

Abstract
Environmental cues are transmitted to the interior of the cell via a complex network of signaling hubs. Receptor tyrosine kinases (RTKs) and trimeric G proteins are 2 such major signaling hubs in eukaryotes. Canonical signal transduction via trimeric G proteins is spatially and temporally restricted, i.e., triggered exclusively at the plasma membrane (PM) by agonist activation of G-protein-coupled receptors (GPCRs) via a process that completes within a few hundred milliseconds. Recently, a rapidly emerging paradigm has revealed a non-canonical pathway for activation of trimeric G proteins by the non-receptor GEF, GIV/Girdin, that has distinctive temporal and spatial features. Such activation can be triggered by multiple growth factor RTKs, can occur at the PM and on internal membranes discontinuous with the PM, and can continue for prolonged periods of time. The molecular mechanisms that govern such non-canonical G protein activation and the relevance of this new paradigm in health and disease is discussed.
Keywords: girdin, GIV, G protein -coupled receptors, growth factor receptor tyrosine kinases, heterotrimeric G proteins
Environmental cues are transmitted to the interior of the cell via a complex network of signaling hubs. Receptor tyrosine kinases (RTKs) and trimeric G proteins are 2 such major signaling hubs in eukaryotes. For several decades these 2 pathways were believed to operate in a discrete mode by transducing signals through their respective downstream intermediates; upon ligand stimulation RTKs propagate the signals to the interior of the cell via adaptor proteins that are recruited to phosphotyrosines on the receptor tail,1 whereas GPCRs, which are 7-transmembrane (TM) receptors with an intrinsic Guanine nucleotide Exchange Factor (GEF) activity, recruit and activate G proteins by triggering the exchange of GDP with GTP.2 However, mounting evidence over time has unfolded a complex array of cross-talk between these 2 pathways. Although transactivation of RTKs by GPCRs is a well-documented and widely-accepted phenomenon, the reverse concept, i.e., transactivation of trimeric G proteins by RTKs has remained controversial. The controversy was fueled largely by the fact that there was no evidence that G proteins and ligand-activated RTKs come within close proximity in cells, nor that RTKs, or any member of the growing family of signal transducing adaptors used by RTKs can serve as GEFs. A lot of these unanswered questions were clarified by the discovery and characterization of Gα-Interacting Vesicle associated protein (GIV; a.k.a Girdin), an unusual molecule that can bind both RTKs and G proteins.3
GIV is a multi-modular signal transducer and a non-receptor GEF for Gαi.4 Working downstream of a variety of growth factors [EGF, IGF, VEGF, Insulin and PDGFR] GIV modulates, i.e., either enhances or suppresses a variety of signaling pathways, all via its ability to activate Gαi in the close proximity of a ligand-activated RTK.3 Multiple studies [summarized in]3 employing a selective GEF-deficient GIV mutant (F1685A) have demonstrated that the signaling network downstream of RTKs in cells with wild-type GIV is a mirror image of the network in cells expressing a GEF-deficient mutant GIV.3 Consistent with its ability to integrate signals downstream of multiple receptors, both at the PM and on other subcellular organelles, GIV modulates diverse cellular processes (Table 1) and GIV-dependent signaling has been implicated in a number of pathophysiologic conditions (Table 2).
Table 1.
| Cellular Process/ Organelle Functions | Effect of GIV's GEF function | Citation |
|---|---|---|
| Cell Migration | “ON” = Enhances, “OFF” = Inhibits | 4,10-16 |
| Golgi Structure and Secretory Function | “ON” = Preserves and enhances, “OFF” = Disrupts, delays | 8 |
| Autophagy | “ON” = Halt and rescue, “OFF” = Initiate and promote | 7 |
| Endosome Maturation | “ON” = Rapid, “OFF” = Slowed | 17 |
| Cell Survival | “ON” = Survive, “OFF” = Apoptosis | 14,18 |
| Cell Polarity | “ON” = Polarity achieved, “OFF” = Loss of polarity | 19,37 |
| Cell Division | Not examined | 20 |
| Endocytosis | Not examined | 21 |
| Cell-cell Junctions, Permeability | Not examined | 22 |
| Neuronal Migration and Differentiation | Not examined | 23,24 |
| Macrophage Chemotaxis | Not examined | 12 |
| Cell Size | Not examined | 25 |
Table 2.
| Disease/ Pathology Investigated | Effect of GIV's GEF function | Receptor(s) Studied | Citation | ||
|---|---|---|---|---|---|
| Cancer Progression | Migration/Invasion | “ON” = Enhances “OFF” = Inhibits | IGF1R, EGFR, Multi-receptor* | 5,9,26-28 | |
| Stemness | Not examined | — | 29 | ||
| Chemoresistance | Not examined | — | 30 | ||
| Tumor-Stroma Interactions | Not examined | PDGFR, TGFβR, CXCR4 | 31 | ||
| Tumor angiogenesis | Not examined | VEGFR | 32 | ||
| Organ Fibrosis (Liver) | Myofibroblast transdifferentiation, collagen production, chemotaxis, mitosis, anti-apoptotic signaling | “ON” = Enhances “OFF” = Inhibits | PDGFR, CCR1, TGFβR | 18 | |
| Dermal Wound Healing | Wound closure | “ON” = Enhances “OFF” = Inhibits | Multi-receptor* | 9 | |
| Nephrotic Syndrome | Podocyte survival after glomerular injury | “ON” = Enhances survival “OFF” = Inhibits survival | VEGFR | 14 | |
| Disorders of Blood Vessels | Neonatal vascular development; Pathologic neovascularization; vein repair; vein graft | Not examined | PDGF, Angiotensin II, VEGF | 33-35 | |
| Neuronal Plasticity, Memory formation | Synaptic plasticity | Not examined | NMDA | 36 | |
Emergence of a New Paradigm; GIV plus RTKs Equals GPCRs
The molecular mechanisms that govern how GIV influences a diverse range of pathophysiologic processes and how it may couple activation of G protein to multiple receptors have come to light only recently,3 and are understood best in the context of a numerous RTKs that signal via GIV. GIV-dependent growth factor signaling appears to rely heavily on the unique modular make-up of its C-terminus (CT), within which 2 unlikely domains coexist (Fig. 1) a previously defined GEF motif via which GIV binds and activates Gi and 2) a ∼110 aa stretch which folds into a SH2-like domain in the presence of phosphotyrosine ligands; the latter is necessary and sufficient to recognize and bind specific sites of autophosphorylation on the receptor tail. The discovery of coexisting SH2-like and GEF modules in-tandem within GIV's C-terminus supported the idea that GIV had the necessary modular make-up to serve as a platform for convergent signaling downstream of multiple RTKs via G proteins. That such a platform is functional in cells was demonstrated only recently using genetically encoded, GIV-derived fluorescent biosensors 5 in living cells for bimolecular fluorescent complementation (BiFC) or Förster resonance energy transfer (FRET) imaging studies, or a combination of both. These studies revealed that the C-terminus of GIV represents the smallest, functionally autonomous unit that retains most key signaling properties of full length GIV: 1) It can bind and activate Gαi in cells in a GEF dependent manner; 2) It retains the properties of receptor recruitment and signal transduction characteristic of full length GIV; and 3) It serves as a bona fide platform for the assembly of RTK-Gαi complexes at the PM and for transactivation of Gαi and suppression of cAMP in response to growth factors. FRET studies in living cells also revealed that although the extent of G protein activation downstream of RTKs and GPCRs appear similar, the spatiotemporal dynamics of non-canonical G protein activation by GIV represents a clear deviation from the dynamics of canonical G protein signaling that is triggered by GPCRs. Canonical signal transduction via trimeric G proteins is spatially and temporally restricted, i.e., triggered exclusively at the PM by agonist activation of GPCRs via a process that completes within a few hundred milliseconds.6 Non-canonical transactivation of trimeric G proteins by RTKs via the GIV-CT platform has distinctive features. Such activation can be triggered by multiple growth factor RTKs as well as other receptors,3 can occur at the PM and on internal membranes discontinuous with the PM7,8 (Table 1), and can continue for prolonged periods of time (several minutes).5 These findings helped substantiate an emerging paradigm in which growth factor RTKs can access and activate G proteins via GIV-CT, much like GPCRs, but with different spatial and temporal dynamics.
Figure 1.
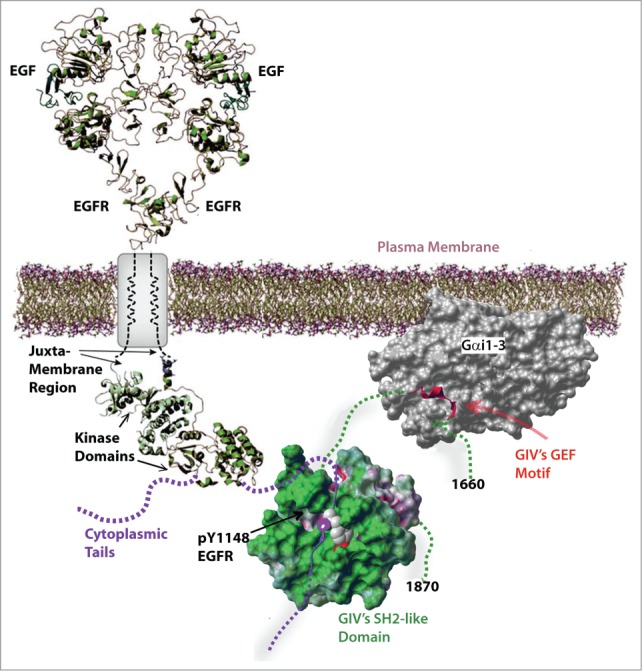
Schematic showing the modular makeup of G-protein coupled growth factor receptor tyrosine kinases. The coexistence of 2 unlikely modules within C-terminus (aa 1660–1870) is critical for the coupling of G proteins to RTKs. An evolutionarily conserved ∼30 aa long α helix that is necessary and sufficient to bind and activate Gαi. A ∼110 aa stretch which is intrinsically disordered in resting state, but is necessary and sufficient to fold into a SH2-like domain (green) exclusively in the presence of phosphotyrosine ligands presented by autophosphorylated cytoplasmic tails (purple interrupted line) of various ligand-activated RTKs. It is the unique coexistence of 2 (GEF and SH2-like) modules that is key, because their collaboration is required and sufficient to assemble RTK-GIV-Gαi complexes at the PM and transactivate Gi.5 Thus, upon growth factor stimulation, GIV's C-terminus enables the assembly of G-protein coupled RTKs, which subsequently leads to the non-canonical transactivation of G proteins. Targeting GIV's GEF function uncouples RTKs from G proteins, prevents non-canonical G protein signaling in response to growth factors, and effectively modulates a variety of cellular processes and pathophysiologic states 9.
Targeting an emerging paradigm
With the emergence of a new paradigm that broadly impacts a variety of disease states (Table 2), the next hurdle was to find a way to target this pathway. The canonical G protein/GPCR pathway has long been the target of small molecule therapeutics accounting for 30% of the launched drug targets, but the unusual spatiotemporal features of non-canonical G protein signaling via RTKs brings forward a unique set of challenges. How to target a pathway that appears to serve as a point of convergence downstream of multiple receptors,3 is functional in multiple cells/tissues and is frequently deregulated in multiple pathophysiologic states,3 and that can activate G proteins at the PM as well as on internal membranes?7,8 Recently, we provided proof-of-concept that such targeting can be achieved by cell-permeable GIV-CT peptides which contain the minimal modular elements of GIV that are necessary and sufficient for activation of Gi downstream of RTKs appended by a TAT-peptide transduction domain (TAT-PTD).9 TAT-GIV-CT peptides could effectively engineer signaling networks and alter cell behavior. In the presence of an intact GEF motif, TAT-GIV-CT peptides enhanced diverse processes in which GIV's GEF function has previously been implicated; e.g., 2D cell migration after scratch-wounding, invasion of cancer cells, and finally, myofibroblast activation and collagen production. Furthermore, topical application of TAT-GIV-CT peptides enhanced wound repair in mice in a GEF-dependent manner. Although cell-permeable peptides allowed exogenous modulation of the RTK-GIV-Gi pathway, it is unlikely that these TAT-appended peptides will serve as marketable pharmacologic agents. But the lessons we learned are invaluable because it appears that GIV-CT peptides may be optimal for potential gene therapy applications to manipulate Gαi activation downstream of multiple growth factors in different cell types and in a diverse array of pathophysiologic conditions. We speculate that these peptides will also modulate other pathophysiologic conditions in which GIV is implicated, but the role of its GEF function is yet to be interrogated (Table 2).3
Future Directions
Despite the insights gained and the rapidity with which the paradigm of G protein-coupled RTKs has shaped up, it is clear that a lot remains unknown. Although homology modeling has proved insightful thus far, obtaining structural insights is expected to greatly facilitate the development of small molecules that can selectively target the GIV:RTK and/or GIV:Gαi interfaces. Targeting such targeted therapy is also expected to pose significant challenges because GIV's GEF function is important for a variety of cell biological processes in diverse cell types.3 Although we have some understanding of how RTKs transactivate G proteins via GIV, how other classes of receptors,3 such as GPCRs, Toll-like receptors (TLRs), Transforming growth factor (TGFβ) receptors also do the same remains unclear. Finally, how non-canonical G protein activation at the PM by GIV-GEF coordinately triggers the same on internal membranes is another unsolved mystery. Thus, it is clear that the emerging paradigm has many unanswered questions. One thing is for sure, with this trajectory, we are in for an exciting new decade ahead.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This work was funded by NIH (R01CA160911 and DK099226).
References
- 1.Schlessinger J. Receptor tyrosine kinases: legacy of the first two decades. Cold Spring Harb Perspect Biol, 2014; 6(3):pii: a008912; PMID:24591517; http://dx.doi.org/ 10.1101/cshperspect.a008912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gilman AG. G proteins: transducers of receptor-generated signals. Annu Rev Biochem, 1987; 56:p. 615-49; http://dx.doi.org/ 10.1146/annurev.bi.56.070187.003151 [DOI] [PubMed] [Google Scholar]
- 3.Garcia-Marcos M, Ghosh P, Farquhar MG. GIV/Girdin transmits signals from multiple receptors by triggering trimeric G protein activation. J Biol Chem, 2015; 290(11):p. 6697-704; http://dx.doi.org/ 10.1074/jbc.R114.613414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Garcia-Marcos M, Ghosh P, Farquhar MG. GIV is a nonreceptor GEF for G alpha i with a unique motif that regulates Akt signaling. Proc Natl Acad Sci U S A, 2009; 106(9):p. 3178-83; PMID:19211784; http://dx.doi.org/ 10.1073/pnas.0900294106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Midde KK, Aznar N, Laederich MB, Ma GS, Kunkel MT, Newton AC, Ghosh P. Multimodular biosensors reveal a novel platform for activation of G proteins by growth factor receptors. Proc Natl Acad Sci U S A, 2015; 112(9):p. E937-46; PMID:25713130; http://dx.doi.org/ 10.1073/pnas.1420140112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lohse MJ, Hein P, Hoffmann C, Nikolaev VO, Vilardaga JP, Bünemann M. Kinetics of G-protein-coupled receptor signals in intact cells. Br J Pharmacol, 2008; 153 Suppl 1:p. S125-32; PMID:18193071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Garcia-Marcos M, Ear J, Farquhar MG, Ghosh P. A GDI (AGS3) and a GEF (GIV) regulate autophagy by balancing G protein activity and growth factor signals. Mol Biol Cell, 2011; 22(5):p. 673-86; PMID:21209316; http://dx.doi.org/ 10.1091/mbc.E10-08-0738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lo IC, Gupta V, Midde KK, Taupin V, Lopez-Sanchez I, Kufareva I, Abagyan R, Randazzo PA, Farquhar MG, Ghosh P. Activation of Galphai at the Golgi by GIV/Girdin Imposes Finiteness in Arf1 Signaling. Dev Cell, 2015; 33(2):p. 189-203; PMID:25865347; http://dx.doi.org/ 10.1016/j.devcel.2015.02.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ma GS, Aznar N, Kalogriopoulos N, Midde KK, Lopez-Sanchez I, Sato E, Dunkel Y, Gallo RL, Ghosh P. Therapeutic effects of cell-permeant peptides that activate G proteins downstream of growth factors. Proc Natl Acad Sci U S A, 2015; 112(20):E2602-10; PMID:25926659; http://dx.doi.org/ 10.1073/pnas.1505543112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Garcia-Marcos M, Ghosh P, Farquhar MG. GIV/ Girdin transmits signals from multiple receptors by triggering trimeric G protein activation. J Biol Chem, 2015; 290(11):6697-704; PMID:25605737; http://dx.doi.org/ 10.1074/jbc.R114.613414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ghosh P, Beas AO, Bornheimer SJ, Garcia-Marcos M, Forry EP, Johannson C, Ear J, Jung BH, Cabrera B, Carethers JM, et al., A G{alpha}i-GIV molecular complex binds epidermal growth factor receptor and determines whether cells migrate or proliferate. Mol Biol Cell, 2010; 21(13):p. 2338-54; PMID:20462955; http://dx.doi.org/ 10.1091/mbc.E10-01-0028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ghosh P, Garcia-Marcos M, Bornheimer SJ, Farquhar MG. Activation of Galphai3 triggers cell migration via regulation of GIV. J Cell Biol, 2008; 182(2):p. 381-93; http://dx.doi.org/ 10.1083/jcb.200712066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ghosh P, Garcia-Marcos M, Farquhar MG. GIV/Girdin is a rheostat that fine-tunes growth factor signals during tumor progression. Cell Adh Migr, 2011; 5(3):p. 237-48; PMID:21546796; http://dx.doi.org/ 10.4161/cam.5.3.15909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang H, Misaki T, Taupin V, Eguchi A, Ghosh P, Farquhar MG. GIV/girdin links vascular endothelial growth factor signaling to Akt survival signaling in podocytes independent of nephrin. J Am Soc Nephrol, 2015; 26(2):p. 314-27; PMID:25012178; http://dx.doi.org/ 10.1681/ASN.2013090985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lopez-Sanchez I, Garcia-Marcos M, Mittal Y, Aznar N, Farquhar MG, Ghosh P. Protein kinase C-theta (PKCtheta) phosphorylates and inhibits the guanine exchange factor, GIV/Girdin. Proc Natl Acad Sci U S A, 2013; 110(14):p. 5510-5; http://dx.doi.org/ 10.1073/pnas.1303392110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Garcia-Marcos M, Ghosh P, Ear J, Farquhar MG. A structural determinant that renders G alpha(i) sensitive to activation by GIV/girdin is required to promote cell migration. J Biol Chem, 2010. 285(17):p. 12765-77; PMID:20157114; http://dx.doi.org/ 10.1074/jbc.M109.045161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Beas AO, Taupin V, Teodorof C, Nguyen LT, Garcia-Marcos M, Farquhar MG. Galphas promotes EEA1 endosome maturation and shuts down proliferative signaling through interaction with GIV (Girdin). Mol Biol Cell, 2012; 23(23):p. 4623-34; http://dx.doi.org/ 10.1091/mbc.E12-02-0133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lopez-Sanchez I, Dunkel Y, Roh YS, Mittal Y, De Minicis S, Muranyi A, Singh S, Shanmugam K, Aroonsakool N, Murray F, et al., GIV/Girdin is a central hub for profibrogenic signalling networks during liver fibrosis. Nat Commun, 2014; 5:p. 4451; PMID:25043713; http://dx.doi.org/ 10.1038/ncomms5451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ohara K, Enomoto A, Kato T, Hashimoto T, Isotani-Sakakibara M, Asai N, Ishida-Takagishi M, Weng L, Nakayama M, Watanabe T, et al., Involvement of Girdin in the determination of cell polarity during cell migration. PLoS One, 2012; 7(5):p. e36681; PMID:22574214; http://dx.doi.org/ 10.1371/journal.pone.0036681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mao JZ, Jiang P, Cui SP, Ren YL, Zhao J, Yin XH, Enomoto A, Liu HJ, Hou L, Takahashi M, et al., Girdin locates in centrosome and midbody and plays an important role in cell division. Cancer Sci, 2012; 103(10):p. 1780-7; PMID:22755556; http://dx.doi.org/ 10.1111/j.1349-7006.2012.02378.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Weng L, Enomoto A, Miyoshi H, Takahashi K, Asai N, Morone N, Jiang P, An J, Kato T, Kuroda K, et al., Regulation of cargo-selective endocytosis by dynamin 2 GTPase-activating protein girdin. EMBO J, 2014; 33(18):p. 2098-112; PMID:25061227; http://dx.doi.org/ 10.15252/embj.201488289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ichimiya H, Maeda K, Enomoto A, Weng L, Takahashi M, Murohara T, et al., Girdin/GIV regulates transendothelial permeability by controlling VE-cadherin trafficking through the small GTPase, R-Ras. Biochem Biophys Res Commun, 2015; 461(2):260-7; PMID:25869066 [DOI] [PubMed] [Google Scholar]
- 23.Enomoto A, Asai N, Namba T, Wang Y, Kato T, Tanaka M, Tatsumi H, Taya S, Tsuboi D, Kuroda K, et al., Roles of disrupted-in-schizophrenia 1-interacting protein girdin in postnatal development of the dentate gyrus. Neuron, 2009; 63(6):p. 774-87; PMID:19778507; http://dx.doi.org/ 10.1016/j.neuron.2009.08.015 [DOI] [PubMed] [Google Scholar]
- 24.Wang Y, Kaneko N, Asai N, Enomoto A, Isotani-Sakakibara M, Kato T, Asai M, Murakumo Y, Ota H, Hikita T, et al., Girdin is an intrinsic regulator of neuroblast chain migration in the rostral migratory stream of the postnatal brain. J Neurosci, 2011; 31(22):p. 8109-22; PMID:21632933; http://dx.doi.org/ 10.1523/JNEUROSCI.1130-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Puseenam A, Yoshioka Y, Nagai R, Hashimoto R, Suyari O, Itoh M, Enomoto A, Takahashi M, Yamaguchi M. A novel Drosophila Girdin-like protein is involved in Akt pathway control of cell size. Exp Cell Res, 2009; 315(19):p. 3370-80; http://dx.doi.org/ 10.1016/j.yexcr.2009.06.019 [DOI] [PubMed] [Google Scholar]
- 26.Dunkel Y, Ong A, Notani D, Mittal Y, Lam M, Mi X, Ghosh P. STAT3 protein up-regulates Galpha-interacting vesicle-associated protein (GIV)/Girdin expression, and GIV enhances STAT3 activation in a positive feedback loop during wound healing and tumor invasion/metastasis. J Biol Chem, 2012; 287(50):p. 41667-83; PMID:23066027; http://dx.doi.org/ 10.1074/jbc.M112.390781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Enomoto A, Murakami H, Asai N, Morone N, Watanabe T, Kawai K, Murakumo Y, Usukura J, Kaibuchi K, Takahashi M. Akt/PKB regulates actin organization and cell motility via Girdin/APE. Dev Cell, 2005; 9(3):p. 389-402; PMID:16139227; http://dx.doi.org/ 10.1016/j.devcel.2005.08.001 [DOI] [PubMed] [Google Scholar]
- 28.Jiang P, Enomoto A, Jijiwa M, Kato T, Hasegawa T, Ishida M, Sato T, Asai N, Murakumo Y, Takahashi M. An actin-binding protein Girdin regulates the motility of breast cancer cells. Cancer Res, 2008; 68(5):p. 1310-8; PMID:18316593; http://dx.doi.org/ 10.1158/0008-5472.CAN-07-5111 [DOI] [PubMed] [Google Scholar]
- 29.Natsume A, Kato T, Kinjo S, Enomoto A, Toda H, Shimato S, Ohka F, Motomura K, Kondo Y, Miyata T, et al., Girdin maintains the stemness of glioblastoma stem cells. Oncogene, 2012; 31(22):p. 2715-24; PMID:22020337; http://dx.doi.org/ 10.1038/onc.2011.466 [DOI] [PubMed] [Google Scholar]
- 30.Zhang YJ, Li AJ, Han Y, Yin L, Lin MB. Inhibition of Girdin enhances chemosensitivity of colorectal cancer cells to oxaliplatin. World J Gastroenterol, 2014; 20(25):p. 8229-36; http://dx.doi.org/ 10.3748/wjg.v20.i25.8229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yamamura Y, Asai N, Enomoto A, Kato T, Mii S, Kondo Y, Ushida K, Niimi K, Tsunoda N, Nagino M, et al., Akt-Girdin signaling in cancer-associated fibroblasts contributes to tumor progression. Cancer Res, 2015. 75(5):p. 813-23; http://dx.doi.org/ 10.1158/0008-5472.CAN-14-1317 [DOI] [PubMed] [Google Scholar]
- 32.Kitamura T, Asai N, Enomoto A, Maeda K, Kato T, Ishida M, Jiang P, Watanabe T, Usukura J, Kondo T, et al., Regulation of VEGF-mediated angiogenesis by the Akt/PKB substrate Girdin. Nat Cell Biol, 2008; 10(3):p. 329-37; PMID:18264090; http://dx.doi.org/ 10.1038/ncb1695 [DOI] [PubMed] [Google Scholar]
- 33.Ito T, Komeima K, Yasuma T, Enomoto A, Asai N, Asai M, Iwase S, Takahashi M, Terasaki H. Girdin and its phosphorylation dynamically regulate neonatal vascular development and pathological neovascularization in the retina. Am J Pathol, 2013; 182(2):p. 586-96; PMID:23195430; http://dx.doi.org/ 10.1016/j.ajpath.2012.10.012 [DOI] [PubMed] [Google Scholar]
- 34.Miyachi H, Mii S, Enomoto A, Murakumo Y, Kato T, Asai N, Komori K, Takahashi M. Role of Girdin in intimal hyperplasia in vein grafts and efficacy of atelocollagen-mediated application of small interfering RNA for vein graft failure. J Vasc Surg, 2014; 60(2):p. 479-489 e5; PMID:23948670; http://dx.doi.org/ 10.1016/j.jvs.2013.06.080 [DOI] [PubMed] [Google Scholar]
- 35.Miyake H, Maeda K, Asai N, Shibata R, Ichimiya H, Isotani-Sakakibara M, Yamamura Y, Kato K, Enomoto A, Takahashi M, et al., The actin-binding protein Girdin and its Akt-mediated phosphorylation regulate neointima formation after vascular injury. Circ Res, 2011; 108(10):p. 1170-9; PMID:21415395; http://dx.doi.org/ 10.1161/CIRCRESAHA.110.236174 [DOI] [PubMed] [Google Scholar]
- 36.Nakai T, Nagai T, Tanaka M, Itoh N, Asai N, Enomoto A, Asai M, Yamada S, Saifullah AB, Sokabe M, et al., Girdin phosphorylation is crucial for synaptic plasticity and memory: a potential role in the interaction of BDNF/TrkB/Akt signaling with NMDA receptor. J Neurosci, 2014; 34(45):p. 14995-5008; PMID:25378165; http://dx.doi.org/ 10.1523/JNEUROSCI.2228-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sasaki K, Kakuwa T, Akimoto K, Koga H, Ohno S. Regulation of epithelial cell polarity by PAR-3 depends on Girdin transcription and Girdin-Gαi3 signaling. J Cell Sci 2015; 128(13):2244-58; http://dx.doi.org/ 10.1242/jcs.160879 [DOI] [PubMed] [Google Scholar]