

Abstract
Despite rapid advances in the field of metal-free, “frustrated Lewis pair” (FLP)-catalyzed hydrogenation, the need for strictly anhydrous reaction conditions has hampered wide-scale uptake of this methodology. Herein, we report that, despite the generally perceived moisture sensitivity of FLPs, 1,4-dioxane solutions of B(C6F5)3 actually show appreciable moisture tolerance and can catalyze hydrogenation of a range of weakly basic substrates without the need for rigorously inert conditions. In particular, reactions can be performed directly in commercially available nonanhydrous solvents without subsequent drying or use of internal desiccants.
Keywords: “frustrated Lewis pairs”, catalytic hydrogenation, water tolerance, solvent effects, metal-free
With the advent of “frustrated Lewis pair” (FLP) chemistry, metal-free catalytic hydrogenation has managed to progress, in less than a decade, from the first example of reversible metal-free H2 activation,1 into an established area of active research.2 In this remarkably short span of time, the substrate scope of FLP hydrogenation methodologies (in which H2 is activated through the cooperative action of a Lewis acid/base pair precluded from forming a strong classical adduct) has expanded from the initial basic imines and aziridines3 to include a wide variety of other functional groups,2 including heterocycles,4 simple alkenes, and alkynes,5 and most recently, aldehydes and ketones.6
As FLP chemistry becomes more established, focus has inevitably begun to shift from early, “proof-of-concept” studies to the aim of developing more practical and widely applicable FLP catalysts. To this end, recent reports have been seen emphasizing the development of FLPs that can operate with low catalyst loadings,5a,7 or with high enantioselectivity.8 Despite these advances, one factor that seriously limits the attractiveness of the FLP catalysts reported to date is their extreme sensitivity (and perceived sensitivity) to moisture9 (as well as many other functional groups),4,9,10 requiring these reagents to be handled and employed under rigorously inert conditions through use of gloveboxes and Schlenk lines. This lack of H2O tolerance represents a significant practical barrier to the uptake of FLP catalysis by the broader chemical community that must be overcome if FLPs are truly to become viable alternatives to transition metal hydrogenation catalysts.
Several attempts have been made to mitigate this problem in recent years. For example, we have reported the use of B(C6Cl5)(C6F5)211 as a bench-stable Lewis acid for FLP chemistry;12 however, this system still required the use of rigorously dry reaction conditions (including freshly distilled, anhydrous solvent) during catalytic hydrogenations. Separately, Ingleson et al. have described the use of the N-methylacridinium cation as a Lewis acid for FLP H2 activation in wet 1,2-dichlorobenzene,13 yet these reaction conditions were not extended to subsequent hydrogenation catalysis. In addition, significant hydrolysis (40%) was observed. More recently, Stephan et al. have reported an FLP-catalyzed deoxygenation of aryl ketones and, although this reaction produces H2O as a byproduct, the use of molecular sieves as a reagent ensures that the reaction mixture remains strictly anhydrous.6c As such, the challenge of developing simple, H2O-tolerant FLP catalytic systems remains an open one.14
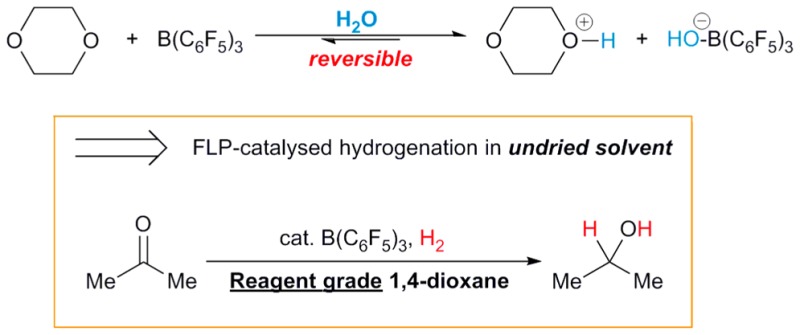
The origins of FLPs’ moisture sensitivity can be understood by considering as an example the archetypal Lewis acid of FLP chemistry: commercially available B(C6F5)3 (1). Because of its high Lewis acidity, complexation of H2O not only is strong, but also leads to significant Brønsted acidification (the pKa of [1·OH2] has been found to be comparable to that of HCl)15 and accordingly, deprotonation by even moderately strong bases (including the amines and phosphines typically employed in FLP chemistry) is irreversible. Under more forcing conditions, the adduct [1·OH2] is also prone to decomposition via B–C bond protonolysis (Scheme 1).16
Scheme 1. Pathways for Deactivation of 1 by H2O.
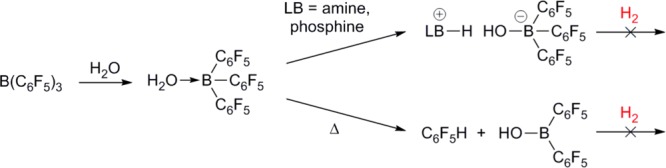
Recently, we6a and Stephan et al.6b have independently reported the hydrogenation of ketones and aldehydes to alcohols, catalyzed by 1 in ethereal solvents (Scheme 2). These reactions are clearly tolerant of hydroxylic functionalities, which is attributed to the weak Brønsted basicity of the ethers employed, which inhibits irreversible deprotonation of the highly acidic [1·ROH] adducts.
Scheme 2. Previously-Reported Hydrogenation of Aldehydes and Ketones Catalyzed by 1.6a,6b.

R = aryl, alkyl; R′ = alkyl, H.
Following on from these results, we reasoned that these systems might also be capable of tolerating the presence of H2O, for similar reasons, which would represent an important advance for the field of FLP chemistry. Initial investigations focused on the hydrogenation of acetone (the simplest substrate previously examined under anhydrous conditions). Gratifyingly, when 1 was replaced with the preformed adduct 1·OH215 under identical conditions, selective hydrogenation to 2-propanol was observed to proceed cleanly and with good turnover number (Table 1, entry 2). Although the rate of reaction is reduced (c.f. Table 1, entry 1), confirming that H2O acts as a significant catalyst poison, inhibition by H2O is nonetheless clearly reversible. At the time, this represented the first example of an FLP-catalyzed hydrogenation that is tolerant of stoichiometric (relative to catalyst) H2O.
Table 1. Metal-free catalytic hydrogenation of acetone in the presence of various amounts of H2O.

Increasing the H2 pressure led to a significant rate enhancement, even at lower catalyst loadings (Table 1, entry 3) and without any detectable catalyst decomposition (ascertained by 19F and 11B NMR spectroscopy). Under these conditions, several more equivalents of H2O could be tolerated, with an attendant decrease in rate yet otherwise no major difference in reaction outcome (Table 1, entries 4 and 5).
On the basis of the ability of this system to tolerate multiple equivalents of water, we reasoned that the use of “undried” solvents ought to be achievable; impressively, the reaction could be performed very effectively in nonanhydrous commercial solvent17even without any need for subsequent drying, degassing, or other purification. Furthermore, doubling the substrate and catalyst concentrations allowed for a significant decrease in reaction time (Table 1, entry 6).
The mechanism by which the hydrogenation is believed to occur is identical to that proposed for the anhydrous reaction, with [1·OH2] acting as an off-cycle resting state (Scheme 3).18 As with previously observed alcohol tolerance, H2O tolerance is attributed to the lack of any strong base, meaning irreversible deprotonation of [1·OH2] does not occur (c.f. Scheme 1). Even so, it seems possible that reversible deprotonation does occur, which could be consistent with the acidity of this adduct (vide supra).19 Evidence for deprotonation comes from 11B NMR spectroscopy: although [1·OH2] alone shows an 11B NMR resonance at 4.6 ppm in the noncoordinating yet highly polar solvent 1,2-difluorobenzene (DFB), addition of 1,4-dioxane leads to a clear upfield shift, to −0.6 ppm with 1 equiv of ethereal base, and −2.1 ppm with 10 equiv (Figure 1).20 Typical nonanhydrous reaction mixtures, which contain [1·OH2] in neat 1,4-dioxane, produce resonances farther upfield still, at −3.0 ppm. For comparison, the 11B NMR shift of [NMe4][1·OH], which contains the “free” conjugate base of [1·OH2], has been reported as −2.1 ppm in CD2Cl2, with similar shifts for related salts.211H NMR analysis also suggests an interaction between [1·OH2] and 1,4-dioxane, with the 1,4-dioxane resonance shifted slightly downfield, from 3.56 ppm in the absence of [1·OH2], to 3.59 ppm in the presence of 1 equiv, indicating overall deshielding. Although displacement of H2O for 1,4-dioxane could also potentially explain these shifts in the NMR spectra, this possibility can be discounted: addition of 1 and 10 equiv of 1,4-dioxane to dry1 in DFB leads to 11B NMR resonances at 9.8 and 5.7 ppm, respectively, and 1,4-dioxane 1H resonances at 3.96 and 3.59 ppm, respectively; in all cases significantly farther downfield than when H2O is also present.
Scheme 3. Proposed Mechanism for Moisture-Tolerant Hydrogenation of Acetone.
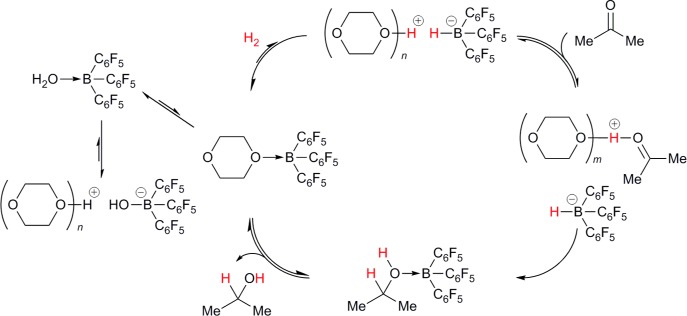
Possible hydrogen bonding of solvent with [1·ROH] and [1·OH2] omitted for clarity.
Figure 1.

Variation in 11B NMR chemical shift of [1·OH2] upon addition of 1,4-dioxane in DFB.
Reversible deprotonation of [1·OH2] and the related [1·ROH] adducts may partially explain the large effect that pressure has been observed to have on both the anhydrous and nonanhydrous reactions. Increasing H2 pressure will increase the solution concentration of H2 and, in turn, the degree of H2 activation. The resulting increase in Brønsted acid concentration should perturb the equilibrium between [1·OH2]/[1·iPrOH] and [1·OH]−/[1·OiPr] – in favor of the more weakly bound neutral adducts, hence facilitating catalytic turnover via ROH/H2O dissociation from 1.
Given that the rationale for H2O tolerance in this system depends primarily on the absence of a strong base, it seemed reasonable that catalytic hydrogenation without the need for rigorously dry conditions might also be achievable for other weakly basic substrates. To this end, a variety of additional substrates (both carbonyls and noncarbonyls) were treated under similar conditions. For most, clean, efficient catalytic reduction was observed without the need for extensive further optimization (Table 2, entries 1–7), clearly indicating the generality of this water-tolerant methodology. As well as using undried solvent, reactions could be performed under open bench conditions without the need for inert atmosphere containment systems22 (although long-term storage of 1 is best carried out in a glovebox). By contrast, the attempted reduction of cyclohexanone under the same conditions was unsuccessful (Table 2, entry 8), which is qualitatively consistent with previous observations made under dry conditions,6a for which higher pressures were required for hydrogenation than for the other carbonyl substrates. Collectively, these results suggest a similar substrate scope for the anhydrous and nonanhydrous reaction protocols.
Table 2. Metal-Free Hydrogenation of Weakly-Basic Substrates in Non-Anhydrous Solvent.
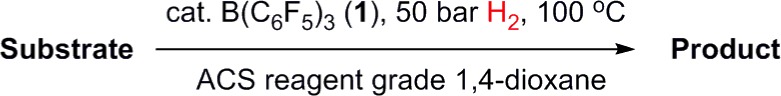
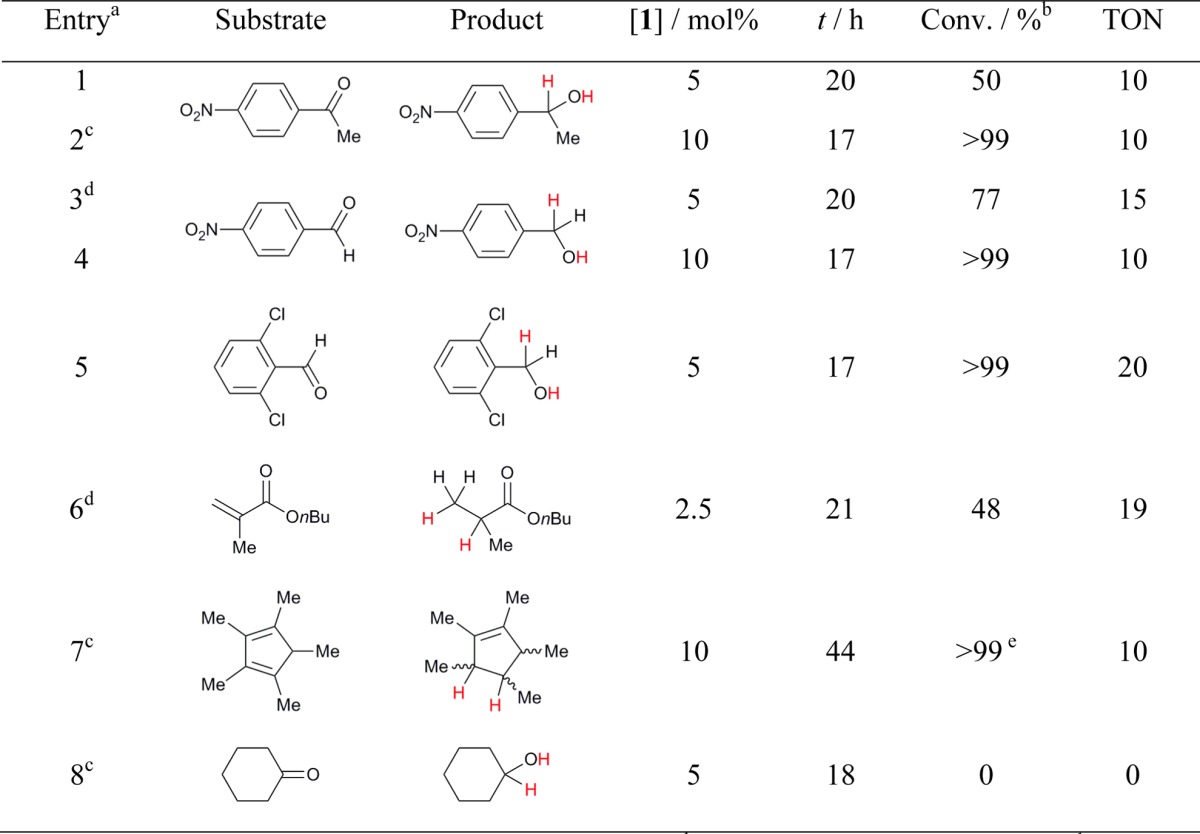
Standard conditions: 4.0 mmol substrate, 0.53 M.
All conversions measured by 1H NMR integration.
2.0 mmol substrate.
8.0 mmol substrate.
Unquantified mixture of isomers.
In addition to the advantages already discussed, moisture-tolerance should also allow FLPs to catalyze reactions that produce H2O. To this end, acetophenone was exposed to our anhydrous reaction conditions. Clean, catalytic reductive deoxygenation to ethylbenzene23 was observed, despite the concomitant formation of H2O (5 equiv relative to catalyst) that necessarily occurs (Scheme 4).
Scheme 4. Hydrogenation of Acetophenone to Ethylbenzene and H2O Catalyzed by 1.
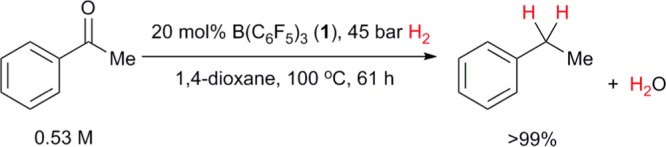
In conclusion, we have demonstrated a number of examples of FLP-catalyzed hydrogenation reactions demonstrating appreciable water tolerance. As a result, a variety of weakly basic substrates can be hydrogenated cleanly and in high yield, in the absence of a transition metal catalyst, in commercial-grade “bench” (undried) solvents. By removing the need for both extensive, laborious drying of reaction solvents and inert atmosphere reaction techniques, this development significantly increases the practicality of the FLP hydrogenation methodology. These findings extend the current reach of FLP hydrogenation catalysis from rigorously anhydrous research laboratory conditions into industrially relevant, commercially available solvent grades and reaction conditions.
Acknowledgments
We would like to thank GreenCatEng, Eli Lilly (Pharmacat consortium), and the EPSRC for providing funding for a Ph.D. studentship (D.J.S.), and Dr. George Britovsek and Prof. Ramon Vilar for access to high-pressure equipment. G.G.W. and A.E.A. thank the Royal Society for financial support through University Research Fellowships. The research leading to these results has received funding from the European Research Council under the ERC Starting Grant Agreement No. 307061 (PiHOMER) and ERC PoC Grant Agreement No. 640988 (FLPower).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscatal.5b01417.
Full experimental details (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Welch G. C.; Juan R. R. S.; Masuda J. D.; Stephan D. W. Science 2006, 314, 1124–1126. 10.1126/science.1134230. [DOI] [PubMed] [Google Scholar]
- For recent reviews, see:; a Stephan D. W.; Erker G. Angew. Chem., Int. Ed. 2015, 54, 6400–6441. 10.1002/anie.201409800. [DOI] [PubMed] [Google Scholar]; b Stephan D. W. Acc. Chem. Res. 2015, 48, 306–316. 10.1021/ar500375j. [DOI] [PubMed] [Google Scholar]
- Chase P. A.; Welch G. C.; Jurca T.; Stephan D. W. Angew. Chem., Int. Ed. 2007, 46, 8050–8053. 10.1002/anie.200702908. [DOI] [PubMed] [Google Scholar]
- Stephan D. W.; Greenberg S.; Graham T. W.; Chase P.; Hastie J. J.; Geier S. J.; Farrell J. M.; Brown C. C.; Heiden Z. M.; Welch G. C.; Ullrich M. Inorg. Chem. 2011, 50, 12338–12348. 10.1021/ic200663v. [DOI] [PubMed] [Google Scholar]
- a Xu X.; Kehr G.; Daniliuc C. G.; Erker G. J. Am. Chem. Soc. 2015, 137, 4550–4557. 10.1021/jacs.5b01623. [DOI] [PubMed] [Google Scholar]; b Chernichenko K.; Madarász Á.; Pápai I.; Nieger M.; Leskelä M.; Repo T. Nat. Chem. 2013, 5, 718–723. 10.1038/nchem.1693. [DOI] [PubMed] [Google Scholar]
- a Scott D. J.; Fuchter M. J.; Ashley A. E. J. Am. Chem. Soc. 2014, 136, 15813–15816. 10.1021/ja5088979. [DOI] [PubMed] [Google Scholar]; b Mahdi T.; Stephan D. W. J. Am. Chem. Soc. 2014, 136, 15809–15812. 10.1021/ja508829x. [DOI] [PubMed] [Google Scholar]; c Mahdi T.; Stephan D. W. Angew. Chem., Int. Ed. 2015, 54, 8511–8514. 10.1002/anie.201503087. [DOI] [PubMed] [Google Scholar]
- Farrell J. M.; Posaratnanathan R. T.; Stephan D. W. Chem. Sci. 2015, 6, 2010–2015. 10.1039/C4SC03675A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Lindqvist M.; Borre K.; Axenov K.; Kótai B.; Nieger M.; Leskelä M.; Pápai I.; Repo T. J. Am. Chem. Soc. 2015, 137, 4038–4041. 10.1021/ja512658m. [DOI] [PubMed] [Google Scholar]; b Zhang Z.; Du H. Org. Lett. 2015, 17, 2816–2819. 10.1021/acs.orglett.5b01240. [DOI] [PubMed] [Google Scholar]; c Ren X.; Li G.; Wei S.; Du H. Org. Lett. 2015, 17, 990–993. 10.1021/acs.orglett.5b00085. [DOI] [PubMed] [Google Scholar]; d Wei S.; Du H. J. Am. Chem. Soc. 2014, 136, 12261–12264. 10.1021/ja507536n. [DOI] [PubMed] [Google Scholar]
- Thomson J. W.; Hatnean J. A.; Hastie J. J.; Pasternak A.; Stephan D. W.; Chase P. A. Org. Process Res. Dev. 2013, 17, 1287–1292. 10.1021/op4000847. [DOI] [Google Scholar]
- a Erős G.; Mehdi H.; Pápai I.; Rokob T. A.; Király P.; Tárkányi G.; Soós T. Angew. Chem., Int. Ed. 2010, 49, 6559–6563. 10.1002/anie.201001518. [DOI] [PubMed] [Google Scholar]; b Erős G.; Nagy K.; Mehdi H.; Pápai I.; Nagy P.; Király P.; Tárkányi G.; Soós T. Chem. - Eur. J. 2012, 18, 574–585. 10.1002/chem.201102438. [DOI] [PubMed] [Google Scholar]; c Greb L.; Daniliuc C. G.; Bergander K.; Paradies J. Angew. Chem., Int. Ed. 2013, 52, 5876–5879. 10.1002/anie.201210175. [DOI] [PubMed] [Google Scholar]
- Ashley A. E.; Herrington T. J.; Wildgoose G. G.; Zaher H.; Thompson A. L.; Rees N. H.; Krämer T.; O’Hare D. J. Am. Chem. Soc. 2011, 133, 14727–14740. 10.1021/ja205037t. [DOI] [PubMed] [Google Scholar]
- Scott D. J.; Fuchter M. J.; Ashley A. E. Angew. Chem., Int. Ed. 2014, 53, 10218–10222. 10.1002/anie.201405531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark E. R.; Ingleson M. J. Angew. Chem., Int. Ed. 2014, 53, 11306–11309. 10.1002/anie.201406122. [DOI] [PubMed] [Google Scholar]
- While this manuscript was under review, Soós et al. reported a moisture-tolerant protocol for FLP-catalysed hydrogenation of aldehydes and ketones using novel boranes specifically designed for that purpose. Their approach (targeted design of new catalysts, rather than adaptation of existing FLP hydrogenation protocols) complements the work reported herein.; Gyömöre Á.; Bakos M.; Földes T.; Pápai I.; Domján A.; Soós T. ACS Catal. 2015, 10.1021/acscatal.5b01299. [DOI] [Google Scholar]
- pKa = 8.4 in MeCN, c.f. 8.5 for HCl:Bergquist C.; Bridgewater B. M.; Harlan C. J.; Norton J. R.; Friesner R. A.; Parkin G. J. Am. Chem. Soc. 2000, 122, 10581–10590. 10.1021/ja001915g. [DOI] [Google Scholar]
- a Bradley D. C.; Harding I. S.; Keefe A. D.; Motevalli M.; Zheng D. H. J. Chem. Soc., Dalton Trans. 1996, 3931–3936. 10.1039/dt9960003931. [DOI] [Google Scholar]; b Ashley A. E.; Thompson A. L.; O’Hare D. Angew. Chem., Int. Ed. 2009, 48, 9839–9843. 10.1002/anie.200905466. [DOI] [PubMed] [Google Scholar]
- ACS reagent grade 1,4-dioxane purchased from Sigma Aldrich was used. Karl Fischer titration indicated 220 ppm/0.022% H2O (w/w) (advertised as up to 0.05%).
- We propose that 1,4-dioxane acts as the main base for hydrogen activation due to the vast excess (12 M) in which it is present; however, in principle, this role could also be played by water or 2-propanol.
- The pKa of protonated 1,4-dioxane has been measured as −2.92 in aqueous solution. Although the aqueous pKa of [1·OH2] has not been measured,15 our results indicate that it may be comparable. See:; Arnett E.; Wu C. Y. J. Am. Chem. Soc. 1960, 82, 4999–5000. 10.1021/ja01503a060. [DOI] [Google Scholar]; .
- It should be noted that these observations could also be consistent with strong hydrogen bonding to the solvent rather than full formal deprotonation; in practice, the true situation might be expected to lie somewhere between these two extremes.
- a Hewavitharanage P.; Danilov E. O.; Neckers D. C. J. Org. Chem. 2005, 70, 10653–10659. 10.1021/jo050695s. [DOI] [PubMed] [Google Scholar]; b Bibal C.; Santini C. C.; Chauvin Y.; Vallee C.; Olivier-Bourbigou H. Dalton Trans. 2008, 37, 2866–2870. 10.1039/b718057h. [DOI] [PubMed] [Google Scholar]
- For all experiments [H2O] > [1], as indicated by control experiments: see SI.
- 1H NMR analysis of an aliquot taken at partial conversion showed formation of styrene (in addition to ethylbenzene and the expected alcohol intermediate), which is consistent with previous observations made at lower pressure.6a At completion, only ethylbenzene was observed, however, indicating that the alkene undergoes further reduction (either directly, or via rehydration to the alcohol).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.