

Abstract
Indolotryptolines are bisindole natural products isolated from microbial and eDNA sources. Here we report the sequence of transformations that convert an indolocarbazole to the indolotryptoline cladoniamide through reconstruction of the four-enzyme cascade in E. coli. This cascade involves, first, conversion of an indolocarbozole to a C4c-C7a cis diol by ClaX1; second, N-methylation by ClaM1; third, rearrangement to the indolotryptoline scaffold by ClaX2; and fourth, installation of an O-methyl group by ClaM3. We furthermore elucidate the origins of minor cladoniamides D-G as the products of non-enzymatic, base-catalyzed opening of the succinimide ring of cladonimiades A-B. Overall, this work reveals the precarious pathway indolocarbazole-derived metabolites must traverse as they are converted into indolotryptoline products and highlights the importance of non-enzymatic chemistry in generating bisindole diversity.
INTRODUCTION
A unique group of bacterially-derived alkaloids are bisindoles, molecules derived from the oxidative dimerization of L-tryptophan.1 Among bisindoles, indolocarbazoles are the most frequently reported structures from bacteria and include examples such as rebeccamycin and staurosporine.2 Other, non-indolocarbazole bisindoles include indolotryptolines, which have a rearranged bisindole core. Cladoniamides A-G (1–7), isolated from a Streptomyces strain, were the first indolotryptolines to be reported with structural information.3 Screening of environmental DNA libraries for chromopyrrolic acid synthase genes has also resulted in identification of gene clusters that when heterologously expressed generate indolotryptolines BE-54017 (8)4 and borregomycin A (9),5 giving a total of three small families of natural indolotryptolines (Figure 1). Cladoniamides A and B are nanomolar cytotoxic agents against colon cancer cell line HCT-116,4,6 whereas cladoniamide G is the active lead analogue against breast cancer cell line MCF-7.3 In vitro studies of BE-54017 show that the compound induces apoptosis in cells overexpressing epidermal growth factor receptor (EGFR), possibly through inhibition of the vacuolar-type H+-ATPase.7 Borregomycin A, by contrast, appears to be a specific inhibitor of kinase CaMKIIδ with an IC50 of 3.4 μM.5 Total synthesis of several indolotryptolines has been reported.7–10
Figure 1.

Indolotryptoline structures and corresponding gene clusters. a) Cladoniamides. b) BE-54017. c) Borregomycin A. d) Cladoniamide (cla), BE-54017 (abe), and borregomycin (bor) gene clusters with flavoenzyme genes X1 and X2 in orange, methyltransferase genes M1 and M3 are in red, indolocarbazole genes O, D, P, and C in blue, halogenase and associated reductase genes in green, regulator and transporter genes in black, and other genes in gray.
The more common indolocarbazole natural products are known to derive from the oxidative dimerization of L-tryptophan derivatives through a four-enzyme cascade that funnels substrates towards indolocarbazole aglycones.2 Early on, with the discovery of the indolotryptoline cladoniamide, it was hypothesized that the rare indolotryptoline molecules might derive from more common indolocarbazole aglycones.3 The pathway was proposed to involve the enzymatic destruction and rearrangement of an indolocarbazole to generate the indolotryptoline scaffold. The first support for this hypothesis came with isolation of the biosynthetic gene cluster for indolotryptoline BE-54017 (6).4 This cluster contains the four genes needed for indolocarbazole formation, along with several new genes. Transposon mutagenesis at the flavoenzyme gene abeX1 locus in the abe cluster led to the accumulation of the indolocarbazole 3-chloroarcyriaflavin A, providing experimental evidence for a link between indolocarbazole and indolotryptoline biosynthesis. Additionally, transposon mutagenesis at the abeX2 locus, another putative flavoenzyme gene, led to the accumulation of an N6, N13-dimethyl-C4c/C7a diol-derivative of an indolocarbazole.4 Collectively, these results suggest that the conversion of an indolocarbazole to an indolotryptoline might involve epoxidation of the C4c/C7a double bond by AbeX1, followed by epoxide hydrolysis, and then enzyme-catalyzed N-methyltransfer reactions. Then, putative flavoenzyme AbeX2 is hypothesized to carry out the subsequent conversion of the indolocarbazole to an indolotryptoline according to the original biogenetic proposal.3,4 Identification and heterologous expression of indolotryptoline cladoniamide (cla) and borregomycin (bor) gene clusters, both of which contain abeX1 and abeX2 homologs and the four indolocarbazole genes, bolstered the claim that the two putative flavoenzyme genes are likely critical to the formation of the indolotryptoline core.5,6,11
These prior studies set the stage for a full exploration of the chemical transformations that enable production of indolotryptolines from indolocarbazole precursors. In this report, we use a combination of deletion mutagenesis, in vitro biochemistry, and biotransformations to establish the pathway leading from an indolocarbazole to an indolotryptoline. We elucidate the unexpected cis arrangement of hydroxyls in the indolocarbazole C4c/C7a diol intermediate and provide the first experimental demonstration that putative flavoenzyme ClaX2 is critical to rearrangement of an indolocarbazole to an indolotryptoline. We also characterize a unique, indolo-oxazino-carbazole metabolite, and we show that this metabolite derives spontaneously from the indolocarbazole diol. Furthermore, we demonstrate that production of the minor cladoniamides D-G (4–7) occurs through non-enzymatic destruction of cladoniamides A-B (1–2) in mild alkaline conditions. Overall, our work reveals the precarious pathway metabolites must traverse from indolocarbazole precursors to indolotryptoline products and emphasizes the importance of non-enzymatic pathways in the generation of bisindole diversity.
RESULTS AND DISCUSSION
Recent work on the biosynthesis of BE-54017 included the generation of several mutants by transposon mutagenesis, however, the N-methyltransferase gene abeM1 was not directly interrogated.4 Therefore, the ability of the corresponding, downstream cladoniamide enzymes to accept non-N6-methylated products was unknown. We thus generated strain S. coelicolor YD52, which has an engineered cla cluster whose claM1 gene was in-frame deleted (Figure S1). Large-scale (12 L) fermentation of S. coelicolor YD52 was performed, and a series of bisindoles were purified from the extract of its culture and characterized based on MS and 1D and 2D NMR analysis (Figures 2a, S2, S9–S13, and S15 and Tables S4, S5 and S7–S9). The major compounds are diols 10, 11, and 12. To determine whether the hydroxyls are cis or trans, we determined the X-ray crystallographic structure of 10, revealing a cis configuration (Figures 2b and Table S6). From S. coelicolor YD52, we also observed a series of compounds (13/14 and 15) whose MS and NMR data are consistent with an indolo[2,3-a][1,3]oxazino[5,6-c]carbazole scaffold. Additionally, demethylxenocladoniamide A (16) was a minor product. Finally, trace amounts of demethylcladoniamides A (18), D (19), and F (20), and demethylxenocladoniamide B (17) were identified by their LC-MS and UV-Vis spectra (Table S3 and Figure S8). The identities and production levels of these metabolites suggest that the likely substrates for ClaM1 are 10–12 and/or 13–15. Furthermore, these results show that downstream cladoniamide enzymes favor the N6-methylated substrates but that non-methylated substrates can (at trace levels) migrate through the entire biosynthetic pathway. We tested selected new compounds for biological activity against human colon cancer cell line HCT-116, and we observed only modest IC50 values of 2.97 μM for 13/14 and 5.65 μM for 15 (Table S10).
Figure 2.
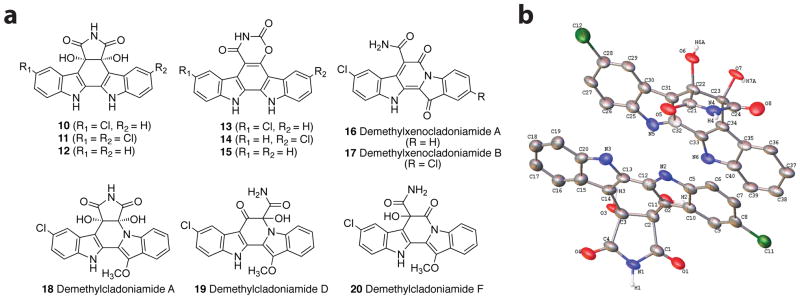
Molecules from S. coelicolor M1146 + cla (ΔclaM1). a) Structures for major (10–15), minor (16), and trace (17–20) compounds. b) ORTEP-style image of compound 10, drawn with 33% probability ellipses. All C-H hydrogen atoms were removed for clarity.
To provide more direct evidence for the transformations catalyzed by cladoniamide enzymes, we sought to reconstruct the cladoniamide pathway in E. coli using synthetic indolocarbazole precursors. We undertook a heterologous biotransformation approach, rather than an in vitro approach, as all efforts to access soluble ClaX1 and ClaX2, including testing of their codon-optimized homologues AbeX1 and AbeX2, were unsuccessful. We reasoned that arcyriaflavin A (21), a commercially available, non-chlorinated indolocarbazole, would be a direct substrate for the first enzyme, ClaX1, based on the production of non-chlorinated metabolites from both cla and abe pathways. We thus fed 21 to the E. coli culture expressing claX1. HPLC analysis revealed the production of two new metabolites, both of which are also produced when E. coli/claX1 is replaced with E. coli/abeX1. As parallel controls, no products were detected when arcyriaflavin A was provided to E. coli strains containing empty vector or claX2 (Figure S3a). These two metabolites were identified to be 12 and 15 by comparison with standard compounds purified from the claM1 deletion mutant. Observation of the indolo-oxazino-carbazole 15 from this biotransformation assay was unexpected. To investigate whether 15 is direct product of ClaX1-catalyzed reaction or instead a decomposed product of 12, we repeated the feeding experiment and analyzed the culture extract at different time points. The result showed the accumulation of large amount of 15 only after longer incubations (12h) (Figure S3b), suggesting that 15 might derive from 12.
To probe the next step in construction of cladoniamide, an in vitro biochemical assay of ClaM1 was performed. ClaM1 was purified as a C-terminal His6-tagged protein from E. coli BL21(DE3). Compounds 12, 15 and 21 were used in the in vitro assay. Incubation of 12 with ClaM1 in the presence of S-adenosyl-L-methionine (SAM) led to the formation of a new product, the molecular mass of which is consistent with that of the N-methyl diol 22 ([M+H]+ ion at m/z 374, [M+TFA-H]− ion at m/z 486) (Figure S4). Additionally, a small amount of 15 formed in all the control and experimental assays, suggesting that 12 partially converted to 15 under the condition we used. No product was observed when we used 21 as a substrate in this assay, confirming that the ClaM1-catalyzed N-methylation occurs after the ClaX1-catalyzed oxidation. Interestingly, 15 can also be accepted as a substrate by ClaM1 and gives a new product. This compound was isolated from a large-scale ClaM1 reaction and confirmed to be the N-methylated indolo-oxazino-carbazole 23 by 1D and 2D NMR (Figure S14 and Tables S7–S8).
To investigate the role of ClaX2, three genes (abeX1, claM1 and claX2) were cloned and assembled in two compatible E. coli vectors for co-expression with all the genes behind the T7 promoter. Since the work described above showed that ClaX1 and AbeX1 are functionally equivalent, we used abeX1 to perform all the subsequent experiments for the convenience of cloning, as this synthetic gene lacks an NdeI restriction site. Successful expression of all three genes was confirmed by SDS-PAGE (Figure S5). Arcyriaflavin A (21) was provided to engineered E. coli strains carrying different combinations of genes, in a similar biotransformation assay as described for claX1. As expected, E. coli/X1M1 produced the N-methyl diol 22 in vivo, whereas E. coli/X1M1X2 produced an additional metabolite (Figure 3a). This compound was identified to be xenocladoniamide C (24) by comparison with a standard compound, and its poor production could be due to the limited soluble ClaX2 pool in the E. coli host. Xenocladoniamides A-C (Figure S6) were previously isolated as major products from a cladoniamide heterologous production system where O-methyltransferase gene claM3 has been inactivated.6 These molecules were proposed to be shunt products that derive from the putative substrates of ClaM3 through non-enzymatic, spontaneous decomposition. Our current data thus linked the X1-M1-X2 reactions with the last step of cladoniamide A-C biosynthesis, performed by ClaM3. Xenocladoniamide C (24) production was also observed when claX2 was replaced with abeX2 in this feeding experiment (data not shown).
Figure 3.

Heterologous reconstitution of the X1-M1-X2-M3 enzymatic cascade in an E. coli host. a) HPLC analysis of the metabolic profiles of (i) E. coli Ctrl3 (empty vectors) + 21, (ii) E. coli X1 (pACYCDuet-abeX1) + 21, (iii) E. coli X1M1 (pACYCDuet-abeX1claM1) + 21, (iv) E. coli X1M1X2 (pACYCDuet-abeX1claM1+ pCOLADuet-claX2) + 21, (v) 24 standard and (vi) 12 standard. Detection wavelength: 348 nm. b) Heterologous production of cladoniamide B (2) by feeding 3,9-dichloro-arcyriaflavin A (◇) to E. coli host carrying the X1-M1-X2-M3 enzymatic cascade. (i) E. coli Ctrl3 (empty vectors), (ii) E. coli X1M1X2M3 (pACYCDuet-abeX1claM1+ pCOLADuet-claX2 + pET22b-claM3), (iii) 2 standard compound, (iv) 3,9-dichloro-arcyriaflavin A (◇) standard compound. Detection wavelength: 348 nm. c) Biosynthetic construction of the cladoniamides, with on-pathway metabolites shown within the box. ClaX2 is critical to ‘flipping’ of the indole ring (in dashed circle) to give the indolotryptoline scaffold of the cladoniamides, and a possible mechanism is shown in the dashed box. Non-enzymatic processes are thought to lead to shunt products 15 and 24, whereas ClaM1 catalyzes the conversion of 15 to 23. Non-chlorinated metabolites are shown, but similar pathways are envisioned for chlorinated metabolites.
To further confirm that the four-enzyme cascade (X1-M1-X2-M3) is sufficient for the conversion of an indolocarbazole precursor to the cladoniamide scaffold, we reconstructed the whole X1M1X2M3 pathway in E. coli. In assays using arcyriaflavin A, we found that the retention time of cladoniamide C is identical to a metabolite (retention time (tR): 20.0 min) from E. coli culture when analyzed by HPLC. Therefore, we used 3,9-dichloro-arcyriaflavin A to feed the engineered E. coli strain, instead of using arcyriaflavin A (21) (Figure S16). HPLC analysis of the ethyl acetate extract of the E. coli/X1M1X2M3 culture revealed the formation of cladoniamide B (2), whereas no cladoniamide B (2) is produced from control experiments (Figure 3b). These results confirm that ClaX1M1X2M3 convert an indolocarbazole precursor into cladoniamides (Figure 3c). Similar functions are expected for the homologs from BE-54017 (AbeX1, AbeM1, AbeX2, and AbeM3) and borregomycin (BorX1, BorM1 and BorX2) biosynthetic pathways.4,5
Having established the biogenetic origins of the major cladoniamides, we turned our attention to the minor metabolites, cladoniamides D-G (4–7). At this point, all the biosynthetic cla genes, with the exception of claY, encoding a putative hydrolase, have defined functions in construction of cladoniamides A-C (1–3). Both a recent study on the abe pathway4 and our data on a claY deletion mutant in Streptomyces uncialis (Figure S7) suggest that ClaY/AbeY are not essential for the biosynthesis of either major or minor cladoniamides. We thus sought to probe whether the formation of cladoniamides D-G is a non-enzymatic consequence. Although we observed that cladoniamide A is stable in methanol at room temperature for ~ 1 year (data not shown), we tested whether it is susceptible to decomposition in either acidic or basic conditions. Previously, we noted that extension of the fermentation time increases the titer of the minor metabolites cladoniamides D-G in S. coelicolor + cla.12 Other studies have shown that during the fermentation process of Streptomyces, the first round of growth is acidogenic and reinitiation of growth coincides with the consumption of organic acids in Streptomyces, resulting in an increased pH.13,14 Thus, we first determined the pH value of the Streptomyces cultures at the end of fermentation, and we found that the pH is ~7.4 for S. albus + cla, and the pH is ~7.9 for S. coelicolor + cla. The S. coelicolor + cla strain is known to produce more minor cladoniamides,6 suggesting that an increase in pH might explain the observed differences in metabolic profile. Thus, pure cladoniamide A (1) and B (2) were added to buffered aqueous solutions (pH 8.0). After 24 hours of incubation, cladoniamides D/F (4/6) and E/G (5/7) were detected from the solution of cladoniamide A (1) and B (2), respectively (Figure 4a). To further investigate the condition for this conversion, cladoniamide A (1) was added to a series of buffered aqueous solutions (Figure 4b). The result shows that opening of the N-methyl-succinimide ring can occur in both neutral and alkaline solutions, and full conversion is observed at pH 8.8 after 24 hours of incubation. Thus, formation of cladoniamides D-G (4–7) is a base-catalyzed, non-enzymatic process (Figure 4c).
Figure 4.

Origins of minor cladoniamides D–G (4–7). a) Conversion of cladoniamide A (1) and B (2) to D (4)/F (6) and E (5)/G (7), respectively. HPLC trace of (i) the crude extract of the S. coelicolor host carrying cla cluster, which produces 1, 2, 4, 5, 6, and 7,6 (ii) incubation of 1 in pure H2O for 24 h, (iii) incubation of 1 in 25 mM Tris-HCl buffer (pH 8.0) for 24 h, (iv) incubation of 2 in pure H2O for 24 h, (v) incubation of 2 in 25 mM Tris-HCl buffer (pH 8.0) for 24 h. b) Conversion of cladoniamide A (1) to D (4)/F (6) in different buffers after 24 h incubation. HPLC trace of (i) incubation of 1 in pure H2O, (ii) incubation of 1 in 25 mM NaOAc buffer (pH 5.3), (ii) incubation of 1 in 25 mM phosphate buffer (pH 6.3), (iv) incubation of 1 in 25 mM phosphate buffer (pH 6.8), (v) incubation of 1 in 25 mM phosphate buffer (pH 7.0), (vi) incubation of 1 in 25 mM Tris-HCl buffer (pH 7.5), (vii) incubation of 1 in 25 mM Tris-HCl buffer (pH 8.0) and (viii) incubation of 1 in 25 mM Tris-HCl buffer (pH 8.8). Detection wavelength: 348 nm. Note that compounds 1–2 and 4–6 have different UV-Vis spectra; see Figure S8. c) Scheme for production of minor cladoniamides in basic conditions.
Prior genetic studies suggested that indolocarbazoles are the precursors to indolotryptolines, and that two putative flavoenzymes are critical to generation of the indolotryptoline core.4 Our heterologous biotransformation work now confirms the role of each enzyme in the pathway, demonstrating the critical role of ClaX1 in forming a C4c/C7a diol of an indolocarbazole and of ClaX2 in mediating formation of the indolotryptoline structure itself. Furthermore, we gained some mechanistic insight into the function of ClaX1 from analysis of 10, purified from the ΔclaM1 strain. Compound 10, which is also a product of ClaX1 catalysis, has its hydroxyls in a cis configuration, in contrast to what would be expected for hydrolysis of an epoxide through an SN2-type mechanism. One scenario to account for the biogenesis of 10 is that ClaX1, a putative flavin monooxygenase, epoxidizes the C4c/C7a double bond of the indolocarbazole. Spontaneous ring-opening of a protonated epoxide would then generate a resonance-stabilized carbocation at either the C4c or C7a carbon, which would then be attacked by water. One possible explanation for the formation of a cis diol, rather than a trans diol, is that the corresponding trans fused 5,6 ring system is strained and therefore less stable. Similarly, trans diol formation through an SN2-type opening of an epoxide is likely disfavored due to formation of a strained trans fused 5,6 ring system. The exact function of ClaX2 is less clear. However, our data now demonstrate that ClaX2, alone, can catalyze the ‘flipping’ of the indole ring to give the indolotryptoline scaffold. Based on the annotation of ClaX2 as a flavin monooxygenase, the mechanism is likely to involve a key epoxidation (or hydroxylation) across the C7b/C12b double bond of the N-methylated indolocarbazole diol, priming the molecule for a spontaneous rearrangement.3 The inability to purify either soluble ClaX1 or ClaX2 has hampered detailed in vitro studies of these enzymes; nonetheless, our heterologous biotransformation is the first unequivocal demonstration of the activity of these enzymes in the indolotryptoline pathway. Flavoenzymes occupy an important place in the biosynthesis of complex alkaloids, with flavoenzymes such as FqzB,15,16 NotB,17 or XiaI18,19 acting to install oxygens in electron-rich substrates to ultimately generate unique chemical structures, and ClaX1 and ClaX2 are likely to similarly install oxygen at key positions to drive formation of the diol-indolocarbazole and indolotryptoline, respectively. In future studies, full characterization of ClaX1 and ClaX2 homologs, likely isolated from other indolotryptoline pathways, will hopefully allow us to investigate the mechanistic details of the chemistry that converts the indolocarbazole to an indolotryptoline.
In this work, we interrogate the formation of indolotryptolines through heterologous biotransformation studies, establishing the order of action of enzymes in converting indolocarbazoles to indolotryptolines. Our studies have revealed the precarious pathway indolocarbazole-derived metabolites must traverse as they are converted into final indolotryptoline products. Indeed, almost every molecule on the pathway, including the major cladoniamide products themselves, is confronted with potential decomposition routes that can derail production of the target metabolites. Our previous genetic studies on claM3 identified the xenocladoniamides as likely spontaneous decomposition products, and our work here, from a different experimental approach, again demonstrates that xenocladoniamides can arise in the absence of O-methylation by ClaM3. Furthermore, we show that the product of ClaX1, the C4c/C7a diol derivative of an indolocarbazole, is also unstable, spontaneously converting to an indolo-oxazino-carbazole over time. Finally, we show that facile, base-catalyzed destruction of succinimide ring of the major cladoniamides can give rise to the minor cladoniamides D-G (4–7). Bisindole metabolites walk a tight-rope as they navigate toward the final indolotryptoline products, threatened at almost every step by their own high reactivity as electron-rich intermediates with appended hydroxyl groups. Indeed, the rich chemical diversity of molecules isolated from the cla and related clusters is likely a result of the action of unique enzyme-catalyzed transformations combined with spontaneous chemical processes.20
MATERIALS AND METHODS
Supplementary Material
Acknowledgments
We are grateful to J. Chen, E. Polishchuk, and X.-Y. Deng for assistance with the cytotoxicity work and to H.-Y. He for feedback on the manuscript. Financial support was provided by CIHR, Genome BC, Grand Challenges Canada, and NSERC. Y.-L. Du is supported by a Michael Smith Foundation for Health Research Fellowship.
Footnotes
ASSOCIATED CONTENT
Supplementary figures and tables, materials and methods. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
References
- 1.Ryan KS, Drennan CL. Divergent pathways in the biosynthesis of bisindole natural products. Chem Biol. 2009;16:351–64. doi: 10.1016/j.chembiol.2009.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sánchez C, Méndez C, Salas JA. Indolocarbazole natural products: occurrence, biosynthesis, and biological activity. Nat Prod Rep. 2006;23:1007–45. doi: 10.1039/b601930g. [DOI] [PubMed] [Google Scholar]
- 3.Williams DE, Davies J, Patrick BO, Bottriell H, Tarling T, Roberge M, Andersen RJ. Cladoniamides A-G, tryptophan-derived alkaloids produced in culture by Streptomyces uncialis. Org Lett. 2008;10:3501–4. doi: 10.1021/ol801274c. [DOI] [PubMed] [Google Scholar]
- 4.Chang FY, Brady SF. Cloning and characterization of an environmental DNA-derived gene cluster that encodes the biosynthesis of the antitumor substance BE-54017. J Am Chem Soc. 2011;133:9996–9999. doi: 10.1021/ja2022653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chang FY, Brady SF. Discovery of indolotryptoline antiproliferative agents by homology-guided metagenomic screening. Proc Natl Acad Sci U S A. 2013;110:2478–2483. doi: 10.1073/pnas.1218073110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Du YL, Ding T, Ryan KS. Biosynthetic O-methylation protects cladoniamides from self-destruction. Org Lett. 2013;15:2538–2541. doi: 10.1021/ol401036f. [DOI] [PubMed] [Google Scholar]
- 7.Kimura T, Kanagaki S, Matsui Y, Imoto M, Watanabe T, Shibasaki M. Synthesis and assignment of the absolute configuration of indenotryptoline bisindole alkaloid BE-54017. Org Lett. 2012;14:4418–21. doi: 10.1021/ol3019314. [DOI] [PubMed] [Google Scholar]
- 8.Loosley BC, Andersen RJ, Dake GR. Total synthesis of cladoniamide G. Org Lett. 2013;15:1152–1154. doi: 10.1021/ol400055v. [DOI] [PubMed] [Google Scholar]
- 9.Ngernmeesri P, Soonkit S, Konkhum A, Kongkathip B. Formal synthesis of (±)-cladoniamide G. Tetrahedron Lett. 2014;55:1621–1624. [Google Scholar]
- 10.Schütte J, Kilgenstein F, Fischer M, Koert U. Unsymmetrical vic-tricarbonyl compounds for the total syntheses of cladoniamide G and cladoniamide F: Unsymmetrical vic-tricarbonyl compounds. Eur J Org Chem. 2014:5302–5311. [Google Scholar]
- 11.Ryan KS. Biosynthetic gene cluster for the cladoniamides, bis-indoles with a rearranged scaffold. PloS One. 2011;6:e23694. doi: 10.1371/journal.pone.0023694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Du YL, Ding T, Patrick BO, Ryan KS. Xenocladoniamide F, minimal indolotryptoline from the cladoniamide pathway. Tetrahedron Lett. 2013;54:5635–5638. [Google Scholar]
- 13.Viollier PH, Minas W, Dale GE, Folcher M, Thompson CJ. Role of acid metabolism in Streptomyces coelicolor morphological differentiation and antibiotic biosynthesis. J Bacteriol. 2001;183:3184–3192. doi: 10.1128/JB.183.10.3184-3192.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Du YL, Shen XL, Yu P, Bai LQ, Li YQ. Gamma-butyrolactone regulatory system of Streptomyces chattanoogensis links nutrient utilization, metabolism, and development. Appl Env Microbiol. 2011;77:8415–26. doi: 10.1128/AEM.05898-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ames BD, Liu X, Walsh CT. Enzymatic processing of fumiquinazoline F: a tandem oxidative-acylation strategy for the generation of multicyclic scaffolds in fungal indole alkaloid biosynthesis. Biochemistry. 2010;49:8564–8576. doi: 10.1021/bi1012029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tsunematsu Y, Ishikawa N, Wakana D, Goda Y, Noguchi H, Moriya H, Hotta K, Watanabe K. Distinct mechanisms for spiro-carbon formation reveal biosynthetic pathway crosstalk. Nat Chem Biol. 2013;9:818–825. doi: 10.1038/nchembio.1366. [DOI] [PubMed] [Google Scholar]
- 17.Li S, Finefield JM, Sunderhaus JD, McAfoos TJ, Williams RM, Sherman DH. Biochemical characterization of NotB as an FAD-dependent oxidase in the biosynthesis of notoamide indole alkaloids. J Am Chem Soc. 2012;134:788–791. doi: 10.1021/ja2093212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li H, Zhang Q, Li S, Zhu Y, Zhang G, Zhang H, Tian X, Zhang S, Ju J, Zhang C. Identification and characterization of xiamycin A and oxiamycin gene cluster reveals an oxidative cyclization strategy tailoring indolosesquiterpene biosynthesis. J Am Chem Soc. 2012;134:8996–9005. doi: 10.1021/ja303004g. [DOI] [PubMed] [Google Scholar]
- 19.Xu Z, Baunach M, Ding L, Hertweck C. Bacterial synthesis of diverse indole terpene alkaloids by an unparalleled cyclization sequence. Angew Chem, Int Ed Engl. 2012;124:10439–10443. doi: 10.1002/anie.201204087. [DOI] [PubMed] [Google Scholar]
- 20.Howard-Jones AR, Walsh CT. Nonenzymatic oxidative steps accompanying action of the cytochrome P450 enzymes StaP and RebP in the biosynthesis of staurosporine and rebeccamycin. J Am Chem Soc. 2007;129:11016–7. doi: 10.1021/ja0743801. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.