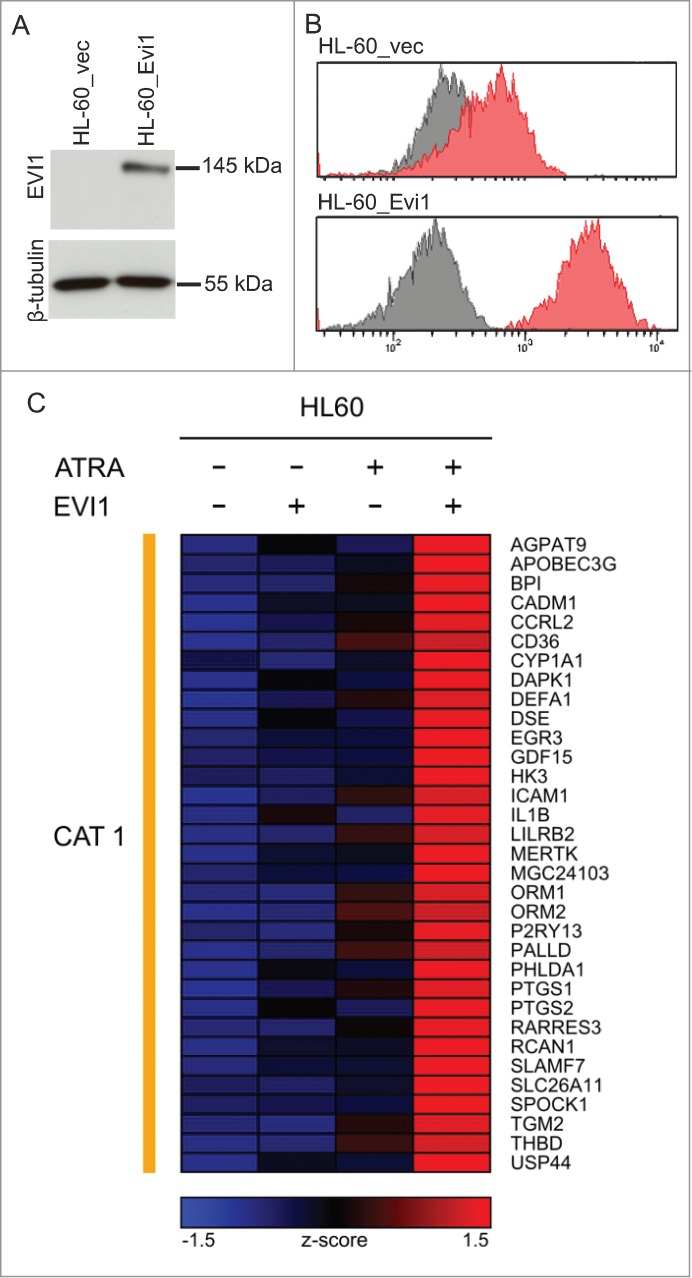
Evi1 and ATRA cooperatively regulate gene expression in HL-60 cells. (A) Immunoblot analysis demonstrating expression of ectopic Evi1 in HL-60_Evi1 cells. (B) Intranuclear staining and flow cytometry showing expression of ectopic Evi1 in HL-60_Evi1 cells on the single cell level. Red, EVI1 antibody; gray, isotype control. (C) Heatmap representing the expression (z-score) of genes whose induction by ATRA was enhanced by, or observed only in the presence of, Evi1 (category 1, CAT1) in HL-60 cells. In this model system, no genes fulfilled the criteria for inclusion into categories 2-4.
To confirm these findings in an independent experimental system, another human myeloid cell line, HL-60, was used. Like U937, HL-60 cells express low to undetectable levels of EVI1 both in the absence and presence of ATRA (data not shown). Transduction with pBMN-mcoEvi1-IRES-GFP (containing a codon optimized version of the murine Evi1 gene, whose gene product exhibits 96% sequence homology to human EVI1) effected constitutive expression of Evi1 as determined by immunoblot analysis (Fig. 2A) and flow cytometry after intranuclear protein staining (Fig. 2B). For genome wide gene expression analysis, HL-60_Evi1 and HL-60_vec cells were treated with ATRA or solvent and used for microarray experiments as described above. At an FDR of 0.05, ATRA up-regulated 1997 and downregulated 2285 genes, of which 325 and 154, respectively, were regulated in the same direction as in U937_vec cells (Fig. 1A; Tables S1C, E). Surprisingly, Evi1 induced only one gene at an FDR of 0.05 (Fig. 1B; Table S1D), possibly due to lower Evi1 expression levels in this system as compared to the U937 based model. Most importantly with respect to the subject of this study, microarray analysis of the HL-60 derivative cell lines uncovered 33 genes that exhibited an Evi1 modulated ATRA response according to the definition explicated above (Fig. 2C). All of these genes were classified into category 1, i.e., their induction by ATRA was enhanced by Evi1, and 10 of them—AGPAT9, APOBEC3G, BPI, CCRL2, EGR3, GDF15, IL1B, LILRB2, P2RY13, and TGM2—were regulated in the same manner in both U937 and HL-60 cells, as also confirmed by qRT-PCR (Fig. 3, Fig. S1).
Figure 3.
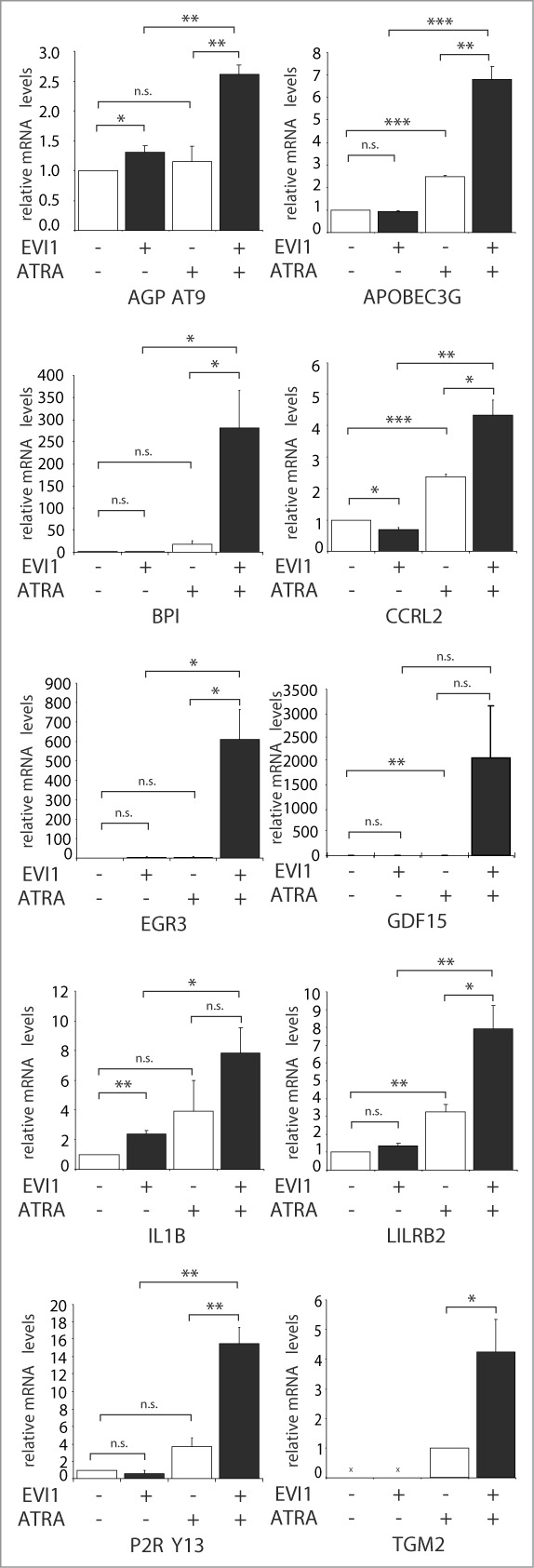
qRT-PCR confirms cooperative gene regulation by EVI1 and ATRA in U937 cells. RNA from U937_vec and U937_EVI1 cells that had been treated with solvent or ATRA for 24 h was reverse transcribed and subjected to qRT-PCR for the indicated genes. Gene expression relative to the housekeeping gene B2M and to solvent treated U937_vec cells was calculated using the ΔΔCt method.88 Results represent means + SEs from at least 3 independent experiments; all of these were also independent of the microarray experiments. In the absence of ATRA, TGM2 mRNA levels were below the detection limit. Significance was calculated using Student's 2-tailed t-test (*, P < 0.05; **, P < 0.01; ***, P < 0.001). Even though results for GDF15 and IL1B were not significant due to variation in induction factors, the expression levels of all genes were reproducibly substantially higher in ATRA treated EVI1 expressing cells as compared to all other conditions. Please note the differences in scale between the different panels.
Enhancement of the ATRA response of GDF15 by EVI1 is not restricted to myeloid cells
The gene whose regulation exhibited by far the most pronounced synergy between ATRA and EVI1 was GDF15, with EVI1 enhancement in the presence of ATRA as high as 185- and 85-fold in U937 and HL-60 cells, respectively (Fig. 3, Fig. S1). GDF15 is a member of the TGF-β superfamily of cytokines. It is processed from a precursor through proteolytic cleavage and secreted into the extracellular space as a disulfide linked homodimer.64,65 To confirm the strongly synergistic regulation of GDF15 by ATRA and EVI1 at the protein level, U937_EVI1 and U937_vec cells were treated with ATRA or DMSO, and the precursor as well as the active form of GDF15 were quantified by immunoblot analysis. Low to undetectable amounts of either form were produced by U937_vec cells irrespective of treatment, as well as by DMSO treated U937_EVI1 cells, but strong signals were observed in, or in the culture supernatants of, U937_EVI1 cells exposed to ATRA (Fig. 4A). An ELISA corroborated the pronounced, EVI1 and ATRA dependent induction of secreted GDF15 (Fig. 4B).
Figure 4.

EVI1 and ATRA synergistically induce GDF15 protein in myeloid and in epithelial cells. (A) U937_vec and U937_EVI1 cells were treated with solvent or ATRA for 3 or 5 days, and the intracellular precursor and secreted mature forms of GDF15 were detected in whole cell extracts (day 3) and culture supernatants (day 5), respectively, by immunoblot analysis. Both forms are disulfide linked homodimers, and the corresponding 35 kDa and 15 kDa monomers are detected after reducing SDS-PAGE. (B) U937_vec and U937_EVI1 cells were treated with solvent or ATRA for 3 or 5 days, and mature GDF15 was detected in culture supernatants by ELISA. Results represent means + SEs from 3 independent experiments. Significance was calculated using Student's 2-tailed t-test (*, P < 0.05; **, P < 0.01; ***, P < 0.001). (C) The nontumorigenic prostate epithelial cell line BPH-1 and its transformed derivative, BPH-1 CAFTD03, were subjected to immunoblot analysis to detect endogenous EVI1. (D) BPH-1 and BPH-1 CAFTD03 cells were treated with 1 or 2 μM ATRA for the indicated times, and intracellular GDF15 levels were determined by immunoblot analysis.
Because of the well documented role of GDF15 in solid tumors,64,65 and because EVI1 has been reported to be overexpressed in a variety of epithelial cancers,13,15,16,66 we asked whether EVI1 and ATRA would also synergize to regulate the expression of this cytokine in epithelial cell lines. Immunoblot analysis showed that the nontumorigenic prostate epithelial cell line BPH-1 expressed low levels of EVI1, while its transformed derivative, BPH-1 CAFTD03, contained substantial amounts of this protein (Fig. 4C). Likewise, basal GDF15 levels were considerably higher in BPH-1 CAFTD03 than in parental cells. Furthermore, GDF15 was substantially induced in BPH-1 CAFTD03 cells as early as 8 h after addition of ATRA, whereas only a weak up-regulation was observed in BPH-1 cells after prolonged ATRA treatment (Fig. 4D). Since ATRA also induced the EVI1 protein in both cell lines (data not shown), the possibility has to be considered that ATRA acted through, rather than cooperating with, EVI1 to up-regulate GDF15. While such an effect may contribute to the induction of GDF15 in this system, it is unlikely to be solely responsible for it, because the strong elevation of GDF15 in BPH-1 CAFTD03 cells already 8 h after ATRA addition cannot readily be explained by a secondary effect. These results therefore suggest that EVI1 and ATRA cooperate to regulate GDF15 not only in haematopoietic , but also in epithelial cells.
EVI1 enhances ATRA induced cell cycle arrest, differentiation, and apoptosis
Because EVI1 modulated the regulation by ATRA of a substantial number of genes, and in fact enhanced, rather than counteracted, it in the large majority of cases (119/124 genes, i.e., 96%, in U937 cells; and 33/33 genes, 100%, in HL-60 cells), we next investigated whether EVI1 would also augment responses to ATRA at the cellular level. U937_EVI1 and U937_vec cells were seeded at equal densities and counted after treatment with ATRA or DMSO for several days. In agreement with our previous report,30 expression of EVI1 by itself did not significantly alter cell numbers in this experimental system. ATRA led to a substantial decrease in viable cell counts, as expected. Notably, this effect was significantly more pronounced in U937_EVI1 than in U937_vec cells, indicating that EVI1 cooperated with ATRA to decrease cellular proliferation (Fig. 5A). This could be due to an interplay between EVI1 and ATRA with regard to the regulation of cell cycle progression, and/or of apoptosis. To address the first possibility, nuclei from ATRA or solvent treated cells were stained with propidium iodide and their DNA content was determined by flow cytometry. In the absence of ATRA, expression of EVI1 did not affect the cell cycle distribution of U937 cells, but in its presence it enhanced both the increase in the G0/G1 population and the decrease in the proportion of cells in S-phase (Fig. 5B; Fig. S2A). Similarly, EVI1 augmented the pro-apoptotic effects of ATRA as revealed by staining with Annexin V and 7AAD (Fig. 5C; Fig. S2B).
Figure 5.
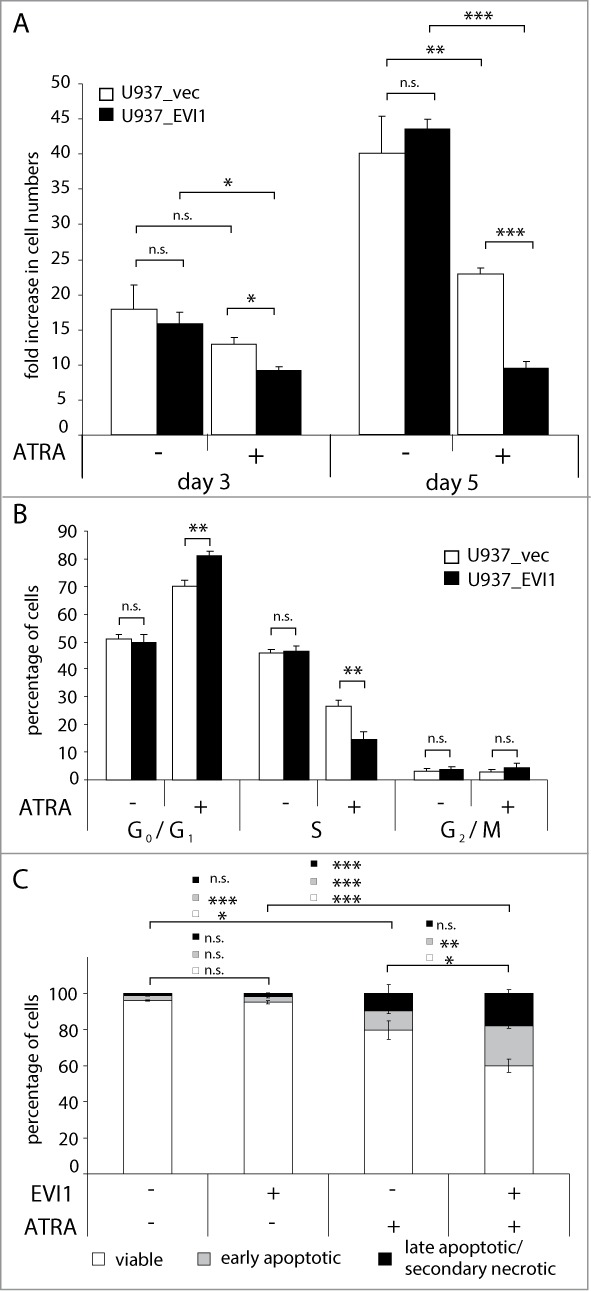
EVI1 enhances ATRA induced cell cycle arrest and apoptosis in U937 cells. (A, B) U937_vec cells (white bars) and U937_EVI1 cells (black bars) were treated with solvent or ATRA and counted at the indicated time points (A), or subjected to cell cycle analysis by propidium iodide staining and flow cytometry on day 5 (B). (C) U937_vec and U937_EVI1 cells were treated with solvent or ATRA for 7 days, stained with Annexin V and 7AAD, and analyzed by flow cytometry. Double negative cells were classified as viable (white portions of bars), Annexin V positive 7AAD negative cells as early apoptotic (gray portions of bars), and double positive cells as late apoptotic/secondary necrotic (black portions of bars).89 Results represent means + SEs from at least 3 independent experiments. Significance was calculated using Student's 2-tailed t-test (*, P < 0.05; **, P < 0.01; ***, P < 0.001).
In HL-60 cells, expression of Evi1 also significantly enhanced the effects of ATRA on apoptosis (Fig. 6A; Fig. S3A). To investigate whether Evi1 also augmented the well characterized differentiation response of HL-60 cells to ATRA,67 ATRA or DMSO treated HL-60_Evi1 and HL-60_vec cells were stained with an antibody for the myeloid differentiation marker CD11b and analyzed by flow cytometry. ATRA increased the mean CD11b fluorescence intensity, and this effect was significantly more pronounced in HL-60_Evi1 than in HL-60_vec cells (Fig. 6B; Fig. S3B). Morphological analysis of Wright-stained cytospin preparations confirmed that Evi1 enhanced the ATRA induced differentiation of HL-60 cells (Fig. 6C). In summary, these data show that EVI1 enhances not only transcriptional, but also biological responses to ATRA in human myeloid cells.
Figure 6.
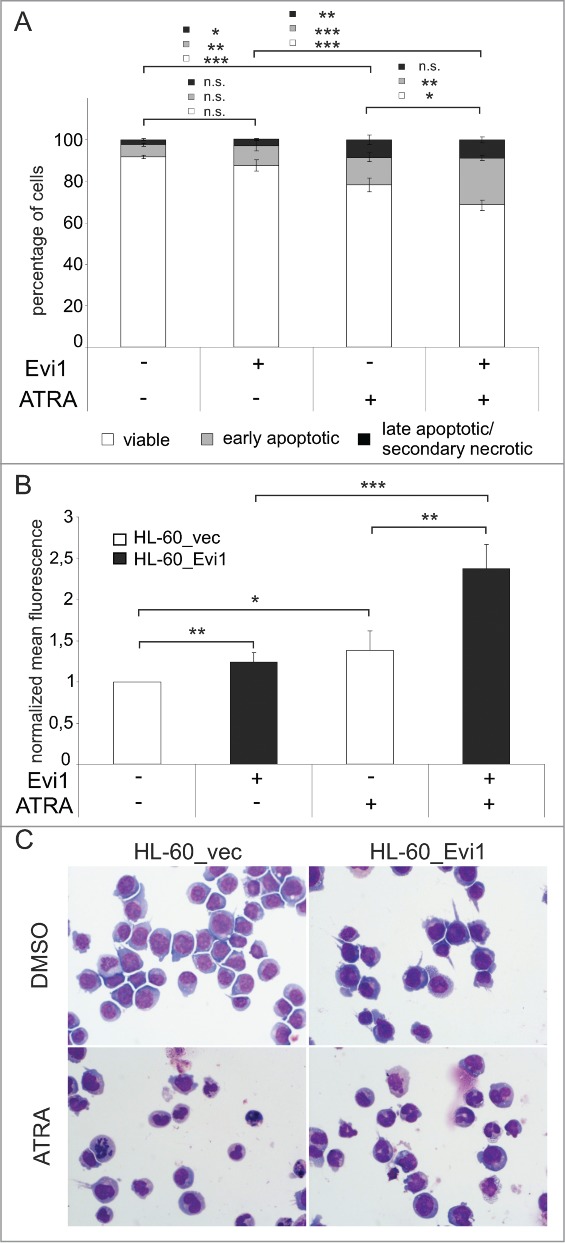
Evi1 enhances ATRA induced apoptosis and differentiation in HL-60 cells. (A) HL-60_vec and HL-60_Evi1 cells were treated with solvent or ATRA for 5 days, stained with Annexin V and 7AAD, and analyzed by flow cytometry. Double negative cells were classified as viable (white portions of bars), Annexin V positive 7AAD negative cells as early apoptotic (gray portions of bars), and double positive cells as late apoptotic/secondary necrotic (black portions of bars).89 Results represent means +/- SEs of 8 independent experiments. (B) HL-60_vec cells (white bars) and HL-60_Evi1 cells (black bars) were treated with solvent or ATRA for 5 days, stained with PE-conjugated CD11b or isotype control antibodies, and subjected to flow cytometry. Results are expressed as the means of the PE fluorescence of CD11b stainings relative to that of the respective isotype controls and to untreated HL-60_vec cells, and represent the means + SEs of 7 independent experiments. Significance was calculated using Student's 2-tailed t-test (*, P < 0.05; **, P < 0.01; ***, P < 0.001). (C) Wright stained cytospin preparations of HL-60_vec and HL-60_Evi1 cells treated with solvent or ATRA for 5 d.
Knockdown of GDF15 counteracts the enhancing effect of EVI1 on ATRA induced cell cycle arrest
GDF15 has been reported to slow cell cycle progression and enhance apoptosis in various tumor cell lines.64,65,68,69 To ask whether it also played a role in the enhancement of the biological effects of ATRA by EVI1, U937_EVI1 and U937_vec cells were infected with retroviral vectors containing shRNAs against GDF15 (shGDF15-1, shGDF15-2) or against renilla luciferase as a control (shRen), and sorted for positivity for the vector encoded marker protein mCherry. qRT-PCR, immunoblot analysis, and ELISA confirmed a strong reduction of GDF15 mRNA and protein levels in ATRA treated U937_EVI1_shGDF15-1 and U937_EVI1_shGDF15-2 cells compared to U937_EVI1_shRen cells (Fig. 7A–C). Cell cycle analysis revealed that experimental down-regulation of GDF15 indeed counteracted the reinforcing effect of EVI1 on the ATRA induced increase and decrease in the G0/G1 and S phase populations, respectively (Fig. 7D).
Figure 7.
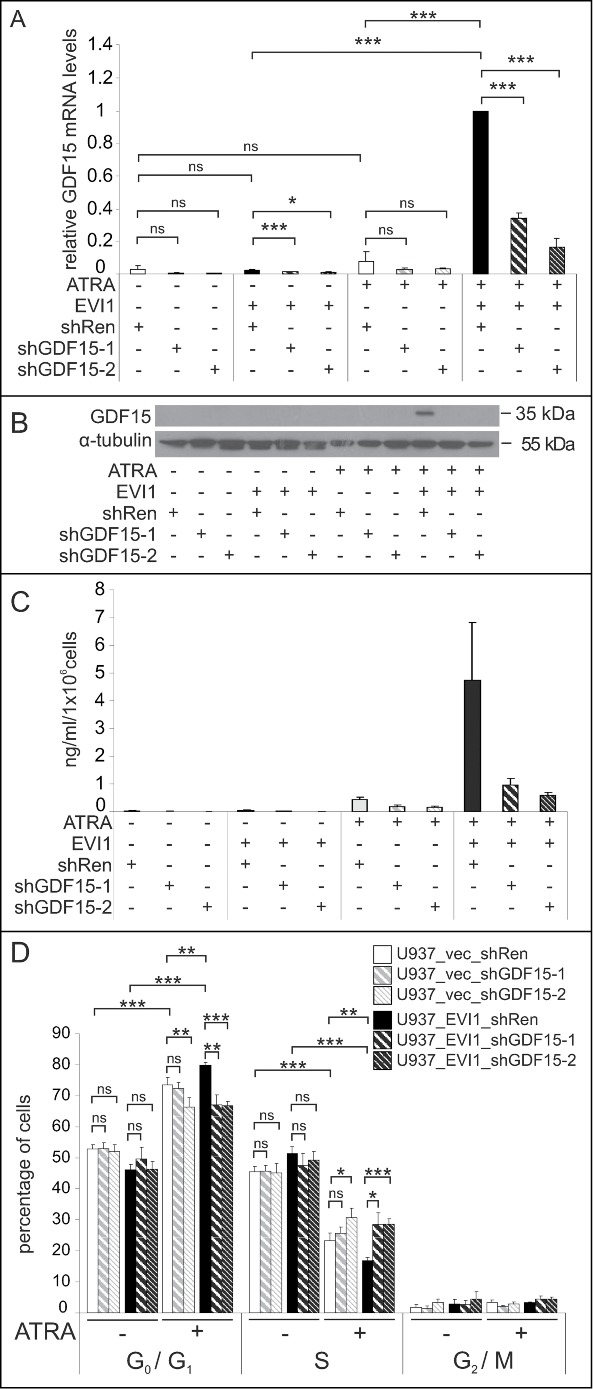
Experimental down-regulation of GDF15 counteracts EVI1 enhancement of the ATRA induced cell cycle arrest. (A) U937_vec and U937_EVI1 cells infected with a control shRNA targeting Renilla luciferase (shRen) or shRNAs against GDF15 (shGDF15-1, shGDF15-2) were treated with solvent or ATRA for 24 h, and GDF15 mRNA levels were measured by qRT-PCR. GDF15 expression was normalized to that of B2M using the ΔΔCt method88 and ATRA treated U937_EVI1_shRen cells as a calibrator. Results represent means + SEs from 3 independent experiments. (B) U937_vec and U937_EVI1 cells containing shRen, shGDF15-1, or shGDF15-2 were treated with solvent or ATRA for 5 days, and the GDF15 precursor was detected by immunoblot analysis. α-tubulin was used as a loading control. (C) U937_vec and U937_EVI1 cells containing shRen, shGDF15-1, or shGDF15-2 were treated with solvent or ATRA for 5 days, and mature GDF15 was detected in culture supernatants by ELISA. Results represent means + SEs from 5 independent experiments. Due to variation in factors results were not significant, but the enhancement of the ATRA induction by EVI1 in shRen infected cells, as well as the knockdown of GDF15 by both shRNAs were consistently seen in all experiments. (D) U937_vec and U937_EVI1 cells containing shRen, shGDF15-1, or shGDF15-2 were treated with solvent or ATRA for 5 d and subjected to cell cycle analysis by propidium iodide staining and flow cytometry. Results represent means + SEs from 7 independent experiments. Significance was calculated using Student's 2-tailed t-test (*, P < 0.05; **, P < 0.01; ***, P < 0.001; ns, not significant).
Discussion
Based on previous data showing that EVI1 enhanced the ATRA induction of the RARβ gene, but inhibited the induction of its own promoter by this agent,50 the present study addressed the questions whether EVI1 modulated transcriptional regulation by ATRA on a more general scale, and whether it also affected biological responses to ATRA. Microarray based genome wide gene expression analyses using 2 different human myeloid cell lines revealed that indeed several dozens of genes fulfilled the stringent criteria applied to define modulation of the ATRA response by EVI1. Notably, EVI1 augmented, rather than counteracted, the ATRA regulation of the large majority of these genes: a positive regulatory interaction between EVI1 and ATRA was observed for 119/124 (96%) and 33/33 (100%) genes with an EVI1 modulated ATRA response in U937 and HL-60 cells, respectively. Of ten genes that exhibited comparable expression patterns in both cell lines, GDF15 displayed the most pronounced synergy between EVI1 and ATRA, with EVI1-dependent differences of the levels of its mRNA in the presence of ATRA around 100-fold. GDF15 is a member of the TGF-β superfamily of cytokines and is secreted into the extracellular space as a disulfide linked homodimer. It has been implicated in numerous pathological processes, including inflammation, atherosclerosis, and cancer.64,65 It was induced by non-steroidal anti-inflammatory drugs (NSAIDs) and by p53, mediated cell cycle arrest and apoptosis of cancer cell lines in vitro, and reduced tumor growth in vivo. However, as with other members of the TGF-β family, there is evidence to suggest that GDF15 turns from a tumor suppressor to a tumor promoter in the course of carcinogenesis.64,65 The data presented in this report imply EVI1, a developmental regulator and an oncogene involved in a variety of tumors,2-5,13-16,66,70 and ATRA as novel regulators of GDF15 in both haematopoietic and epithelial cells, and show that GDF15 is required for the reinforcing effect of EVI1 on ATRA induced cell cycle arrest. In addition to providing new information about the regulation and function of this clinically highly relevant cytokine, these results suggest that cooperative interactions between EVI1 and ATRA are not restricted to haematopoietic cells. Observations that EVI1 enhanced the ATRA induction of a RARE-driven reporter gene in NTERA-2 human teratocarcinoma cells50 and in MCF7 breast cancer cells (BS, unpublished observations) further support the conclusion that EVI1 is able to modulate gene regulation in response to ATRA not only in myeloid cells, but in a variety of different cell types.
Functional interactions between EVI1 and other sequence specific transcription factors, e.g., RUNX1/AML1,45 PU.1,46 GATA1,44 SMAD3,47-49 and FOS42 have been reported previously. Interestingly, EVI1 inhibited the function of almost all of these factors, and a cooperative relationship, as observed here with respect to ATRA regulated transcription, was proposed only in the case of FOS.42
Of the numerous transcription factors that have been shown to interact with the retinoic acid receptors, STAT5 and MN1 may be considered as particularly interesting in the context of AML. Constitutive activation of STAT5 was observed in a large proportion of patients with AML, may contribute to its pathobiology in several ways, and has recently been proposed to be associated with shortened survival.71-73 Through mechanisms that were not fully elucidated but may involve increased DNA binding by the RAR/RXR heterodimer, IL-3, acting via STAT5, augmented the ATRA induction of a reporter gene driven by the RARβ RARE in haematopoietic cells.74
High mRNA levels of MN1, which codes for a transcriptional regulator,75 were associated with a poor prognosis in patients with cytogenetically normal AML.76,77 MN1 synergized with ATRA to induce transcription from a retroviral long-terminal repeat sequence,75 but when the effects of MN1 on ATRA regulated transcription were investigated on a genome wide scale, comparable numbers of genes exhibited an ATRA response that was augmented or counteracted by MN1, respectively.78 Experimental expression of MN1 in murine bone marrow cells strongly diminished the proliferation inhibiting and differentiation promoting effects of ATRA.79 Accordingly, only patients with low, but not those with high, MN1 expression benefitted from the addition of ATRA to antileukemic therapy in one study,79 albeit this was not confirmed in an independent trial.61
In contrast to MN1, which mostly inhibited the actions of ATRA, the data presented in this report show that EVI1 not only augmented transcriptional responses to this agent, but also enhanced several of its biological effects in human myeloid cells. Since EVI1 reinforced ATRA regulated gene expression in non-haematopoietic cell types as well, it will be of interest to determine whether it also cooperates with this agent in more of the numerous biological processes controlled by it. Furthermore, our results have potential clinical ramifications: the finding that EVI1 enhanced the effects of ATRA on the proliferation, differentiation, and apoptosis of myeloid cells raises the possibility that AML patients overexpressing EVI1, who have a poor prognosis under current therapeutic regimens,2-5,17,18 may benefit from the addition of ATRA to chemotherapy. Because EVI1 mRNA levels are elevated only in around 10% of patients with AML, large clinical trials will be required to address this question in an informative manner.
A second context in which the property of EVI1 to enhance responsiveness to ATRA may be of relevance is that of APL. The inclusion of ATRA, which promotes APL blast differentiation, into the therapeutic regimen has greatly improved the prognosis of patients suffering from this subtype of AML.60 Even though by far outweighed by its benefits, ATRA treatment also carries the risk of a specific and potentially life-threatening complication, differentiation syndrome (DS). DS occurs in about 20—25% of APL patients, yet few if any risk factors have been non-controversially defined.80,81 Pulmonary infiltration by maturing APL blasts plays a major role in DS pathogenesis,80,81 and required the action of ATRA on both the infiltrated tissue and the APL cells themselves in a mouse model.82 Interestingly, EVI1 was upregulated during ATRA therapy in human APL patients83 (and BS, unpublished results), raising the possibility that it may augment the therapeutic effects of this agent. Alternatively or additionally, EVI1 may cooperate with ATRA to evoke DS. Future studies will address the potential beneficial or deleterious roles of EVI1 in APL.
Materials and Methods
Cell culture, retroviral infections, and ATRA treatment
The human myeloid cell lines U937 and HL-60 and their derivatives were grown in RPMI medium (Life Technologies) containing 10% (v/v) Fetal Bovine Serum (FBS, Life Technologies) and 1x Penicillin-Streptomycin-Glutamine (PSG, Life Technologies). The human prostate epithelial cell lines BPH-1 and BPH-1 CAFTD03 were cultured in RPMI medium with 5% FBS and 1x PSG.84,85 Phoenix cells were grown in DMEM medium (Life Technologies) with 10% FBS and 1x PSG.
U937_EVI1 cells, which constitutively overexpress EVI1 through insertion of the retroviral vector pBMN-EVI1-IRES-GFP, and the respective empty vector control cells, U937_vec, have been described previously.30 EVI1 levels in U937_EVI1 cells are comparable to those in HNT-34 cells, which express high amounts of endogenous EVI1 due to a rearrangement of its gene locus in chromosome band 3q26.86
For overexpression of Evi1 in HL-60 cells, a codon optimized version of murine Evi1 (mcoEvi1, whose product is 93% identical and 96% similar to human EVI1; kind gift of Jean-Yves-Metais, NHLBI, NIH) was cloned into pBMN-IRES-GFP (Addgene) using the BamHI and EcoRI restriction sites to yield pBMN-mcoEvi1-IRES-GFP. To increase the infection efficiency with this vector, HL-60 cells were first transduced with a vector driving expression of the ecotropic receptor EcoR, pEcoR-IRES-lyt2 (kindly provided by Herbert Strobl, Institute of Immunology, Medical University of Vienna). Lyt2-positive, EcoR expressing HL-60 cells (HL-60e) were then infected with pBMN-mcoEvi1-IRES-GFP or empty vector and sorted for green fluorescent protein (GFP) positivity. Because Evi1 expression in this system declined over time (data not shown), cells were used for experiments only within a period of 15 d after each transduction.
Experimental downregulation of GDF15 in U937_vec and U937_EVI1 cells was achieved by infection with LEPC based vectors87 containing shRNAs directed against the human GDF15 gene (shGDF15-1, TGCTGTTGACAGTGAGCGCGGAACTGTGTATTTATTTAAATAGTGAAGCCACAGATGTATTTAAATAAATACACAGTTCCATGCCTACTGCCTCGGA, and shGDF15-2, TGCTGTTGACAGTGAGCGCTCCCATGGTGCTCATTCAAAATAGTGAAGCCACAGATGTATTTTGAATGAGCACCATGGGATTGCCTACTGCCTCGGA; Mirimus). An shRNA directed against renilla luciferase (shRen, TGCTGTTGACAGTGAGCGCAGGAATTATAATGCTTATCTATAGTGAAGCCACAGATGTATAGATAAGCATTATAATTCCTATGCCTACTGCCTCGGA; Mirimus) was used as a control. Successfully infected cells were sorted based on positivity for the vector encoded mCherry marker protein.
Retroviral infections were carried out using standard procedures (http://www.stanford.edu/group/nolan/retroviral_systems/phx.html) and Phoenix-gp cells as producer line for transduction of HL-60, U937_EVI1, and U937_vec cells. HL-60e cells were infected with virus produced from Phoenix-Eco cells. Cell sorting was performed on a Beckman Coulter MoFloAstrios using Summit V6.1 software.
Logarithmically growing cells were treated with 1 or 2 μM ATRA (Sigma; 10 mM stock in dimethyl sulfoxide, DMSO) or corresponding amounts of DMSO for 8 h to 7 days, as indicated. If treatment periods exceeded 3 days, DMSO treated cells were diluted as necessary to avoid saturation.
RNA extraction and microarray analyses
For microarray analyses, 3 logarithmically growing biological replicate cultures of U937 or HL-60 cells with or without ectopic expression of EVI1 were treated with ATRA or solvent for 24 h. RNA was prepared using QIAshredder homogenizers, the RNeasy Mini Kit, and the RNase free DNase set (all from Qiagen) according to the manufacturer's instructions. Samples were further processed and hybridized to GeneChip Genome U133 Plus 2.0 arrays (Affymetrix) by a service provider (KFB, Regensburg, Germany). For primary data analysis, the robust multi-array average (RMA) algorithm was used. Statistical analysis was performed using the software package R, including the limma package for the identification of differentially expressed genes. Genes were considered to be regulated by ATRA or EVI1 if they were differentially expressed between the relevant samples at a false discovery rate (FDR)63 of <0 .05. To identify genes whose regulation by ATRA was modified by EVI1, the following relations between samples were defined: the fold-change (FC) between samples not expressing ectopic EVI1 and treated with ATRA or solvent was termed ATRA regulation (Ar); the FC between solvent treated cells expressing or not expressing EVI1 was termed EVI1 regulation (Er); and the FC between ATRA treated cells expressing or not expressing EVI1 was termed EVI1 modulation (Em). Based on these relations, 4 categories of genes whose ATRA regulation was modified at least 2-fold by EVI1 were defined. Category 1 contained genes whose induction by ATRA was enhanced by, or observed only in the presence of, EVI1. Genes in this category were not repressed by ATRA (logFCAr ≥0 ), their expression was positively modulated at least 2-fold by EVI1 in a statistically significant manner (logFCEm ≥1 at P < 0.05), and their modulation by EVI1 in the presence of ATRA was at least the square of their regulation by EVI1 in the absence of ATRA (logFCEm ≥2 *logFCEr). Category 2 contained genes whose repression by ATRA was counteracted by EVI1. These genes were significantly repressed by ATRA by at least 2-fold (logFCAr ≤−1 at P < 0.05) and significantly increased by EVI1 in the presence of ATRA by at least 2-fold (logFCEm ≥1 at P < 0.05). In addition, the increase by EVI1 in the presence of ATRA was at least the square of the induction by EVI1 in the absence of ATRA (logFCEm ≥2 *logFCEr). Category 3 was composed of genes whose significant, at least 2-fold induction by ATRA was counteracted by EVI1 in a manner analogous to that described for category 2: logFCAr ≥1 at P < 0.05 and logFCEm ≤−1 at P < 0.05 and logFCEm ≤2 *logFCEr. Category 4 comprised genes whose repression by ATRA was enhanced by, or was observed only in the presence of, EVI1: logFCAr ≤0 and logFCEm ≤−1 at P < 0.05 and logFCEm ≤2 *logFCEr.
Quantitative real-time reverse transcriptase PCR (qRT-PCR)
To confirm the gene expression patterns observed on the arrays, and to assess the efficacy of the GDF15 knockdown, cells were cultured in the same manner as described in the previous section in at least 3 independent experiments. RNA for qRT-PCR was extracted using Trizol reagent (Life Technologies) according to the manufacturer's instructions. cDNA synthesis was performed using M-MLV reverse transcriptase and random hexamer primers (both from Life Technologies). qRT-PCR was carried out with TaqMan Gene Expression Mastermix (Life Technologies) and TaqMan Gene Expression Assays (AGPAT9: Hs00262010_m1, APOBEC3G: Hs00222415_m1, β-2- microglobulin (B2M): 4333766F, BPI: Hs01552756_m1, CCRL2: Hs00243702_s1, EGR3: Hs00231780_m1, GDF15: Hs00171132_m1, LILRB2: Hs01629548_s1, IL1B: Hs01555410_m1, P2RY13: Hs03043902_s1, TGM2: Hs00190278_m1; Life Technologies) on a StepOnePlus Real Time PCR system (Applied Biosystems), and analyzed using the ΔΔCt method,88 with normalization to the housekeeping gene B2M.
Immunoblot analysis
Protein extracts were prepared by boiling cells in 2x sample buffer (125 mM Tris-HCl pH 6.8, 4% (w/v) SDS, 22% (v/v) Glycerol, 0.1 mM EDTA, 0.5% (v/v) ß-mercaptoethanol, bromophenol blue). Alternatively, cells were lysed in RIPA buffer (50 mM Tris-HCl pH 7.4, 1% NP-40, 0.25% sodium deoxycholate, 150 mM NaCl, protease inhibitors (Serva) and phosphatase inhibitor cocktail set II (Merck)), protein concentrations determined using a detergent-compatible protein assay (Bio-Rad), and cell lysates diluted to the same concentrations mixed with 3x SDS loading buffer (150 mM Tris-HCl pH 6.8, 3% SDS, 0.03% bromophenol blue, 30% glycerol, 3% β-mercaptoethanol). Conditioned media was concentrated 20-fold by centrifugation (12 000 g, 10 min, 4°C) through filter units (UFC800396, 5k, MWCO, Millipore) prior to addition of 3x SDS loading buffer. Protein samples were incubated at 95°C for 5 min, separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), and blotted onto PVDF membranes (Pall Corporation or Millipore) using standard procedures.
Antibody hybridizations were performed in 1% or 5% skim milk in Tris-Buffered Saline/0.1% (v/v) Tween-20 (TBST). Antibodies against EVI1 (C50E12, Cell Signaling), GDF15 (07–217, Millipore), β-tubulin (TUB 2.1, Sigma), and α-tubulin (T9026, Sigma) were used at concentrations of 1:1,000, 1:1,000, 1:2,500, and 1:4,000, respectively. Horseradish peroxidase (HRP) conjugated secondary antibodies goat anti-rabbit IgG (111035008, Jackson ImmunoResearch), goat anti-mouse IgG (115035008, Jackson ImmunoResearch), donkey anti-rabbit IgG (NA934, GE Healthcare), and sheep anti-mouse IgG1 (NA931, Amersham) were diluted 1:50,000, 1:50,000, 1:3,000, and 1:4,000, respectively. Signals were detected using SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific) or Immobilon Western HRP Substrate (Millipore), and visualized on X–ray films (Agfa or GE Healthcare). Equal loading of cell culture supernatants was verified by non-specific amidoblack stain.
Intranuclear staining of EVI1
For intranuclear staining of EVI1, 1.5×106 exponentially growing cells per sample were harvested and blocked with 1% (w/v) bovine serum albumin (BSA; Sigma) and 1.25% (v/v) Beriglobin (CSL-Behring) in phosphate buffered saline (PBS) for 15 min on ice. Cells were fixed and permeabilized by incubation in 1 ml 2% (w/v) paraformaldehyde (Sigma) in PBS for 10 min at 37°C, followed by dropwise addition of 9 ml of ice cold methanol. Samples were blocked again, split into 3 aliquots that were incubated with 10 ng of EVI1 antibody (C50E10, Cell Signaling), isotype control antibody (DA1E, Cell Signaling), or no antibody, respectively, and stained with 20 ng of Alexa Fluor 647 conjugated goat anti rabbit antibody (A-21246, Life Technologies). All antibody incubations were carried out in 60 μl of 1% (w/v) BSA in PBS for 1 h on ice. Each step of the protocol was followed by a washing step with either PBS (before fixation) or wash buffer (1% (w/v) BSA in PBS; after fixation). Data acquisition and analysis was performed on a LSR Fortessa using FACSDiva software (BD Biosciences).
Enzyme-linked immunosorbent assay (ELISA)
Quantification of GDF15 in cell culture supernatants was accomplished using a commercially available ELISA kit (DY957, R&D Systems) according to the manufacturer's instructions. Supernatants were supplemented with protease inhibitor cocktail (cOmplete, Roche) and serially diluted down to 1:25 or 1:100 to obtain values within the linear range of the assay. Samples from at least 3 independent experiments were analyzed in technical duplicate. Results represent means + standard errors of concentrations in pg/ml normalized to 1 × 106 cells.
Analysis of proliferation, cell cycle distribution, apoptosis, and differentiation
For proliferation, cell cycle, apoptosis, and differentiation experiments, exponentially growing cells were seeded at a density of 1×105 cells per ml and treated with 1 μM ATRA or the corresponding amount of DMSO for 5-7 d as indicated. Cell numbers were determined using a Casy Counter (Roche). For cell cycle analysis, 5×105 cells per sample were harvested, washed, resuspended in 1 ml of 0.5% (v/v) Tween-20 in 0.5 M citric acid, and passed 3 times through an 18 G injection needle. The isolated nuclei were pelleted and stained by incubation with 0.05 mg/ml propidium iodide and 0.1 mg/ml RNase in PBS. Apoptosis was analyzed by labeling 2×105 cells per sample with Annexin V and 7AAD using the Annexin V-APC Kit (BD Biosciences) according to the manufacturer's instructions. Cells were classified as viable (Annexin V-/7AAD-), apoptotic (Annexin V + /7AAD-) and late apoptotic/secondary necrotic (Annexin V + /7AAD+) as described.89 To stain for the differentiation marker CD11b, 5×105 cells per sample were washed with PBS and incubated with 1.1 μl PE conjugated CD11b (D12, BD Biosciences) or isotype control (Mouse IgG2a,κ, X39, BD Biosciences) antibody in 110 μl PBS for 30 min on ice. Flow cytometric analyses were carried out on an LSR Fortessa SORP using FACSDiva software (BD Biosciences). Cell cycle distribution was quantitatively evaluated using ModFit LT software (Verity Software House). Morphological analysis was performed on cells that had been applied to glass slides using cytospin centrifugation and stained according to a modified Wright procedure.
Funding Statement
This work was supported by the Austrian Science Foundation, grant no P21401-B12 to RW, the Ministry of Health of the Czech Republic, grant no IGA MZD NT13573–4/2012 to KS, the Czech Science Foundation, grant no GA CR P301/12/P407 to ES, and the European Regional Development Fund, grant FNUSA-ICRC (no. CZ.1.05/1.1.00/02.0123) to KS.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
pEcoR-IRES-lyt2 and a vector containing a codon optimized version of the murine Evi1 gene were kindly provided by Herbert Strobl, Institute of Immunology, Medical University of Vienna, Austria, and Jean-Yves Metais and Cynthia Dunbar, NHLBI, NIH, USA, respectively. Ernst Müllner of the Medical University of Vienna is gratefully acknowledged for his outstanding commitment as a member of the thesis committee of BS.
Supplemental Materials
Supplemental data for this article can be accessed on the publisher's website.
References
- 1. Morishita K, Parker D, Mucenski M, Jenkins N, Copeland N, Ihle J. Retroviral activation of a novel gene encoding a zinc finger protein in IL-3-dependent myeloid leukemia cell lines. Cell 1988; 54:831-40; PMID: 2842066; http://dx.doi.org/ 10.1016/S0092-8674(88)91175-0 [DOI] [PubMed] [Google Scholar]
- 2. Haas K, Kundi M, Sperr W, Esterbauer H, Ludwig W, Ratei R, Koller E, Gruener H, Sauerland C, Fonatsch C, et al. Expression and prognostic significance of different mRNA 5'-end variants of the oncogene EVI1 in 266 patients with de novo AML: EVI1 and MDS1/EVI1 overexpression both predict short remission duration. Genes Chromosomes Cancer 2008; 47:288-98; PMID: 18181178; http://dx.doi.org/ 10.1002/gcc.20532 [DOI] [PubMed] [Google Scholar]
- 3. Lugthart S, van Drunen E, van Norden Y, van Hoven A, Erpelinck C, Valk P, Beverloo H, Lowenberg B, Delwel R. High EVI1 levels predict adverse outcome in acute myeloid leukemia: prevalence of EVI1 overexpression and chromosome 3q26 abnormalities underestimated. Blood 2008; 111:4329-37; PMID: 18272813; http://dx.doi.org/ 10.1182/blood-2007-10-119230 [DOI] [PubMed] [Google Scholar]
- 4. Groschel S, Lugthart S, Schlenk R, Valk P, Eiwen K, Goudswaard C, van Putten W, Kayser S, Verdonck L, Lubbert M, et al. High EVI1 expression predicts outcome in younger adult patients with acute myeloid leukemia and is associated with distinct cytogenetic abnormalities. J Clin Oncol 2010; 28:2101-7; PMID: 20308656; http://dx.doi.org/ 10.1200/JCO.2009.26.0646 [DOI] [PubMed] [Google Scholar]
- 5. Vazquez I, Maicas M, Cervera J, Agirre X, Marin-Bejar O, Marcotegui N, Vicente C, Lahortiga I, Gomez-Benito M, Carranza C, et al. Down-regulation of EVI1 is associated with epigenetic alterations and good prognosis in patients with acute myeloid leukemia. Haematologica 2011; 96:1448-56; PMID: 21750091; http://dx.doi.org/ 10.3324/haematol.2011.040535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Russell M, List A, Greenberg P, Woodward S, Glinsmann B, Parganas E, Ihle J, Taetle R. Expression of EVI1 in myelodysplastic syndromes and other hematologic malignancies without 3q26 translocations. Blood 1994; 84:1243-8; PMID: 8049440 [PubMed] [Google Scholar]
- 7. Dreyfus F, Bouscary D, Melle J, Ribrag V, Guesnu M, Varet B. Expression of the Evi-1 gene in myelodysplastic syndromes. Leukemia 1995; 9:203-5; PMID: 7845018 [PubMed] [Google Scholar]
- 8. Thol F, Yun H, Sonntag A, Damm F, Weissinger E, Krauter J, Wagner K, Morgan M, Wichmann M, Gohring G, et al. Prognostic significance of combined MN1, ERG, BAALC, and EVI1 (MEBE) expression in patients with myelodysplastic syndromes. Ann Hematol 2012; 91:1221-33; PMID: 22488406; http://dx.doi.org/ 10.1007/s00277-012-1457-7 [DOI] [PubMed] [Google Scholar]
- 9. Ogawa S, Kurokawa M, Tanaka T, Tanaka K, Hangaishi A, Mitani K, Kamada N, Yazaki Y, Hirai H. Increased Evi-1 expression is frequently observed in blastic crisis of chronic myelocytic leukemia. Leukemia 1996; 10:788-94; PMID: 8656673 [PubMed] [Google Scholar]
- 10. Daghistani M, Marin D, Khorashad J, Wang L, May P, Paliompeis C, Milojkovic D, De Melo V, Gerrard G, Goldman J, et al. EVI-1 oncogene expression predicts survival in chronic-phase CML patients resistant to imatinib treated with second-generation tyrosine kinase inhibitors. Blood 2010; 116:6014-7; PMID: 20855863; http://dx.doi.org/ 10.1182/blood-2010-01-264234 [DOI] [PubMed] [Google Scholar]
- 11. Paquette R, Nicoll J, Chalukya M, Elashoff D, Shah N, Sawyers C, Spiteri E, Nanjangud G, Rao P. Frequent EVI1 translocations in myeloid blast crisis CML that evolves through tyrosine kinase inhibitors. Cancer Genet 2011; 204:392-7; PMID: 21872826; http://dx.doi.org/ 10.1016/j.cancergen.2011.06.002 [DOI] [PubMed] [Google Scholar]
- 12. Konantz M, Andre M, Ebinger M, Grauer M, Wang H, Grzywna S, Rothfuss O, Lehle S, Kustikova O, Salih H, et al. EVI-1 modulates leukemogenic potential and apoptosis sensitivity in human acute lymphoblastic leukemia. Leukemia 2013; 27:56-65; PMID: 22828445; http://dx.doi.org/ 10.1038/leu.2012.211 [DOI] [PubMed] [Google Scholar]
- 13. Nanjundan M, Nakayama Y, Cheng K, Lahad J, Liu J, Lu K, Kuo W, Smith-McCune K, Fishman D, Gray J, et al. Amplification of MDS1/EVI1 and EVI1, located in the 3q26.2 amplicon, is associated with favorable patient prognosis in ovarian cancer. Cancer Res 2007; 67:3074-84; PMID: 17409414; http://dx.doi.org/ 10.1158/0008-5472.CAN-06-2366 [DOI] [PubMed] [Google Scholar]
- 14. Patel J, Appaiah H, Burnett R, Bhat-Nakshatri P, Wang G, Mehta R, Badve S, Thomson M, Hammond S, Steeg P, et al. Control of EVI-1 oncogene expression in metastatic breast cancer cells through microRNA miR-22. Oncogene 2010; 30:1290-301; PMID: 21057539; http://dx.doi.org/ 10.1038/onc.2010.510 [DOI] [PubMed] [Google Scholar]
- 15. Deng X, Cao Y, Liu Y, Li F, Sambandam K, Rajaraman S, Perkins A, Fields A, Hellmich M, Townsend C Jr, et al. Overexpression of Evi-1 oncoprotein represses TGF-beta signaling in colorectal cancer. Mol Carcinog 2013; 52:255-64; PMID: 22161860; http://dx.doi.org/ 10.1002/mc.21852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tanaka M, Suzuki H, Shibahara J, Kunita A, Isagawa T, Yoshimi A, Kurokawa M, Miyazono K, Aburatani H, Ishikawa S, et al. EVI1 oncogene promotes KRAS pathway through suppression of microRNA-96 in pancreatic carcinogenesis. Oncogene 2014; 33:2454-63; PMID: 23752186; http://dx.doi.org/ 10.1038/onc.2013.204 [DOI] [PubMed] [Google Scholar]
- 17. Ho P, Alonzo T, Gerbing R, Pollard J, Hirsch B, Raimondi S, Cooper T, Gamis A, Meshinchi S. High EVI1 expression is associated with MLL rearrangements and predicts decreased survival in paediatric acute myeloid leukaemia: a report from the children's oncology group. Br J Haematol 2013; 162:670-7; PMID: 23826732; http://dx.doi.org/ 10.1111/bjh.12444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Groschel S, Schlenk R, Engelmann J, Rockova V, Teleanu V, Kuhn M, Eiwen K, Erpelinck C, Havermans M, Lubbert M, et al. Deregulated expression of EVI1 defines a poor prognostic subset of MLL-rearranged acute myeloid leukemias: a study of the German-Austrian Acute Myeloid Leukemia Study Group and the Dutch-Belgian-Swiss HOVON/SAKK Cooperative Group. J Clin Oncol 2013; 31:95-103; PMID: 23008312; http://dx.doi.org/ 10.1200/JCO.2011.41.5505 [DOI] [PubMed] [Google Scholar]
- 19. Stein S, Ott M, Schultze-Strasser S, Jauch A, Burwinkel B, Kinner A, Schmidt M, Kramer A, Schwable J, Glimm H, et al. Genomic instability and myelodysplasia with monosomy 7 consequent to EVI1 activation after gene therapy for chronic granulomatous disease. Nat Med 2010; 16:198-204; PMID: 20098431; http://dx.doi.org/ 10.1038/nm.2088 [DOI] [PubMed] [Google Scholar]
- 20. Buonamici S, Li D, Chi Y, Zhao R, Wang X, Brace L, Ni H, Saunthararajah Y, Nucifora G. EVI1 induces myelodysplastic syndrome in mice. J Clin Invest 2004; 114:713-9; PMID: 15343390; http://dx.doi.org/ 10.1172/JCI21716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jin G, Yamazaki Y, Takuwa M, Takahara T, Kaneko K, Kuwata T, Miyata S, Nakamura T. Trib1 and Evi1 cooperate with Hoxa and Meis1 in myeloid leukemogenesis. Blood 2007; 109:3998-4005; PMID: 17227832; http://dx.doi.org/ 10.1182/blood-2006-08-041202 [DOI] [PubMed] [Google Scholar]
- 22. Watanabe-Okochi N, Kitaura J, Ono R, Harada H, Harada Y, Komeno Y, Nakajima H, Nosaka T, Inaba T, Kitamura T. AML1 mutations induced MDS and MDS/AML in a mouse BMT model. Blood 2008; 111:4297-308; PMID: 18192504; http://dx.doi.org/ 10.1182/blood-2007-01-068346 [DOI] [PubMed] [Google Scholar]
- 23. Sitailo S, Sood R, Barton K, Nucifora C. Forced expression of the leukemia-associated gene EVI1 in ES cells: a model for myeloid leukemia with 3q26 rearrangements. Leukemia 1999; 13:1639-45; PMID: 10557037; http://dx.doi.org/ 10.1038/sj.leu.2401585 [DOI] [PubMed] [Google Scholar]
- 24. Kurokawa M, Mitani K, Yamagata T, Takahashi T, Izutsu K, Ogawa S, Moriguchi T, Nishida E, Yazaki Y, Hirai H. The evi-1 oncoprotein inhibits c-Jun N-terminal kinase and prevents stress-induced cell death. EMBO J 2000; 19:2958-68; PMID: 10856240; http://dx.doi.org/ 10.1093/emboj/19.12.2958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kilbey A, Alzuherri H, McColl J, Cales C, Frampton J, Bartholomew C. The Evi1 proto-oncoprotein blocks endomitosis in megakaryocytes by inhibiting sustained cyclin-dependent kinase 2 catalytic activity. Br J Haematol 2005; 130:902-11; PMID: 16156860; http://dx.doi.org/ 10.1111/j.1365-2141.2005.05709.x [DOI] [PubMed] [Google Scholar]
- 26. Yuasa H, Oike Y, Iwama A, Nishikata I, Sugiyama D, Perkins A, Mucenski M, Suda T, Morishita K. Oncogenic transcription factor Evi1 regulates hematopoietic stem cell proliferation through GATA-2 expression. EMBO J 2005; 24:1976-87; PMID: 15889140; http://dx.doi.org/ 10.1038/sj.emboj.7600679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Liu Y, Chen L, Ko T, Fields A, Thompson E. Evi1 is a survival factor which conveys resistance to both TGFbeta- and taxol-mediated cell death via PI3K/AKT. Oncogene 2006; 25:3565-75; PMID: 16462766; http://dx.doi.org/ 10.1038/sj.onc.1209403 [DOI] [PubMed] [Google Scholar]
- 28. Laricchia-Robbio L, Nucifora G. Significant increase of self-renewal in hematopoietic cells after forced expression of EVI1. Blood Cells Mol Dis 2008; 40:141-7; PMID: 17913523; http://dx.doi.org/ 10.1016/j.bcmd.2007.07.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yamakawa N, Kaneda K, Saito Y, Ichihara E, Morishita K. The increased expression of integrin alpha6 (ITGA6) enhances drug resistance in EVI1(high) leukemia. PLoS One 2012; 7:e30706; PMID: 22295105; http://dx.doi.org/ 10.1371/journal.pone.0030706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rommer A, Steinmetz B, Herbst F, Hackl H, Heffeter P, Heilos D, Filipits M, Steinleitner K, Hemmati S, Herbacek I, et al. EVI1 inhibits apoptosis induced by antileukemic drugs via upregulation of CDKN1A/p21/WAF in human myeloid cells. PLoS One 2013; 8:e56308; PMID: 23457546; http://dx.doi.org/ 10.1371/journal.pone.0056308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kustikova O, Schwarzer A, Stahlhut M, Brugman M, Neumann T, Yang M, Li Z, Schambach A, Heinz N, Gerdes S, et al. Activation of Evi1 inhibits cell cycle progression and differentiation of hematopoietic progenitor cells. Leukemia 2013; 27:1127-38; PMID: 23212151; http://dx.doi.org/ 10.1038/leu.2012.355 [DOI] [PubMed] [Google Scholar]
- 32. Morishita K, Parganas E, Matsugi T, Ihle J. Expression of the Evi-1 zinc finger gene in 32Dc13 myeloid cells blocks granulocytic differentiation in response to granulocyte colony-stimulating factor. Mol Cell Biol 1992; 12:183-9; PMID: 1370341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kreider B, Orkin S, Ihle J. Loss of erythropoietin responsiveness in erythroid progenitors due to expression of the Evi-1 myeloid-transforming gene. Proc Natl Acad Sci U S A 1993; 90:6454-8; PMID: 8341654; http://dx.doi.org/ 10.1073/pnas.90.14.6454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kazama H, Kodera T, Shimizu S, Mizoguchi H, Morishita K. Ecotropic viral integration site-1 is activated during, and is sufficient for, neuroectodermal P19 cell differentiation. Cell Growth and Differentiation 1999; 10:565-73; PMID: 10470856 [PubMed] [Google Scholar]
- 35. Shimizu S, Nagasawa T, Katoh O, Komatsu N, Yokota J, Morishita K. EVI1 Is Expressed in Megakaryocyte Cell Lineage and Enforced Expression of EVI1 in UT-7/GM Cells Induces Megakaryocyte Differentiation. Biochem Biophys Res Commun 2002; 292:609-16; PMID: 11922610; http://dx.doi.org/ 10.1006/bbrc.2002.6693 [DOI] [PubMed] [Google Scholar]
- 36. Konrad T, Karger A, Hackl H, Schwarzinger I, Herbacek I, Wieser R. Inducible expression of EVI1 in human myeloid cells causes phenotypes consistent with its role in myelodysplastic syndromes. J Leukoc Biol 2009; 86:813-22; PMID: 19605700; http://dx.doi.org/ 10.1189/jlb.0109042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Karakaya K, Herbst F, Ball C, Glimm H, Kramer A, Loffler H. Overexpression of EVI1 interferes with cytokinesis and leads to accumulation of cells with supernumerary centrosomes in G0/1 phase. Cell Cycle 2012; 11:3492-503; PMID: 22894935; http://dx.doi.org/ 10.4161/cc.21801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Steinleitner K, Rampetsreiter P, Koffel R, Ramanathan G, Mannhalter C, Strobl H, Wieser R. EVI1 and MDS1/EVI1 Expression During Primary Human Hematopoietic Progenitor Cell Differentiation into Various Myeloid Lineages. Anticancer Res 2012; 32:4883-9; PMID: 23155256 [PMC free article] [PubMed] [Google Scholar]
- 39. Nayak K, Kuila N, Das Mohapatra A, Panda A, Chakraborty S. EVI1 targets DeltaNp63 and upregulates the cyclin dependent kinase inhibitor p21 independent of p53 to delay cell cycle progression and cell proliferation in colon cancer cells. Int J Biochem Cell Biol 2013; 45:1568-76; PMID: 23665236; http://dx.doi.org/ 10.1016/j.biocel.2013.04.032 [DOI] [PubMed] [Google Scholar]
- 40. De Weer A, Van der Meulen J, Rondou P, Taghon T, Konrad T, De Preter K, Mestdagh P, Van Maerken T, Van Roy N, Jeison M, et al. EVI1-mediated down regulation of miR449A is essential for the survival of EVI1 positive leukaemic cells. Br J Haematol 2011; 154:337-48; PMID: 21569010; http://dx.doi.org/ 10.1111/j.1365-2141.2011.08737.x [DOI] [PubMed] [Google Scholar]
- 41. Pradhan A, Mohapatra A, Nayak K, Chakraborty S. Acetylation of the proto-oncogene EVI1 abrogates Bcl-xL promoter binding and induces apoptosis. PLoS One 2011; 6:e25370; PMID: 21980434; http://dx.doi.org/ 10.1371/journal.pone.0025370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bard-Chapeau E, Jeyakani J, Kok C, Muller J, Chua B, Gunaratne J, Batagov A, Jenjaroenpun P, Kuznetsov V, Wei C, et al. Ecotopic viral integration site 1 (EVI1) regulates multiple cellular processes important for cancer and is a synergistic partner for FOS protein in invasive tumors. Proc Natl Acad Sci U S A 2012; 109:2168-73; PMID: 22308434; http://dx.doi.org/ 10.1073/pnas.1119229109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Glass C, Wuertzer C, Cui X, Bi Y, Davuluri R, Xiao Y, Wilson M, Owens K, Zhang Y, Perkins A. Global Identification of EVI1 Target Genes in Acute Myeloid Leukemia. PLoS One 2013; 8:e67134; PMID: 23826213; http://dx.doi.org/ 10.1371/journal.pone.0067134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Laricchia-Robbio L, Fazzina R, Li D, Rinaldi C, Sinha K, Chakraborty S, Nucifora G. Point mutations in two EVI1 Zn fingers abolish EVI1-GATA1 interaction and allow erythroid differentiation of murine bone marrow cells. Mol Cell Biol 2006; 26:7658-66; PMID: 16954386; http://dx.doi.org/ 10.1128/MCB.00363-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Senyuk V, Sinha K, Li D, Rinaldi C, Yanamandra S, Nucifora G. Repression of RUNX1 activity by EVI1: a new role of EVI1 in leukemogenesis. Cancer Res 2007; 67:5658-66; PMID: 17575132; http://dx.doi.org/ 10.1158/0008-5472.CAN-06-3962 [DOI] [PubMed] [Google Scholar]
- 46. Laricchia-Robbio L, Premanand K, Rinaldi C, Nucifora G. EVI1 Impairs myelopoiesis by deregulation of PU.1 function. Cancer Res 2009; 69:1633-42; PMID: 19208846; http://dx.doi.org/ 10.1158/0008-5472.CAN-08-2562 [DOI] [PubMed] [Google Scholar]
- 47. Kurokawa M, Mitani K, Irie K, Matsuyama T, Takahashi T, Chiba S, Yazaki Y, Matsumoto K, Hirai H. The oncoprotein Evi-1 represses TGF-beta signalling by inhibiting Smad3. Nature 1998; 394:92-6; PMID: 9665135; http://dx.doi.org/ 10.1038/27945 [DOI] [PubMed] [Google Scholar]
- 48. Izutsu K, Kurokawa M, Imai Y, Maki K, Mitani K, Hirai H. The corepressor CtBP interacts with Evi-1 to repress transforming growth factor beta signaling. Blood 2001; 97:2815-22; PMID: 11313276; http://dx.doi.org/ 10.1182/blood.V97.9.2815 [DOI] [PubMed] [Google Scholar]
- 49. Alliston T, Ko T, Cao Y, Liang Y, Feng X, Chang C, Derynck R. Repression of bone morphogenetic protein and activin-inducible transcription by Evi-1. J Biol Chem 2005; 280:24227-37; PMID: 15849193; http://dx.doi.org/ 10.1074/jbc.M414305200 [DOI] [PubMed] [Google Scholar]
- 50. Bingemann S, Konrad T, Wieser R. Zinc finger transcription factor ecotropic viral integration site 1 is induced by all-trans retinoic acid (ATRA) and acts as a dual modulator of the ATRA response. FEBS J 2009; 276:6810-22; PMID: 19843176; http://dx.doi.org/ 10.1111/j.1742-4658.2009.07398.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Niederreither K, Subbarayan V, Dolle P, Chambon P. Embryonic retinoic acid synthesis is essential for early mouse post-implantation development. Nature Genetics 1999; 21:444-8; PMID: 10192400; http://dx.doi.org/ 10.1038/7788 [DOI] [PubMed] [Google Scholar]
- 52. Zile M. Function of vitamin A in vertebrate embryonic development. J Nutr 2001; 131:705-8; PMID: 11238746 [DOI] [PubMed] [Google Scholar]
- 53. Wei L. Retinoid receptors and their coregulators. Annu Rev Pharmacol Toxicol 2003; 43:47-72; PMID: 12142470; http://dx.doi.org/ 10.1146/annurev.pharmtox.43.100901.140301 [DOI] [PubMed] [Google Scholar]
- 54. Bastien J, Rochette-Egly C. Nuclear retinoid receptors and the transcription of retinoid-target genes. Gene 2004; 328:1-16; PMID: 15019979; http://dx.doi.org/ 10.1016/j.gene.2003.12.005 [DOI] [PubMed] [Google Scholar]
- 55. de The H, Vivanco-Ruiz M, Tiollais P, Stunnenberg H, Dejean A. Identification of a retinoic acid responsive element in the retinoic acid receptor beta gene. Nature 1990; 343:177-80; PMID: 2153268; http://dx.doi.org/ 10.1038/343177a0 [DOI] [PubMed] [Google Scholar]
- 56. Chanda B, Ditadi A, Iscove N, Keller G. Retinoic Acid signaling is essential for embryonic hematopoietic stem cell development. Cell 2013; 155:215-27; PMID: 24074870; http://dx.doi.org/ 10.1016/j.cell.2013.08.055 [DOI] [PubMed] [Google Scholar]
- 57. Purton L, Bernstein I, Collins S. All-trans retinoic acid delays the differentiation of primitive hematopoietic precursors (lin(−)c-kit(+)Sca-1(+)) while enhancing the terminal maturation of committed granulocyte monocyte progenitors. Blood 1999; 94:483-95; PMID: 10397716 [PubMed] [Google Scholar]
- 58. Purton L, Bernstein I, Collins S. All-trans retinoic acid enhances the long-term repopulating activity of cultured hematopoietic stem cells. Blood 2000; 95:470-7; PMID: 10627451 [PubMed] [Google Scholar]
- 59. Collins S. The role of retinoids and retinoic acid receptors in normal hematopoiesis. Leukemia 2002; 16:1896-905; PMID: 12357341; http://dx.doi.org/ 10.1038/sj.leu.2402718 [DOI] [PubMed] [Google Scholar]
- 60. Mi J, Li J, Shen Z, Chen S, Chen Z. How to manage acute promyelocytic leukemia. Leukemia 2012; 26:1743-51; PMID: 22422168; http://dx.doi.org/ 10.1038/leu.2012.57 [DOI] [PubMed] [Google Scholar]
- 61. Burnett A, Hills R, Green C, Jenkinson S, Koo K, Patel Y, Guy C, Gilkes A, Milligan D, Goldstone A, et al. The impact on outcome of the addition of all-trans retinoic acid to intensive chemotherapy in younger patients with nonacute promyelocytic acute myeloid leukemia: overall results and results in genotypic subgroups defined by mutations in NPM1, FLT3, and CEBPA. Blood 2010; 115:948-56; PMID: 19965647; http://dx.doi.org/ 10.1182/blood-2009-08-236588 [DOI] [PubMed] [Google Scholar]
- 62. Aytekin M, Vinatzer U, Musteanu M, Raynaud S, Wieser R. Regulation of the expression of the oncogene EVI1 through the use of alternative mRNA 5'-ends. Gene 2005; 356:160-8; PMID: 16014322; http://dx.doi.org/ 10.1016/j.gene.2005.04.032 [DOI] [PubMed] [Google Scholar]
- 63. Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Statist Soc B 1995; 57:289-300. [Google Scholar]
- 64. Breit S, Johnen H, Cook A, Tsai V, Mohammad M, Kuffner T, Zhang H, Marquis C, Jiang L, Lockwood G, et al. The TGF-beta superfamily cytokine, MIC-1/GDF15: a pleotrophic cytokine with roles in inflammation, cancer and metabolism. Growth Factors 2011; 29:187-95; PMID: 21831009; http://dx.doi.org/ 10.3109/08977194.2011.607137 [DOI] [PubMed] [Google Scholar]
- 65. Vanhara P, Hampl A, Kozubik A, Soucek K. Growth/differentiation factor-15: prostate cancer suppressor or promoter? Prostate Cancer Prostatic Dis 2012; 15:320-8; PMID: 22370725; http://dx.doi.org/ 10.1038/pcan.2012.6 [DOI] [PubMed] [Google Scholar]
- 66. Brooks D, Woodward S, Thompson F, Dos Santos B, Russell M, Yang J, Guan X, Trent J, Alberts D, Taetle R. Expression of the zinc finger gene EVI-1 in ovarian and other cancers. Br J Cancer 1996; 74:1518-25; PMID: 8932329; http://dx.doi.org/ 10.1038/bjc.1996.583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Drexler H, Quentmeier H, MacLeod R, Uphoff C, Hu Z. Leukemia cell lines: in vitro models for the study of acute promyelocytic leukemia. Leuk Res 1995; 19:681-91; PMID: 7500643; http://dx.doi.org/ 10.1016/0145-2126(95)00036-N [DOI] [PubMed] [Google Scholar]
- 68. Lee S, Yamaguchi K, Kim J, Eling T, Safe S, Park Y, Baek S. Conjugated linoleic acid stimulates an anti-tumorigenic protein NAG-1 in an isomer specific manner. Carcinogenesis 2006; 27:972-81; PMID: 16286461; http://dx.doi.org/ 10.1093/carcin/bgi268 [DOI] [PubMed] [Google Scholar]
- 69. Lim J, Woo S, Min K, Park E, Jang J, Seo B, Iqbal T, Lee T, Kim S, Choi Y, et al. Rottlerin induces apoptosis of HT29 colon carcinoma cells through NAG-1 upregulation via an ERK and p38 MAPK-dependent and PKC delta-independent mechanism. Chem Biol Interact 2012; 197:1-7; PMID: 22410117; http://dx.doi.org/ 10.1016/j.cbi.2012.02.003 [DOI] [PubMed] [Google Scholar]
- 70. Wieser R. The oncogene and developmental regulator EVI1: expression, biochemical properties, and biological functions. Gene 2007; 396:346-57; PMID: 17507183; http://dx.doi.org/ 10.1016/j.gene.2007.04.012 [DOI] [PubMed] [Google Scholar]
- 71. Spiekermann K, Pau M, Schwab R, Schmieja K, Franzrahe S, Hiddemann W. Constitutive activation of STAT3 and STAT5 is induced by leukemic fusion proteins with protein tyrosine kinase activity and is sufficient for transformation of hematopoietic precursor cells. Exp Hematol 2002; 30:262-71; PMID: 11882364; http://dx.doi.org/ 10.1016/S0301-472X(01)00787-1 [DOI] [PubMed] [Google Scholar]
- 72. Schepers H, van Gosliga D, Wierenga A, Eggen B, Schuringa J, Vellenga E. STAT5 is required for long-term maintenance of normal and leukemic human stem/progenitor cells. Blood 2007; 110:2880-8; PMID: 17630355; http://dx.doi.org/ 10.1182/blood-2006-08-039073 [DOI] [PubMed] [Google Scholar]
- 73. Brady A, Gibson S, Rybicki L, Hsi E, Saunthararajah Y, Sekeres M, Tiu R, Copelan E, Kalaycio M, Sobecks R, et al. Expression of phosphorylated signal transducer and activator of transcription 5 is associated with an increased risk of death in acute myeloid leukemia. Eur J Haematol 2012; 89:288-93; PMID: 22725130; http://dx.doi.org/ 10.1111/j.1600-0609.2012.01825.x [DOI] [PubMed] [Google Scholar]
- 74. Si J, Collins S. IL-3-induced enhancement of retinoic acid receptor activity is mediated through Stat5, which physically associates with retinoic acid receptors in an IL-3-dependent manner. Blood 2002; 100:4401-9; PMID: 12393611; http://dx.doi.org/ 10.1182/blood-2001-12-0374 [DOI] [PubMed] [Google Scholar]
- 75. van Wely K, Molijn A, Buijs A, Meester-Smoor M, Aarnoudse A, Hellemons A, den Besten P, Grosveld G, Zwarthoff E. The MN1 oncoprotein synergizes with coactivators RAC3 and p300 in RAR-RXR-mediated transcription. Oncogene 2003; 22:699-709; PMID: 12569362; http://dx.doi.org/ 10.1038/sj.onc.1206124 [DOI] [PubMed] [Google Scholar]
- 76. Heuser M, Beutel G, Krauter J, Dohner K, von Neuhoff N, Schlegelberger B, Ganser A. High meningioma 1 (MN1) expression as a predictor for poor outcome in acute myeloid leukemia with normal cytogenetics. Blood 2006; 108:3898-905; PMID: 16912223; http://dx.doi.org/ 10.1182/blood-2006-04-014845 [DOI] [PubMed] [Google Scholar]
- 77. Langer C, Marcucci G, Holland K, Radmacher M, Maharry K, Paschka P, Whitman S, Mrozek K, Baldus C, Vij R, et al. Prognostic importance of MN1 transcript levels, and biologic insights from MN1-associated gene and microRNA expression signatures in cytogenetically normal acute myeloid leukemia: a cancer and leukemia group B study. J Clin Oncol 2009; 27:3198-204; PMID: 19451432; http://dx.doi.org/ 10.1200/JCO.2008.20.6110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Meester-Smoor M, Janssen M, Grosveld G, de Klein A, van IW, Douben H, Zwarthoff E. MN1 affects expression of genes involved in hematopoiesis and can enhance as well as inhibit RAR/RXR-induced gene expression. Carcinogenesis 2008; 29:2025-34; PMID: 18632758; http://dx.doi.org/ 10.1093/carcin/bgn168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Heuser M, Argiropoulos B, Kuchenbauer F, Yung E, Piper J, Fung S, Schlenk R, Dohner K, Hinrichsen T, Rudolph C, et al. MN1 overexpression induces acute myeloid leukemia in mice and predicts ATRA resistance in patients with AML. Blood 2007; 110:1639-47; PMID: 17494859; http://dx.doi.org/ 10.1182/blood-2007-03-080523 [DOI] [PubMed] [Google Scholar]
- 80. Rego E, De Santis G. Differentiation syndrome in promyelocytic leukemia: clinical presentation, pathogenesis and treatment. Mediterr J Hematol Infect Dis 2011; 3:e2011048; PMID: 22110898; http://dx.doi.org/ 10.4084/mjhid.2011.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Rogers J, Yang D. Differentiation syndrome in patients with acute promyelocytic leukemia. J Oncol Pharm Pract 2011; 18:109-14; PMID: 21364078; http://dx.doi.org/ 10.1177/1078155211399163 [DOI] [PubMed] [Google Scholar]
- 82. Ninomiya M, Kiyoi H, Ito M, Hirose Y, Naoe T. Retinoic acid syndrome in NOD/scid mice induced by injecting an acute promyelocytic leukemia cell line. Leukemia 2004; 18:442-8; PMID: 14749706; http://dx.doi.org/ 10.1038/sj.leu.2403284 [DOI] [PubMed] [Google Scholar]
- 83. Xi Z, Russell M, Woodward S, Thompson F, Wagner L, Taetle R. Expression of the Zn finger gene, EVI-1, in acute promyelocytic leukemia. Leukemia 1997; 11:212-20; PMID: 9009083; http://dx.doi.org/ 10.1038/sj.leu.2400547 [DOI] [PubMed] [Google Scholar]
- 84. Hayward S, Dahiya R, Cunha G, Bartek J, Deshpande N, Narayan P. Establishment and characterization of an immortalized but non-transformed human prostate epithelial cell line: BPH-1. In Vitro Cell Dev Biol Anim 1995; 31:14-24; PMID: 7535634; http://dx.doi.org/ 10.1007/BF02631333 [DOI] [PubMed] [Google Scholar]
- 85. Hayward S, Wang Y, Cao M, Hom Y, Zhang B, Grossfeld G, Sudilovsky D, Cunha G. Malignant transformation in a nontumorigenic human prostatic epithelial cell line. Cancer Res 2001; 61:8135-42; PMID: 11719442 [PubMed] [Google Scholar]
- 86. Hamaguchi H, Suzukawa K, Nagata K, Yamamoto K, Yagasaki F, Morishita K. Establishment of a novel human myeloid leukemia cell line (HNT-34) with t(3;3)(q21;q26), t(9;22)(q34;q11) and the expression of EVI1 gene, p210 and p190 BCR/ABL chimaeric transcripts from a patient with AML after MDS with 3q21q26 syndrome. Br J Haematol 1997; 98:399-407; PMID: 9266939; http://dx.doi.org/ 10.1046/j.1365-2141.1997.2143029.x [DOI] [PubMed] [Google Scholar]
- 87. Fellmann C, Hoffmann T, Sridhar V, Hopfgartner B, Muhar M, Roth M, Lai D, Barbosa I, Kwon J, Guan Y, et al. An optimized microRNA backbone for effective single-copy RNAi. Cell Rep 2013; 5:1704-13; PMID: 24332856; http://dx.doi.org/ 10.1016/j.celrep.2013.11.020 [DOI] [PubMed] [Google Scholar]
- 88. Livak K, Schmittgen T. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001; 25:402-8; PMID: 11846609; http://dx.doi.org/ 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
- 89. Logue S, Elgendy M, Martin S. Expression, purification and use of recombinant annexin V for the detection of apoptotic cells. Nat Protoc 2009; 4:1383-95; PMID: 19730422; http://dx.doi.org/ 10.1038/nprot.2009.143 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.