

Abstract
Within regenerating tissues, aging is characterized by a progressive general deterioration of organ function, thought to be driven by the gradual depletion of functional adult stem cells. Although there are probably multifactorial mechanisms that result in compromized stem cell functionality with advancing age, the accumulation of DNA damage within the stem cell compartment is likely to make a major contribution to this process. However, the physiologic source of DNA damage within the different tissue specific stem cell compartments remains to be determined, as does the fate of stem cells exposed to such damage. Using the haematopoietic system as a model organ, we have recently shown that certain forms of physiologic stress, such as infection-associated inflammation and extensive blood loss, leads to the induction of biologically relevant levels of DNA damage in haematopoietic stem cells (HSCs) by dramatically increasing the proliferative index of this normally quiescent cell population.1 We were also able to demonstrate that such stress-associated DNA damage was sufficient to completely deplete HSCs and promote severe aplastic anemia (SAA) in the Fanconi anemia (FA) knockout mouse model, which has compromized replication-associated DNA repair. In this “Extra Views” article, we extend this previous work to show that FA mice do not spontaneously develop a haematopoietic phenotype consistent with SAA, even at extreme old age. This suggests that HSC quiescence restricts the acquisition of DNA damage during aging and preserves the functional integrity of the stem cell pool. In line with this hypothesis, we provide an extended time course analysis of the response of FA knockout mice to chronic inflammatory stress and show that enforced HSC proliferation leads to a highly penetrant SAA phenotype, which closely resembles the progression of the disease in FA patients.
Keywords: aging, aplastic anemia, bone marrow failure, cell cycle, DNA damage, Fanconi anemia, Haematopoietic stem cells, inflammation, quiescence, stress
Introduction
HSCs accumulate DNA damage with age and recent genome sequencing studies underscore this fact by demonstrating that HSCs from normal aged individuals harbor hundreds of somatic mutations per genome.2 This progressive acquisition of DNA damage within the HSC compartment is thought to be a major contributing factor toward the gradual functional decline of the haematopoietic system with increasing age. In line with this hypothesis, a number of studies have observed that the age-related acquisition of somatic mutations correlates with a dramatically reduced clonal repertoire of HSCs sustaining ongoing hematopoiesis within elderly humans.3-6 Despite this likely causal link between DNA damage and HSC dysfunction, relatively little is known about the predominant physiologic source of this damage. Indeed, most of our knowledge of the HSC DNA damage response (DDR) has been gained from studies that employ ionizing radiation or chemotherapy as agonists to provoke DNA damage, which may not relate to the type and magnitude of damage that is normally encountered within the HSC pool for the majority of individuals.
Replication stress has been proposed to act as a universal mechanism through which somatic cells progressively acquire DNA damage with age.7,8 With regards to adult stem cell biology, it is interesting to note that the lifetime risk of developing cancer in a given tissue strongly correlates with the estimated proliferative index of the stem cell population responsible for sustaining that tissue, presumably due to increased mutational burden resulting from replication stress.9 However, many adult stem cell populations are characterized by their long-term quiescent status and this is certainly the case for HSCs, where it has been calculated that under standard laboratory conditions, so-called long-term (LT)-HSCs in mice may divide as few as 5 times during the lifetime of the animal and only infrequently contribute to the production of mature blood cells.10-12 One would predict that this long-term quiescent status would act to limit replication stress-induced damage in LT-HSCs and perhaps preserve their genomic and functional integrity. This may explain why replicative history appears to inversely correlate with HSC functional potency in mice.12,13
The recent revelation that the infection-mediated inflammatory response can induce HSCs to transiently enter into cycle in vivo, led us to speculate that this phenomenon could link physiologic stress with elevated proliferation-induced DNA damage and tissue degeneration by significantly increasing the proliferative index of LT-HSCs in vivo.14-16 By using a range of different agonists to mimic processes such as viral infection or blood loss in mice, we were able to force LT-HSCs out of quiescence in vivo and observe that this universally led to the induction of DNA damage within the stem cell compartment.1 Although the level of DNA damage within the LT-HSC compartment was quite modest compared to that observed when high dose irradiation or chemotherapy are used on mice, chronic treatment with pro-inflammatory agonists led to a profound reduction in the number of functional LT-HSCs combined with a myeloid differentiation bias that resembled the haematopoietic phenotype of aged mice. The causal role of DNA damage in this stress-induced HSC attrition was established by using a mouse model with a clinically relevant defect in the cellular DDR. Mice that harbor inactivating deletions within genes involved in the FA signaling pathway have a cellular defect in resolving certain forms of DNA damage that result in replication fork arrest, such as DNA interstrand crosslinks.17 When mice with a targeted deletion of the FA gene, Fanca, were subject to physiologic stress agonists as described above, the degree of DNA damage observed in LT-HSC was significantly higher than in their wild type (WT) counterparts, demonstrating that the FA pathway is involved in the resolution of stress-induced DNA damage within the LT-HSC compartment in vivo. In line with this observation, chronic stress exposure resulted in an almost complete exhaustion of the LT-HSC pool within Fanca−/− mice leading to eventual bone marrow failure (BMF). This completely recapitulates the highly penetrant SAA that is a defining characteristic of the disease seen in patients who have germline inactivating mutations in FA pathway members.18 Taken together, these data support an important role for physiologic stress as a biologically relevant source of DNA damage within the LT-HSC compartment in vivo and suggest that cumulative exposure to such stress can induce age-associated phenotypes within the haematopoietic system.
In this “Extra Views” article we will provide additional data extending the work described in our recent research article linking inflammation, DNA damage and HSC aging and will also discuss the broader implications of these findings.1
Results
Haematopoietic phenotype of WT and Fanca−/− mice with increasing age
Of the 16 FA genes that have been identified in patients to date and whose loss of function has been found to be causative of the biochemical defect in DNA interstrand crosslink repair, FANCA is the most frequently mutated.18-20 Cells from mice with targeted deletions of the Fanca gene have exactly the same DNA repair defect as cells from FA patients and these mice do demonstrate some of the developmental defects that are heterogeneously manifest in patients such as growth retardation, germ cell defects, micropthalmia and craniofacial abnormalities.21,22 However, Fanca−/− mice have not been reported to spontaneously develop the SAA that is a defining characteristic of this disease in humans. This uncoupling of the cellular DNA repair defect from the clinically prevalent phenotype of BMF is a general feature of all single gene deletion mouse models of FA and has been a major hindrance in understanding why the pathway is required for the effective maintenance of hematopoiesis (reviewed in17,23). This has led to speculation about the utility of the mouse model in the study of this disease. In an attempt to determine whether any haematopoietic phenotype would become manifest in Fanca−/− mice in extreme old age, we analyzed hematologic parameters of young (6 months old), middle aged (12 months old) and aged (24 months old) Fanca−/− and WT mice that had been maintained under specified pathogen free laboratory conditions for their entire life. Flow cytometry-based analysis of mature cells in the peripheral blood from these animals showed that there was no significant difference in the relative production of B-, T- and myeloid cells between WT and Fanca−/− mice, regardless of age (Fig. 1A). Both Fanca−/− and WT mice demonstrated an equivalent age-dependent skewing toward increased myeloid output that has been previously documented in the mouse model of hematopoiesis.24,25 Evaluation of peripheral blood cell counts showed that Fanca−/− and WT mice had similar numbers of circulating leukocytes, erythrocytes and thrombocytes at each time point analyzed (Fig. 1B). Characterization of more immature haematopoietic cells in the bone marrow (BM) also yielded no evidence of abnormal hematopoiesis in Fanca−/− mice, with both the total femur cellularity and relative frequencies of lymphoid and myeloid cells showing no significant difference between Fanca−/− and WT mice in both the young and aged setting (Fig. 1C and D). Taken together, these data indicate that, unlike FA patients, Fanca−/− mice do not spontaneously develop SAA nor any milder defect in the production of mature blood cells under standard laboratory conditions, even at time points close to the normal maximum life expectancy of experimental animals.
Figure 1.

Analysis of mature blood cells in the peripheral blood and bone marrow of young and aged WT and Fanca−/− mice. (A and B) Peripheral blood cell analysis of WT (black) and Fanca−/− (gray) mice at 0.5 year, 1 and 2 y showing A) the percentage of myeloid (Gr-1+, Mac-1+), T-cells (CD3e+) and B-cells (B220+); B) the white blood cell (WBC), red blood cell (RBC) and platelet (PLT) count. (C and D) Analysis of BM of 0.5 year, 1 and 2 y old WT (black) and Fanca−/− (gray) mice showing (C) the percentage of myeloid (Gr-1+, Mac-1+), T-cells (CD3e+) and B-cells (B220+); (D) the absolute number of BM cells per femur. Mean ± standard deviation (s.d.) is shown for n = 3 −6 mice per group. Not significant (ns) = P > 0.05, unpaired t-test,
We next shifted our analysis to the study of the more primitive HSC and progenitor (HSC/P) compartments within the BM. In contrast to the results obtained when mature blood cells were considered, the absolute numbers of BM populations corresponding to committed progenitors (lineage (lin)−, c-Kit+, Sca-1−), HSCs plus multipotent progenitors (MPPs; lin−, c-Kit+, Sca1+), MPPs alone (Lin−, c-Kit+, Sca1+, CD48+) and LT-HSCs alone (Lin−, c-Kit+, Sca1+, CD48+, CD150+, CD34−) were all significantly decreased in 6-month old Fanca−/− mice compared to their WT counterparts (Fig. 2A-D). However, at one year of age, the comparative differences between all of these populations had normalized across Fanca−/− and WT mice and, by 2 y of age, the LT-HSC compartment was significantly overrepresented within the BM of Fanca−/− mice compared to WT. These findings may relate to previous observations detailing the abnormal embryonic development of FA deficient HSCs, which may account for the comparatively lower absolute numbers of HSC/Ps in young Fanca−/− mice.26-28 Nonetheless, it is clear that this phenotype is not exacerbated during normal aging of mice within the laboratory environment, but instead appears to be rescued with increased age. While the age-dependent accumulation of immunophenotypically-defined HSCs is a well-documented feature of aged mice, the molecular and cellular processes responsible for the increased accumulation of LT-HSCs in the BM of Fanca−/− mice are not yet described and could be reflective of an accelerated aging phenotype.29,30 Regardless, the relatively modest differences in HSC/P numbers found between the BM of Fanca−/− and WT mice do not translate into abnormal production of mature blood cells at any of the time points indicated and certainly do not recapitulate the time-dependent attrition of the haematopoietic system that is seen in the majority of FA patients.
Figure 2.
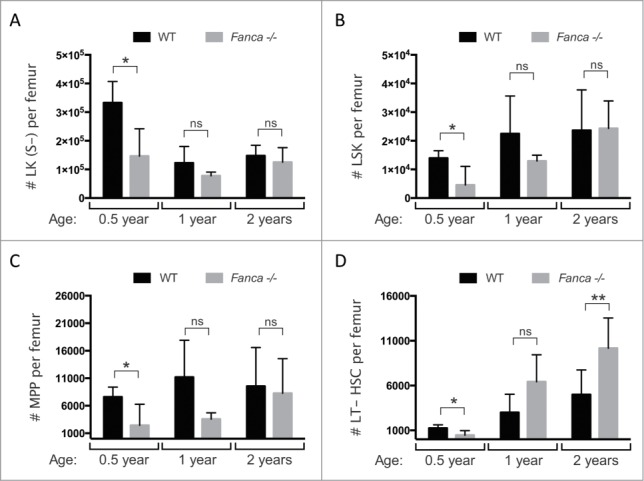
Evaluation of HSC/Ps in young and aged WT and Fanca−/− mice. Absolute cell counts per femur of (A) committed progenitors: Lineage−, c-Kit+, Sca-1− (LK(S-)); (B) HSCs and MPPs: Lineage−, c-Kit+, Sca-1+ (LSK); (C) MPPs: Lineage-, c-Kit+, Sca-1+, CD48+ and; (D) LT-HSCs: Lineage-, c-Kit+, Sca-1+, CD48−, CD150+, CD34− in 0.5, 1 and 2 y old WT and Fanca−/− mice. Mean ± s.d is shown for n = 3 −6 mice per group. *p < 0.05, ** p < 0.01, not significant (ns) = p > 0.05, unpaired t-test.
Haematopoietic phenotype of WT and Fanca−/− mice following chronic inflammatory stress
Since cells from FA knockout mouse models demonstrate equivalent DNA repair defects to FA patient cells, yet never develop BMF, it suggests that either the DNA repair defect is not causative of the haematopoietic phenotype seen in patients or that the source of DNA damage that patients are exposed to is not present in experimental mice. The observation that FA-deficient murine HSCs can be depleted by in vivo treatment with the DNA interstrand crosslinking agent MMC would appear to support the latter hypothesis, but is clearly of limited physiologic relevance to BMF in patients.31 A large number of studies have generated data supporting a role for pro-inflammatory cytokines as mediators of BMF in patients (reviewed in17,32). However, the relationship of this phenomenon to the defective DDR was previously unclear and it had not been formally demonstrated that chronic inflammation could precipitate SAA in any FA knockout mouse model.32 Given the predominant quiescent status of LT-HSCs in the experimental mouse model, the involvement of the FA pathway in DNA replication associated repair and the recent revelation that certain pro-inflammatory cytokines could force LT-HSCs into active cell cycle in vivo, we hypothesized that the FA pathway could be an important route via which DNA damage induced by replication stress is resolved in HSCs that have been forced to exit quiescence in vivo in response to stress.12-16 We recently demonstrated that this is indeed the case, with LT-HSCs from Fanca−/− deficient mice showing significantly increased levels of DNA damage compared to their WT counterparts when the double stranded RNA mimetic polyinosinic:polycytidylic acid (pI:pC) was used to mimic a viral infection, thus promoting a type I interferon pro-inflammatory response.1 Elevated levels of DNA damage were also observed in LT-HSCs when mice were exposed to a range of alternative pro-proliferative agonists, such as recombinant thrombopoietin, granulocyte colony stimulating factor and serial bleeding. Importantly, this compromized in vivo DDR also correlated with an accelerated loss of functional LT-HSCs in Fanca−/− mice compared to WT control animals following chronic treatment with pI:pC. This treatment regimen eventually resulted in an exhaustion of the HSC/P pool within the majority of Fanca−/− mice, leading to the onset of SAA.1 A long-term follow up of mice subject to serial treatment with pI:pC revealed that more than 70% of Fanca−/− mice succumbed to SAA as defined by profound BM aplasia in combination with peripheral blood cytopenias in at least 2 compartments from leukocytes, erythrocytes and/or thrombocytes (Fig. 3A-D). WT mice exposed to the same pI:pC treatment regimen never developed SAA, nor did age-matched Fanca−/− or WT mice serially injected with phosphate buffered saline (PBS). In fact, pI:pC-treated WT mice demonstrated a mild but significant leukocytosis, which we postulate may be the result of an overcompensated production of leukocytes following sustained stress (Fig. 3D). To our knowledge, this is the first demonstration that single gene FA knockout mice can reproducibly phenocopy the progressive BMF that is observed in the majority of FA patients. Taken together, our data demonstrate that environmental stress can precipitate a biologically relevant dose of replication-induced DNA damage in LT-HSCs in vivo by forcing these cells out of their predominant quiescent status. We also provide a novel mechanistic link between pro-inflammatory cytokines and DNA damage, which would potentially explain the role of such factors in the etiology of BMF in FA patients.
Figure 3.

Extended time-course analysis of incidence of SAA in Fanca−/− mice in response to chronic inflammatory stress. (A) Schematic representation of the pI:pC treatment schedule which was repeated up to 8 times. (B) Cumulative BMF expressed as percentage in WT and Fanca−/− mice. n = 12 mice were analyzed in the WT and Fanca−/− control groups, and n = 18 mice were included in both WT and Fanca−/− pI:pC treated groups. Statistical significance was assessed by log-rank (Mantel-Cox) test. ***p < 0.001. (C) Representative hematoxylin and eosin staining of femoral sections from WT or Fanca−/− mice after cessation of treatment. (D) Peripheral blood cell count analysis of the WT and Fanca−/− control groups after cessation of treatment, and the Fanca−/− pI:pC treated group at the time the mice displayed SAA. White blood cell = WBC, Platelet = PLT. Individual mice are represented with circles and mean ± s.d is shown. *=p < 0.05, ***=p < 0.001, non significant (ns) = p > 0.05, unpaired t-test. Data comprises an extended time course of experiments originally published in.1
Discussion
Over the course of a lifetime, an organism will be exposed to multiple rounds of hematologic stress, such as during infection or following extensive blood loss. Such events drive LT-HSCs into active cell cycle and this is likely essential for the recovery and maintenance of the haematopoietic system in the face of such challenge. Our data support a model where this transient stress-induced activation of HSCs does not come without a cost, in that the HSC pool will progressively be exposed to increasing amounts of DNA damage with each successive round of activation out of quiescence (Fig. 4). Thus, we propose that under homeostatic conditions, the low proliferative index of LT-HSCs will restrict DNA damage and preserve the genomic and functional integrity of the stem cell pool. In the event of a defective DDR, such as in the case of FA, quiescence will limit the levels of DNA damage in LT-HSCs, so that ineffective repair only results in the attrition of a minor fraction of the stem cell pool. In contrast, during chronic stress, the resulting increase in LT-HSC proliferative index will generate elevated levels of DNA damage. In a DNA repair proficient cell, much of this damage will be repaired, thereby preventing extensive depletion of the HSC pool. However, in FA cells, the loss of a high fidelity DNA repair pathway will mean that high rates of replication stress-induced DNA damage will translate into elevated levels of HSC loss with each round of challenge. Cumulative rounds of stress with increasing age will therefore prematurely exhaust the HSC pool in the setting of FA, while in the repair proficient setting, the HSC pool will be depleted but will still be sufficient to sustain the ongoing production of mature blood cells.
Figure 4.

Schematic model representing putative relationship between HSC replicative history, DNA damage and cellular attrition. Graphical illustration of the putative association between cumulative HSC divisional history, levels of DNA damage and HSC attrition. Four conditions are considered: Homeostatic HSCs with normal or compromized DNA repair and HSCs during stress hematopoiesis, again with normal or compromized repair. The blue area represents cumulative HSC divisions with time; the orange area represents the acquisition of DNA damage in HSCs due to replication stress; and the red area represents the loss of functional HSCs induced by DNA damage, for example due to apoptosis. Under homeostatic conditions, the prevalent quiescent status of HSCs restricts DNA damage acquisition and therefore maintains the functional stem cell pool, even in the presence of a compromized DNA repair system. Under conditions of stress, HSCs suffer increased levels of DNA damage and this results in elevated HSC attrition. In the case of defective DNA repair, HSC attrition is accelerated in response to the same level of DNA damage as WT cells.
Our in vivo characterization of LT-HSC replication status as a critical mediator of DNA damage in FA cells would appear to directly correlate with several in vitro studies identifying the FA pathway as essential for the efficient resolution of DNA ultrafine bridges that can arise as a result of DNA replication stress.33-35 Whether these ultrafine bridges occur in HSCs in vivo and what would impact on their production remains to be determined. However, it is known that DNA lesions can provoke replication stress, either by directly impeding the progression of the replication fork, or by acting as a pre-cursor for a DNA repair intermediate that can stall the replication fork.8 Such stalled forks can then collapse resulting in DNA double strand breaks. Our recent work suggested a role for metabolic reactive oxygen species (ROS) as an important source of DNA adducts in WT LT-HSCs during stress hematopoiesis.1 We could demonstrate that elevated metabolic ROS, present in actively cycling LT-HSCs, were causative for DNA double strand breaks and that this correlated with increased de novo generation of the ROS-induced 8-oxo-2′-deoxyguanosine adduct on DNA. This de novo induction of ROS-induced DNA damage in WT LT-HSCs as they entered into cycle would appear to directly contradict the concept that stem cells accumulate DNA damage during quiescence and then resolve this damage as they start to proliferate.36 However, we have not formally established whether these ROS-induced adducts were responsible for the increased levels of DNA double strand breaks seen in Fanca−/− LT-HSCs compared to WT LT-HSCs during stress hematopoiesis. In fact, we do not exclude that the FA pathway may be involved in replication-coupled repair of alternative types of DNA lesion not associated with ROS, that may already exist in quiescent cells or which are generated de novo upon cell cycle entry. Nonetheless, our data does demonstrate that DNA replication is required for such lesions to be converted into DNA double strand breaks in both Fanca−/− and WT LT-HSCs. Regarding the identification of such alternative DNA lesions and their potential source, the extremely low frequency of HSCs and unknown levels of relevant DNA lesions within these cells restricts the application of technologies such as mass spectrometry as a broad discovery tool. However, the Patel laboratory has recently demonstrated that reactive aldehydes can synergize with a defective FA repair pathway to result in elevated DNA double strand breaks.37,38 Although the exact identity of the DNA lesion caused by elevated reactive aldehyde levels is currently not known, coordinate genetic ablation of the FA pathway and the aldehyde detoxifying enzyme Aldh2 in mice led to the spontaneous onset of acute lymphoblastic leukemia in the majority of experimental animals, with the remainder demonstrating BMF.37,38 The clinical relevance of this observation has been demonstrated in a subsequent study showing that FA patients with a dominant negative variant allele of ALDH2 demonstrated a more accelerated progression of BMF than FA patients who had normal ALDH2 function.39 Given the data identifying reactive aldehydes as a biologically relevant source of DNA damage in vivo, it would be intriguing to interrogate whether DNA damage induced by metabolic reactive aldehydes alters in response to physiologic stress and whether such lesions can synergize with increased LT-HSC proliferation to elicit rapid depletion of the stem cell pool.
Finally, one might speculate that similar stress-associated DNA damage mechanisms may operate in other regenerating adult tissues that harbor quiescent stem cell populations, although the pro-proliferative agonists are likely to be different to those characterized in the haematopoietic system. Indeed, this concept may shed new light on the etiology of disease in other tissues that demonstrate an abnormal phenotype in the context of germ line or somatic loss of function of the Fanconi pathway, such as skin, breast, ovary and bone.
Methods
Animals
Mice were housed in individually ventilated cages, specified pathogen free, within the DKFZ animal facility. All experimental procedures were approved by the Animal Care and Use Committees of the German Regierungspräsidium Karlsruhe für Tierschutz und Arzneimittelüberwachung. Wild-type C57BL/6J mice were obtained from Harlan Laboratories. Fanca−/− mice have been previously described.22 Mice were 8–16 weeks of age at the point at which experimental studies were initiated. To provoke in vivo cycling of HSCs, mice were injected with 5 mg kg−1 pI:pC intraperitoneally (i.p.) (InvivoGen, catalog # tlrl-pic-5). Peripheral blood cell numbers were evaluated using a Hemavet 950 FS veterinary blood cell counting machine (Drew Scientific). A diagnosis of BMF/SAA in treated mice was defined as bi- or tri- lineage (leukocytes, erythrocytes and/or thrombocytes) cytopenias in the peripheral blood in addition to BM aplasia. Individual mice were excluded from analysis when a diagnosis of SAA could not be confirmed, for example when autopsy was not be performed in a timely manner, resulting in a decomposition of tissue morphology and precluding evaluation of peripheral blood cell counts.
Identification and purification of murine haematopoietic stem and progenitor cells
Murine lineage-depleted BM cells were isolated as described previously.40 In brief, low-density mononuclear cells (LDMNCs) were purified by density gradient centrifugation using Histopaque 1083 (Sigma-Aldrich, catalog # 10831) and then stained with a panel of rat anti-mouse biotin-conjugated lineage markers (CD5/53–7.3, CD8a/53–6.7, CD11b/M1/70, B220/RA3–6B2, Gr1/RB6–8C5, Ter119/Ter-119, all BD Bioscience). The resulting lineage-depleted cells were then stained with a panel of antibodies (CD117-APC/2B8, Sca1-APC-Cy7/D7, CD150-PE-Cy5/TC15–12F12.2, CD48-PB/HM48–1, CD34-FITC/RAM34, all BD Bioscience) to identify LT-HSCs (lineage negative (lin−), c-Kit+, Sca-1+, CD48−, CD150+, CD34−), ST-HSCs (lin−, c-Kit+, Sca-1+, CD48−, CD150+, CD34+) MPPs (lin−, c-Kit+, Sca-1+, CD48+), LSKs (lin−, c-Kit+, Sca-1+) and LKs (lin−, c-Kit+, Sca-1−) by flow cytometry analysis. All FACS samples were analyzed with a LSRII, LSR-Fortessa, FACSAria I, II or III flow cytometer (BD Bioscience). Dead cells were excluded by using 7-amino actinomycin D staining at a final concentration of 5 μg/ml (7AAD, Invitrogen).
Staining of mature cells in peripheral blood and bone marrow
Peripheral blood and BM cells were simultaneously stained with antibodies against lineage-specific cell surface markers (CD3e-FITC/145–2C11, CD11b-APC/M1/70, Gr-1-APC/RB6–8C5, B220-PE/RA3–6B2, all BD Bioscience). After red blood cells lysis with ACK lysing buffer, cells were additionally washed with PBS and then FACS analyzed.
Hematoxylin and eosin (H&E) staining
Tibiae were fixed for one day in formalin, decalcified for 5 d in 0,5 M EDTA (Ethylenediaminetetraacetic acid) buffer (pH 7,2) at 4°C and paraffin-embedded (Histo Star, Thermo Scientific). Embedded bones were cut with a Microtome (Microm HM 355S, Thermo Scientific). BM sections were de-paraffinized, rehydrated and H&E staining was performed using a standard protocol [Fischer et al., 2008]
Statistical analysis
Analyses were always carried out in comparison to the control group. Two-sided unpaired t-tests were used for pair-wise comparisons. For the incidence of BMF, a log-rank Mantel-Cox test was performed. Statistical analysis was performed using GraphPad Prism 6.0d software (GraphPad Software, Inc., SanDiego, CA, http://www.graphpad.com).
Funding
DW, AL and MDM were supported by the BioRN Leading-Edge Cluster “Cell-Based and Molecular Medicine” funded by the German Federal Ministry of Education and Research, and the Dietmar Hopp Foundation. PK was funded by a fellowship from the Helmholtz International Graduate School and RB by a post-doctoral fellowship from the DKFZ.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank the Animal Laboratory Services Deutsches Krebsforschungszentrum (DKFZ) Core Facility and the Imaging and Cytometry DKFZ Core Facility, in particular Ann Atzberger and Klaus Hexel. We would also like to express our gratitude to Vanessa Vogel for technical support.
References
- 1.Walter D, Lier A, Geiselhart A, Thalheimer FB, Huntscha S, Sobotta MC, Moehrle B, Brocks D, Bayindir I, Kaschutnig P, et al.. Exit from dormancy provokes DNA-damage-induced attrition in haematopoietic stem cells. Nature 2015; 520:549-52; PMID:25707806; http://dx.doi.org/ 10.1038/nature14131 [DOI] [PubMed] [Google Scholar]
- 2.Welch JS, Ley TJ, Link DC, Miller CA, Larson DE, Koboldt DC, Wartman LD, Lamprecht TL, Liu F, Xia J, et al.. The origin and evolution of mutations in acute myeloid leukemia. Cell 2012; 150:264-78; PMID:22817890; http://dx.doi.org/ 10.1016/j.cell.2012.06.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Busque L, Patel JP, Figueroa ME, Vasanthakumar A, Provost S, Hamilou Z, Mollica L, Li J, Viale A, Heguy A, et al.. Recurrent somatic TET2 mutations in normal elderly individuals with clonal hematopoiesis. Nature Genet 2012; 44:1179-81; PMID:23001125; http://dx.doi.org/ 10.1038/ng.2413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Genovese G, Kahler AK, Handsaker RE, Lindberg J, Rose SA, Bakhoum SF, Chambert K, Mick E, Neale BM, Fromer M, et al.. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med 2014; 371:2477-87; PMID:25426838; http://dx.doi.org/ 10.1056/NEJMoa1409405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Holstege H, Pfeiffer W, Sie D, Hulsman M, Nicholas TJ, Lee CC, Ross T, Lin J, Miller MA, Ylstra B, et al.. Somatic mutations found in the healthy blood compartment of a 115-yr-old woman demonstrate oligoclonal hematopoiesis. Genome Res 2014; 24:733-42; PMID:24760347; http://dx.doi.org/ 10.1101/gr.162131.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, Lindsley RC, Mermel CH, Burtt N, Chavez A, et al.. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med 2014; 371:2488-98; PMID:25426837; http://dx.doi.org/ 10.1056/NEJMoa1408617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Flach J, Bakker ST, Mohrin M, Conroy PC, Pietras EM, Reynaud D, Alvarez S, Diolaiti ME, Ugarte F, Forsberg EC, et al.. Replication stress is a potent driver of functional decline in ageing haematopoietic stem cells. Nature 2014; 512:198-202; PMID:25079315; http://dx.doi.org/ 10.1038/nature13619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zeman MK, Cimprich KA. Causes and consequences of replication stress. Nat Cell Biol 2014; 16:2-9; PMID:24366029; http://dx.doi.org/ 10.1038/ncb2897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tomasetti C, Vogelstein B. Cancer etiology. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science 2015; 347:78-81; PMID:25554788; http://dx.doi.org/ 10.1126/science.1260825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Busch K, Klapproth K, Barile M, Flossdorf M, Holland-Letz T, Schlenner SM, Reth M, Hofer T, Rodewald HR. Fundamental properties of unperturbed haematopoiesis from stem cells in vivo. Nature 2015; 518:542-6; PMID:25686605; http://dx.doi.org/ 10.1038/nature14242 [DOI] [PubMed] [Google Scholar]
- 11.Sun J, Ramos A, Chapman B, Johnnidis JB, Le L, Ho YJ, Klein A, Hofmann O, Camargo FD. Clonal dynamics of native haematopoiesis. Nature 2014; 514:322-7; PMID:25296256; http://dx.doi.org/ 10.1038/nature13824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wilson A, Laurenti E, Oser G, van der Wath RC, Blanco-Bose W, Jaworski M, Offner S, Dunant CF, Eshkind L, Bockamp E, et al.. Hematopoietic stem cells reversibly switch from dormancy to self-renewal during homeostasis and repair. Cell 2008; 135:1118-29; PMID:19062086; http://dx.doi.org/ 10.1016/j.cell.2008.10.048 [DOI] [PubMed] [Google Scholar]
- 13.Qiu J, Papatsenko D, Niu X, Schaniel C, Moore K. Divisional history and hematopoietic stem cell function during homeostasis. Stem Cell Reports 2014; 2:473-90; PMID:24749072; http://dx.doi.org/ 10.1016/j.stemcr.2014.01.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baldridge MT, King KY, Boles NC, Weksberg DC, Goodell MA. Quiescent haematopoietic stem cells are activated by IFN-gamma in response to chronic infection. Nature 2010; 465:793-7; PMID:20535209; http://dx.doi.org/ 10.1038/nature09135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Essers MA, Offner S, Blanco-Bose WE, Waibler Z, Kalinke U, Duchosal MA, Trumpp A. IFNalpha activates dormant haematopoietic stem cells in vivo. Nature 2009; 458:904-8; PMID:19212321; http://dx.doi.org/ 10.1038/nature07815 [DOI] [PubMed] [Google Scholar]
- 16.Takizawa H, Regoes RR, Boddupalli CS, Bonhoeffer S, Manz MG. Dynamic variation in cycling of hematopoietic stem cells in steady state and inflammation. J Exp Med 2011; 208:273-84; PMID:21300914; http://dx.doi.org/ 10.1084/jem.20101643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Geiselhart A, Lier A, Walter D, Milsom MD. Disrupted Signaling through the Fanconi Anemia Pathway Leads to Dysfunctional Hematopoietic Stem Cell Biology: Underlying Mechanisms and Potential Therapeutic Strategies. Anemia 2012; 2012:265790; PMID:22675615; http://dx.doi.org/ 10.1155/2012/265790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kutler DI, Singh B, Satagopan J, Batish SD, Berwick M, Giampietro PF, Hanenberg H, Auerbach AD. A 20-year perspective on the International Fanconi Anemia Registry (IFAR). Blood 2003; 101:1249-56; PMID:12393516; http://dx.doi.org/ 10.1182/blood-2002-07-2170 [DOI] [PubMed] [Google Scholar]
- 19.Longerich S, Li J, Xiong Y, Sung P, Kupfer GM. Stress and DNA repair biology of the Fanconi anemia pathway. Blood 2014; 124:2812-9; PMID:25237197; http://dx.doi.org/ 10.1182/blood-2014-04-526293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Walden H, Deans AJ. The Fanconi anemia DNA repair pathway: structural and functional insights into a complex disorder. Annu Rev Biophys 2014; 43:257-78; PMID:24773018; http://dx.doi.org/ 10.1146/annurev-biophys-051013-022737 [DOI] [PubMed] [Google Scholar]
- 21.Cheng NC, van de Vrugt HJ, van der Valk MA, Oostra AB, Krimpenfort P, de Vries Y, Joenje H, Berns A, Arwert F. Mice with a targeted disruption of the Fanconi anemia homolog Fanca. Hum Mol Genet 2000; 9:1805-11; PMID:10915769; http://dx.doi.org/ 10.1093/hmg/9.12.1805 [DOI] [PubMed] [Google Scholar]
- 22.Wong JC, Alon N, McKerlie C, Huang JR, Meyn MS, Buchwald M. Targeted disruption of exons 1 to 6 of the Fanconi Anemia group A gene leads to growth retardation, strain-specific microphthalmia, meiotic defects and primordial germ cell hypoplasia. Hum Mol Genet 2003; 12:2063-76; PMID:12913077; http://dx.doi.org/ 10.1093/hmg/ddg219 [DOI] [PubMed] [Google Scholar]
- 23.Parmar K, D'Andrea A, Niedernhofer LJ. Mouse models of Fanconi anemia. Mutat Res 2009; 668:133-40; PMID:19427003; http://dx.doi.org/ 10.1016/j.mrfmmm.2009.03.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sudo K, Ema H, Morita Y, Nakauchi H. Age-associated characteristics of murine hematopoietic stem cells. J Exp Med 2000; 192:1273-80; PMID:11067876; http://dx.doi.org/ 10.1084/jem.192.9.1273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rossi DJ, Bryder D, Zahn JM, Ahlenius H, Sonu R, Wagers AJ, Weissman IL. Cell intrinsic alterations underlie hematopoietic stem cell aging. Proc Natl Acad Sci U S A 2005; 102:9194-9; PMID:15967997; http://dx.doi.org/ 10.1073/pnas.0503280102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kamimae-Lanning AN, Goloviznina NA, Kurre P. Fetal origins of hematopoietic failure in a murine model of Fanconi anemia. Blood 2013; 121:2008-12; PMID:23315168; http://dx.doi.org/ 10.1182/blood-2012-06-439679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tulpule A, Lensch MW, Miller JD, Austin K, D'Andrea A, Schlaeger TM, Shimamura A, Daley GQ. Knockdown of Fanconi anemia genes in human embryonic stem cells reveals early developmental defects in the hematopoietic lineage. Blood 2010; 115:3453-62; PMID:20089964; http://dx.doi.org/ 10.1182/blood-2009-10-246694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ceccaldi R, Parmar K, Mouly E, Delord M, Kim JM, Regairaz M, Pla M, Vasquez N, Zhang QS, Pondarre C, et al.. Bone marrow failure in Fanconi anemia is triggered by an exacerbated p53/p21 DNA damage response that impairs hematopoietic stem and progenitor cells. Cell Stem Cell 2012; 11:36-49; PMID:22683204; http://dx.doi.org/ 10.1016/j.stem.2012.05.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Woolthuis CM, de Haan G, Huls G. Aging of hematopoietic stem cells: Intrinsic changes or micro-environmental effects? Curr Opin Immunol 2011; 23:512-7; PMID:21664115; http://dx.doi.org/ 10.1016/j.coi.2011.05.006 [DOI] [PubMed] [Google Scholar]
- 30.Gekas C, Graf T. CD41 expression marks myeloid-biased adult hematopoietic stem cells and increases with age. Blood 2013; 121:4463-72; PMID:23564910; http://dx.doi.org/ 10.1182/blood-2012-09-457929 [DOI] [PubMed] [Google Scholar]
- 31.Battaile KP, Bateman RL, Mortimer D, Mulcahy J, Rathbun RK, Bagby G, Fleming WH, Grompe M. In vivo selection of wild-type hematopoietic stem cells in a murine model of Fanconi anemia. Blood 1999; 94:2151-8; PMID:10477746 [PubMed] [Google Scholar]
- 32.Garaycoechea JI, Patel KJ. Why does the bone marrow fail in Fanconi anemia? Blood 2014; 123:26-34; PMID:24200684; http://dx.doi.org/ 10.1182/blood-2013-09-427740 [DOI] [PubMed] [Google Scholar]
- 33.Vinciguerra P, Godinho SA, Parmar K, Pellman D, D'Andrea AD. Cytokinesis failure occurs in Fanconi anemia pathway-deficient murine and human bone marrow hematopoietic cells. J Clin Invest 2010; 120:3834-42; PMID:20921626; http://dx.doi.org/ 10.1172/JCI43391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chan KL, Palmai-Pallag T, Ying S, Hickson ID. Replication stress induces sister-chromatid bridging at fragile site loci in mitosis. Nat Cell Biol 2009; 11:753-60; PMID:19465922; http://dx.doi.org/ 10.1038/ncb1882 [DOI] [PubMed] [Google Scholar]
- 35.Naim V, Rosselli F. The FANC pathway and BLM collaborate during mitosis to prevent micro-nucleation and chromosome abnormalities. Nat Cell Biol 2009; 11:761-8; PMID:19465921; http://dx.doi.org/ 10.1038/ncb1883 [DOI] [PubMed] [Google Scholar]
- 36.Beerman I, Seita J, Inlay MA, Weissman IL, Rossi DJ. Quiescent hematopoietic stem cells accumulate DNA damage during aging that is repaired upon entry into cell cycle. Cell Stem Cell 2014; 15:37-50; PMID:24813857; http://dx.doi.org/ 10.1016/j.stem.2014.04.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Garaycoechea JI, Crossan GP, Langevin F, Daly M, Arends MJ, Patel KJ. Genotoxic consequences of endogenous aldehydes on mouse haematopoietic stem cell function. Nature 2012; 489:571-5; PMID:22922648; http://dx.doi.org/ 10.1038/nature11368 [DOI] [PubMed] [Google Scholar]
- 38.Langevin F, Crossan GP, Rosado IV, Arends MJ, Patel KJ. Fancd2 counteracts the toxic effects of naturally produced aldehydes in mice. Nature 2011; 475:53-8; PMID:21734703; http://dx.doi.org/ 10.1038/nature10192 [DOI] [PubMed] [Google Scholar]
- 39.Hira A, Yabe H, Yoshida K, Okuno Y, Shiraishi Y, Chiba K, Tanaka H, Miyano S, Nakamura J, Kojima S, et al.. Variant ALDH2 is associated with accelerated progression of bone marrow failure in Japanese Fanconi anemia patients. Blood 2013; 122:3206-9; PMID:24037726; http://dx.doi.org/ 10.1182/blood-2013-06-507962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Milsom MD, Schiedlmeier B, Bailey J, Kim MO, Li D, Jansen M, Ali AM, Kirby M, Baum C, Fairbairn LJ, et al.. Ectopic HOXB4 overcomes the inhibitory effect of tumor necrosis factor-{α} on Fanconi anemia hematopoietic stem and progenitor cells. Blood 2009; 113:5111-20; PMID:19270262; http://dx.doi.org/ 10.1182/blood-2008-09-180224 [DOI] [PMC free article] [PubMed] [Google Scholar]