

Abstract
While it has been widely established that defective fork restart after exposure to stress results in increased genomic instability, the importance of fork protection during stalling for safeguarding genomic integrity has recently been fully appreciated. BRCA2, Breast tumor suppressor, has dual functionality promoting not only DNA repair but also preventing DNA lesions at stalled forks. In response to replication stress, BRCA2 recruits RAD51 onto nascent DNA at stalled forks, protecting nascent DNA from nucleolitic cleavage. Phosphorylation of the BRCA2 C-terminal RAD51 binding site by CDK2 promotes RAD51 filament disassembly, leading to nucleolitic cleavage of newly synthesized DNA and compromised fork integrity. Recently we uncovered how the core Hippo pathway components RASSF1A, MST2 and LATS1 regulate CDK2 activity towards BRCA2, in response to fork stalling. In complex with LATS1, CDK2 exhibits reduced kinase activity which results in low levels of pBRCA2-S3291 and stable RAD51 filaments protecting nascent DNA from MRE11 cleavage. In the absence of the RASSF1A/MST2/LATS1/CDK2 pathway increased resection of newly synthesized DNA leads to chromosomal instability and malignant transformation. This function of RASSF1A in stalled replication fork protection adds to the role of RASSF1A as a tumor suppressor and builds up evidence for RASSF1A status and its prognostic and predictive value in cancer.
Introduction
Genomic instability is a major characteristic of most tumors however, is often detected in premalignant lesions, indicating that is also a driving force of cancer transformation.1-3 Over the last decade different mechanisms have been suggested to explain the source of genome instability, including defective DNA damage repair,4 oxidative stress,5 errors in chromosomal segregation6 and telomere attrition.7 However, these mechanisms contribute mainly to chromosomal changes, which appear in more advanced stages of cancer development.8 The most attractive model to explain the induction of genomic instability in early stages of cancer development is stress caused to DNA replication as a result of oncogene activation and subsequent loss of sufficient nucleotide pools to support normal DNA replication, resulting in fork stalling.9-11
Observations made more than 10 y ago showed that induction of replication stress with hydroxyurea (HU) in cells that lacked BRCA2 resulted in collapse of replication forks and double strand breaks, possibly due to inability to resolve secondary structures.12 Recently though, using detailed DNA fiber analysis in response to replication stress induced by short treatment with HU, BRCA2 was characterized as a central component of a distinct replication fork protection pathway.13 The CDK2 mediated phosphorylation of the C-terminal segment (TR2 domain) of BRCA2 was shown to abrogate the formation of protective RAD51 nucleofilaments at nascent DNA at stalled forks. Most importantly, in the absence of BRCA2 or in the presence of the phosphomimetic BRCA2-S3291A mutant, nascent DNA gets resected from the MRE11 nuclease resulting in accumulation of single stranded DNA gaps and chromosomal aberrations.13,14
In our recent study, we further elucidated the molecular mechanism that governs replication fork protection during stress. We demonstrated pathway crosstalk between BRCA2-RAD51 fork protection and the Hippo pathway,15 a kinase cascade involved in cell proliferation, survival and apoptosis. Specifically we show that LATS1, a central kinase of the Hippo pathway interacts with CDK2 only after treatment with replication stress agents resulting in decreased CDK2 kinase activity and low levels pBRCA2-S3291. Most importantly LATS1 function in replication fork protection is kinase independent and depends on interaction with CDK2 in a complex with no cyclins. In the absence of LATS1, increased CDK2 activity toward BRCA2 promotes the disruption of RAD51 filaments and exposes nascent DNA to nucleolitic attack. RASSF1A tumor suppressor has a central role in the Hippo pathway activation in response to stress. In absence of RASSF1A, LATS1 fails to interact with CDK2 resulting in unstable RAD51 nucleofilaments and nascent DNA resection.15
Repair Proteins Protecting Stalled Forks
Schlacher and colleagues showed that components of Homologous Recombination (HR) function in a separate pathway that protects rather than repairs damage at stalled replication forks.13 BRCA2 promotes RAD51 loading on nascent DNA exposed at stalled forks protecting it from extensive nucleolitic cleavage. MRE11 presence at stalled forks is known to be necessary for fork restart,16 as controlled nucleolitic activity at stalled forks is hypothesized to be required for enlargement of small ssDNA gaps created by the discontinuous fork progression, facilitating amplification of checkpoint signaling (Fig. 1). Thus, Rad51 presence at stalled forks is believed to function as a protective barrier that limits the extent of DNA resection, therein maintaining genome stability.13,14,17
Figure 1.

RASSF1A/LATS1/CDK2 pathway upon fork stalling. In response to ATR activation RASSF1A gets targeted at Serine 131 triggering LATS1 phosphorylation and interaction with CDK2. Low levels of pBRCA2-S3291 due to decreased kinase activity of CDK2 fraction bound to LATS1 secure RAD51 filament formation at stalled forks. RAD51 filaments protect nascent DNA from MRE11 nucleolitic activity that is recruited to the fork in parallel with other DNA repair factors to promote fork restart after the removal of the stalling agent. RASSF1A inactivation due to promoter methylation or the SNP at the 133A site leads to increased pBRCA2-S3291, unstable RAD51 filaments and uncontrolled nascent DNA resection. Red circles: RPA, green circles: RAD51, packman: MRE11.
In addition to BRCA2, several other components of the HR machinery and DNA repair proteins are necessary for the formation of stable RAD51 nucleofilamets upon fork stalling.18 FANCD2, known for promoting break formation and translesion synthesis in response to intra-strand crosslinks, functions epistatically with BRCA2 and RAD51 in the stabilization of stalled forks. BRCA1, breast cancer suppressor which interacts with several FANC proteins, including FANCD2 and BRCA2 is also part of the replication fork protection pathway.18 Interestingly, Schlacher et al. found that BRACA1/2 and FANCD2 are dispensable for fork restart after the removal of the stalling agent18 despite the necessity of RAD51 for fork recovery.16,19 Conversely, BLM, a RecQ helicase that promotes dissolution of Holliday junction intermediates after HR repair of DNA double strand breaks,20 is not required for the protection pathway but is necessary for fork restart.18 In a more recent report though, it was proposed that FANCD2 does have a role in fork recovery via BLM regulation.21 WRN, another RecQ helicase has been found to be required for fork progression rather than restart after the removal of the stalling agent,22 while potential function of WRN in the replication fork protection pathway has not been addressed (Fig. 1). Thus, further studies are necessary to clarify if repair proteins recruited at stalled forks function in protection as well as restart, or whether is a ‘work load’ distribution among them.
Overexpression of a mutant RAD51 with impaired ATPase activity that results in stable filaments can rescue nascent DNA shortening at stalled forks in cells lacking BRCA2.13 Moreover, overexpression of wild type RAD51 can alleviate resection in Fanconi Anemia (FA) deficient cells.18 Previous studies have also shown that RAD51 can compensate for some aspects of a BRCA2 gene defects, including the impaired radiation responsiveness evident in BRCA2 mutant cells.23
PARP1, is necessary for MRE11 relocation and efficient restart of collapsed forks as a result of long HU treatments, 24 however is dispensable for MRE11 recruitment at transiently stalled forks without evidence of double strand breaks.17 Formation of PAR polymers at stalled forks also prevent resection although potentially through a distinct mechanism.17 The full understanding of the rules that govern replication fork protection and whether BRCA2/RAD51 pathway alone is sufficient, or not, for protection of stalled replication forks could offer a significant therapeutic potential in the future.
BRCA2 C-Terminus in the Regulation of Stalled Fork Protection: LATS1 Promotes Fork Stability
Detailed BRCA2 domain mapping showed that the DNA binding domain of BRCA2 is dispensable for fork protection, however interaction with RAD51 is required.13 BRCA2 has 2 interaction domains with RAD51. The first contains 8 centrally localized BRC repeats, which are known to bind RAD51 monomers in a 1:1 complex between monomeric RAD51 and BRC repeat.25 The second domain (TR2 domain) lays in the far C-terminus of BRCA2, binds oligomeric RAD51 and stabilises the nucleofilaments.26,27 RAD51 interaction with the BRCA2 C-terminus is regulated by CKDs. In a normal cell cycle, increased levels of pBRCA2-S3291 are evident in G2/M transition after activation of CDK1 resulting in disassembly of RAD51 oligomers to allow DNA segregation.28 In response to DNA damage during S phase, levels of pBRCA2-S3291 further decrease to permit fork protection and recombination activity.28
Mutations in S3291 appear to be dispensable for HR, however BRCA2-S3291A is unable to rescue increased MRE11 resection in nascent DNA in response to HU induced stress in V-C8 cells that lack functional BRCA2, indicating the significance of CDK2 activity regulation for protection of stalled forks.13
In our recent study we offered further mechanistic insight in the regulation of BRCA2-S3291 phosphorylation by CDK2 in response to ATR activation, where we showed that the Hippo pathway has a major role in that via activation of the tumor suppressor LATS1.15
LATS1 is a Serine/Threonine kinase that phosphorylates transcriptional co activator and canonical Hippo pathway substrate, YAP on HX(R/H/K)XX(S/T) consensus sites regulating YAP nuclear/cytoplasmic shuttling and differential complex formation between YAP and transcription factors.29-32 LATS1 activation depends on the MST1/2 (Hippo orthologues in mammals), which mediate LATS1 phosphorylation at S909 and T1079 leading to conformational changes and kinase activity stimulation.33 Intriguingly, LATS1 was identified in a proteomic analysis for novel pathways that respond to ATM/ATR stimulation and maintain genomic stability, offering a new insight on the function of the Hippo pathway.34
LATS1 interaction with CDKs has been reported upon entry to mitosis, however the signal that drives this interaction had been missing.35 Activated LATS1 was shown to interact specifically with CDK1 in the G2/M transition in a complex which had compromised kinase activity toward histone H1 substrate. It was suggested that LATS1 interaction with CDK1 was dependent on its phosphorylation but the responsible kinase was not determined.35
In our study we found that regulation of BRCA2 C-terminal phosphorylation requires activation by MST2 but does not depend on LATS1 kinase activity or its canonical substrate YAP.15 In response to replication stress MST2 targets LATS1 offering the necessary conformational change to allow interaction with CDK2. Subsequent sequestration of CDK2 from its active complex with cyclins leads to decreased TR2 domain phosphorylation in BRCA2 and RAD51 nucleofilament stabilization on nascent DNA exposed at stalled/regressed forks.15 Detailed mapping for LATS1/CDK2 interaction showed that binding to CDK2 depends on the N-terminus of LATS115. The first 200 amino acids necessary for interaction with CDK2 contain LATS Conserved Domain 1 (LCD1) (aa12-167), one of the 2 conserved domains in the N-terminus between LATS1 and LATS2 orthologues. Lats1LCD1−/− mice show increased chromosomal instability and tumorigenicity,36 indicative of the LCD1 domain importance for LATS1 tumor suppressive function. Deletion of LCD1 from LATS2 also abolishes its tumor suppressor activity on NIH3T3/v- ras cells.37 Therefore, it would be interesting to test if regulation of CDK2 is a conserved function between the orthologues in response to MST1/2 activation or is a distinct function of LATS1.
In Lats1−/− MEFS treated with HU, significantly increased levels of pBRCA2-S3291 led to nascent DNA resection. Treatment of Lats1−/− MEFS with the MRE11 inhibitor mirin rescued shortening of nascent DNA tracks, indicating that LATS1 directly regulates BRCA2 mediated RAD51 stabilization at stalled forks.15
LATS1 ablation has been shown to result in increased tumorigenicity both in mice and flies.38,39 Moreover, analysis of the mutation distribution across different domains of LATS1 from patient samples in the Catalog of Somatic Mutation in Cancer (COSMIC) database, showed mutations at several conserved sites in LCD1.40 Our recent study offers a new insight in LATS1 tumor suppressive function and also supports a kinase independent role of LATS1 in addition to its kinase mediated regulation of YAP.15
RASSF1A Inactivation, Genomic Stability and Cancer Predisposition
Deregulation of the tumor suppressive ‘Hippo pathway’ results in tissue overgrowth and carcinogenesis in model systems, but its role in human cancer is less clear. The mammalian Hippo kinase orthologues, MST1/2, are tightly controlled by the RASSF family, of which RASSF1A is widely described to be epigenetically inactivated, displaying increased risk of onset and poor prognosis across all major sporadic malignancies. RASSF1A is one of the 7 different isoforms that rise from the RASSF1 gene at the 3p21.3 locus, a segment of the genome highly populated by tumor suppressor genes.41–43 RASSF1A is a Ras effector that possesses tumor suppressive properties functioning as a scaffold protein, since it lacks enzymatic activity. Genetically ablated mice for Rassf1A demonstrate an enhanced predisposition to develop spontaneous tumors and this effect is amplified in a p53 null background or after inactivation of APC (Adenomatus Polyposis Coli). 44–46 Moreover, Rassf1A−/− and Rassf1A−/+ mice show increased tumor multiplicity and tumor size after treatment with chemical carcinogens.44
RASSF1A regulates Hippo pathway pro-apoptotic activity in response to Fas receptor activation and double strand breaks.30,47 In response to these stimuli RASSF1A sequesters MST2 from the inhibitory complex with Raf1, allowing MST2 dimerization and autophosphorylation on the Thr residues within the activation loop.47,48 MST2 increased kinase activity in response to RASSF1A overexpression leads to LATS1 phosphorylation and kinase activity activation that has been shown to result in preferential YAP binding to p73 and transcription of the proapoptotic target gene PUMA.30
RASSF1A has a unique SQ site at S131 that is targeted by ATM in response to double strand breaks resulting in RASSF1A dimerization. RASSF1A dimer has a greater affinity for MST2, possibly due to an altered orientation of the SARAH interaction domains, compared to the monomer.30 RASSF1A activation in response to ionizing radiation leads in apoptotic transcription stimulation via the main Hippo pathway output, YAP and the DNA damage responsive factor p73 (Fig. 2).
Figure 2.
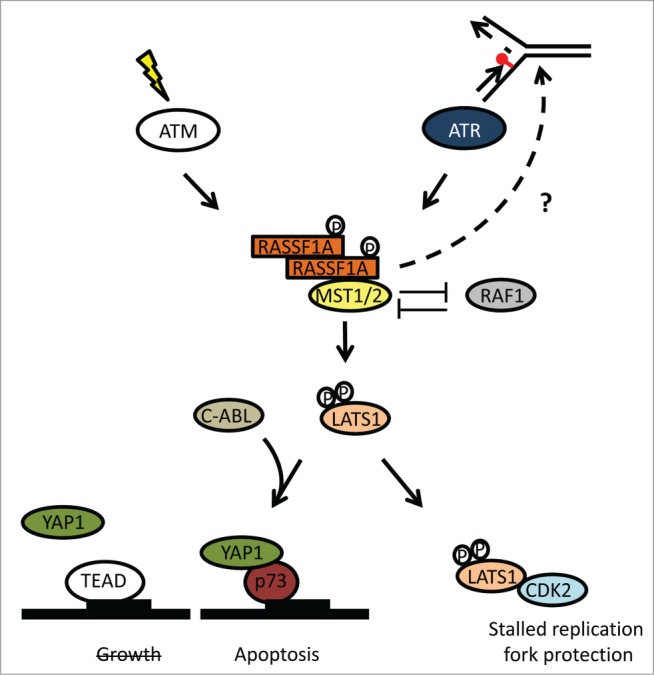
RASSF1A regulation of the Hippo pathway in response to DNA damage. Double strand breaks leading to ATM activation or stalled replication forks that trigger ATR activity result in phosphorylation of RASSF1A at S131. Phosphorylated RASSF1A forms dimers that bind to MST2 abrogating inhibitory interaction with RAF1. Activated MST2 binds and phosphorylates LATS1 offering the necessary conformational change for CDK2 interaction that lead to decreased pBRCA2-S3291. LATS1 phosphorylation also stimulates its kinase activity toward YAP1 leading to decreased association with the TEAD transcription factors that promote cell growth and increased association with p73 inducing apoptosis, potentially in synergy with c-ABL tyrosine kinase.
We showed that RASSF1A is also necessary for Hippo pathway activation in response to replication stress. After HU treatment in the absence of RASSF1A, MST2 fails to target LATS1, and stimulate secondary structure changes that allow interaction with CDK2. Uncontrolled CDK2 kinase activity leads to increased levels of pBRCA2-S3291, unstable RAD51 nucleofilaments and increased nucleolitic cleavage of nascent DNA at stalled forks. The end result is widespread chromosomal aberrations and increased genomic instability, similar to BRCA mutant cells.15 Silencing of the RASSF1A/LATS1 cascade also leads to defective fork restart and progression after stress removal.15 Weather this is BRCA2 dependent or contributes independently in the genomic instability evident in the absence of the cascade has to be examined.
Epigenetic silencing of RASSF1A is probably the most frequently described epigenetic inactivation event so far in human cancers. Hypermethylation of the RASSF1A promoter CpG Island has been reported in early stages of many different cancer types including lung, breast, prostate, glioma, neuroblastoma and kidney cancer and several recent studies have illustrated the diagnostic and prognostic potential of RASSF1A methylation. In lung cancers, which are the most frequent cancer type worldwide, RASSF1A is hypermethylated in 72–100% of Small Cell Lung Cancer (SCLC) tumor specimens, in 30–45% of resected Non-SCLC tissues and in 36–88% of NSCLC cell lines respectively.49-51 Moreover, RASSF1A methylation has been negatively correlated in a number of studies with disease free and overall survival.52-54 In our study, in a lung adenocarcinoma cohort of 188 patients from the Cancer Genome Atlas (TCGA) database we found increased correlation between RASSF1A methylation and Copy Number Alterations (CNA).15 A recent study also found a positive correlation of RASSF1 promoter methylation and increased CNA in breast cancer patients.55 These finding highlight the significance of RASSF1A/LATS1/CDK2 axis for protection of genomic stability and promote the idea of using replication stress agents as a therapeutic approach to target patients with high RASSF1A methylation in a synthetically lethal approach. In agreement with this, NSCL patients showed increased sensitivity in the replication stress inducing agent, Gemcitabine further supporting the potential use of RASSF1A as biomarker NSCL cancer patients.56
Loss of RASSF1A expression is largely attributed to promoter hypermethylation or LOH at the 3p21.3 locus, as somatic mutations of RASSF1A are rare.57 However, Single Nucleotide Polymorphisms (SNPs) inherited in germline DNA, often affect gene regulation and as with familial syndromes, can influence susceptibility to a variety of disease including cancer. rs2073498, represents a relative frequent alteration in the RASSF1 sequence in European and Asian populations where 10–20% of individuals carry at least one allele variant. While the majority of SNPs represent changes in intergenic regions within exon regions result in alterations in the coding sequence. The rs2073498 SNP located in exon 3 alters the coding sequence at amino acid position 133 of isoform A, next to the ATM activation site, that results in the conversion of a phylogenetically conserved Alanine (encoded by the major G allele) to Serine (encoded by the minor T allele) which is unable to get phosphorylated by ATM in response to damage.58 Serine substitution of the genetically conserved Alanine at S133 next to the ATM phospho-site, possibly results in loss of a helical turn and secondary structure disruption leading to impaired ATM targeting.58
Re-expression of RASSF1A-A133S in cells that lack RASSF1A is unable to restore LATS1/CDK2 interaction and rescue the DNA shortening at stalled forks.15 We showed that ATR targets RASSF1A at S131 but not the polymorphic mutant which like RASSF1A loss of expression correlates with increased cancer susceptibility and worse disease outcome in lung, breast, prostate, head and neck, liver and soft tissues sarcomas.57-60 Schagdarsurengin et al. showed a higher frequency of the A133S polymorphism in 141 Caucasian females with breast cancer compared with 70 healthy individuals.61 More importantly in a study where the age of breast cancer onset among the BRCA1/2 mutation carriers with or without A133S was compared, minor allele carriers were diagnosed 6 y earlier than those with wild-type RASSF1A.59 Although subsequently questioned,62 there is increasing evidence for association between the polymorphic mutant and familiar breast cancer, and the inconsistencies may simply reflect additional parameters not accounted depending on BRCA mutation or cancer subtypes. Thus, the presence of an A133S polymorphism along with a BRCA1/2 mutation in the same individual may result as a BRCA2 Loss of Heterozygosity like event leading to early disease onset.
Conclusion and Unresolved Questions
Hippo pathway is a developmental pathway known to signal to YAP/TAZ transcription factors and regulate their activity.63 Many components of the Hippo pathway have been shown to possess tumor suppressive functions independently of the cascade. Our recent study showed that the Hippo pathway functions independently of YAP to promote genome stability at stalled replication forks in response to RASSF1A activation.15 RASSF1A is one of the most commonly inactivated genes in premalignant and malignant lesions. We highlight here that RASSF1A activation in response to stress not only result in regulation of the transcriptome via YAP, but also abrogates nascent DNA resection exhibiting a protective role at the fork (Fig. 2). Apart from increased DNA resection RASSF1A inactivation leads to defective fork restart and decreased velocity indicating that RASSF1A could have additional roles at the fork.15 BRCA2 depletion despite RAD51 destabilization does not lead to restart problems after stress removal.13 Recently RASSF1A was shown to interact with nuclear XPA promoting its repair activity in Nucleotide Excision Repair (NER) showing a nuclear role of RASSF1A in DNA repair.64 Moreover NER in combination with fork processing can catalyze blockage removal and fork restart.65 It is possible for RASSF1A to have a fork-localized function independent of control of LATS1/CDK2 interaction. In conclusion with our recent study we offer a new aspect on how the Hippo pathway can function in maintenance of genome integrity and protection from malignant transformation.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011; 144:646–74; PMID:21376230; http://dx.doi.org/ 10.1016/j.cell.2011.02.013 [DOI] [PubMed] [Google Scholar]
- 2.Klausner RD. The fabric of cancer cell biology - Weaving together the strands. Cancer Cell 2002; 1:3–10; PMID:12086880; http://dx.doi.org/ 10.1016/S1535-6108(02)00020-X [DOI] [PubMed] [Google Scholar]
- 3.Negrini S, Gorgoulis VG, Halazonetis TD. Genomic instability - an evolving hallmark of cancer. Nat Rev Mol Cell Bio 2010; 11:220–8; PMID:20177397; http://dx.doi.org/ 10.1038/nrm2858 [DOI] [PubMed] [Google Scholar]
- 4.Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature 2009; 461:1071–8; PMID:19847258; http://dx.doi.org/ 10.1038/nature08467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bartkova J, Hamerlik P, Stockhausen MT, Ehrmann J, Hlobilkova A, Laursen H, Kalita O, Kolar Z, Poulsen HS, Broholm H, et al.. Replication stress and oxidative damage contribute to aberrant constitutive activation of DNA damage signalling in human gliomas. Oncogene 2010; 29:5095–102; PMID:20581868; http://dx.doi.org/ 10.1038/onc.2010.249 [DOI] [PubMed] [Google Scholar]
- 6.Gisselsson D. Classification of chromosome segregation errors in cancer. Chromosoma 2008; 117:511–9; PMID:18528701; http://dx.doi.org/ 10.1007/s00412-008-0169-1 [DOI] [PubMed] [Google Scholar]
- 7.Artandi SE, Chang S, Lee SL, Alson S, Gottlieb GJ, Chin L, DePinho RA. Telomere dysfunction promotes non-reciprocal translocations and epithelial cancers in mice. Nature 2000; 406:641–5; PMID:10949306; http://dx.doi.org/ 10.1038/35020592 [DOI] [PubMed] [Google Scholar]
- 8.Gorgoulis VG, Vassiliou LVF, Karakaidos P, Zacharatos P, Kotsinas A, Liloglou T, Venere M, DiTullio RA, Kastrinakis NG, Levy B, et al.. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature 2005; 434:907–13; PMID:15829965; http://dx.doi.org/ 10.1038/nature03485 [DOI] [PubMed] [Google Scholar]
- 9.Bester AC, Roniger M, Oren YS, Im MM, Sarni D, Chaoat M, Bensimon A, Zamir G, Shewach DS, Kerem B. Nucleotide deficiency promotes genomic instability in early stages of cancer development. Cell 2011; 145:435–46; PMID:21529715; http://dx.doi.org/ 10.1016/j.cell.2011.03.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Di Micco R, Fumagalli M, Cicalese A, Piccinin S, Gasparini P, Luise C, Schurra C, Garre M, Nuciforo PG, Bensimon A, et al.. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature 2006; 444:638–42; PMID:17136094; http://dx.doi.org/ 10.1038/nature05327 [DOI] [PubMed] [Google Scholar]
- 11.Bartkova J, Rezaei N, Liontos M, Karakaidos P, Kletsas D, Issaeva N, Vassiliou LVF, Kolettas E, Niforou K, Zoumpourlis VC, et al.. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 2006; 444:633–7; PMID:17136093; http://dx.doi.org/ 10.1038/nature05268 [DOI] [PubMed] [Google Scholar]
- 12.Lomonosov M, Anand S, Sangrithi M, Davies R, Venkitaraman AR. Stabilization of stalled DNA replication forks by the BRCA2 breast cancer susceptibility protein. Gene Dev 2003; 17:3017–22; PMID:14681210; http://dx.doi.org/ 10.1101/gad.279003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schlacher K, Christ N, Siaud N, Egashira A, Wu H, Jasin M. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11 (vol 145, pg 529, 2011). Cell 2011; 145: 993-; PMID:21565612; http://dx.doi.org/ 10.1016/j.cell.2011.05.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hashimoto Y, Ray Chaudhuri A, Lopes M, Costanzo V. Rad51 protects nascent DNA from Mre11-dependent degradation and promotes continuous DNA synthesis. Nat Struct Mol Biol 2010; 17:1305–11; PMID:20935632; http://dx.doi.org/ 10.1038/nsmb.1927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pefani DE, Latusek R, Pires I, Grawenda AM, Yee KS, Hamilton G, van der Weyden L, Esashi F, Hammond EM, O'Neill E. RASSF1A-LATS1 signalling stabilizes replication forks by restricting CDK2-mediated phosphorylation of BRCA2. Nat Cell Biol 2014; 16:962–71; PMID:25218637; http://dx.doi.org/ 10.1038/ncb3035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hashimoto Y, Puddu F, Costanzo V. RAD51- and MRE11-dependent reassembly of uncoupled CMG helicase complex at collapsed replication forks. Nat Struct Mol Biol 2012; 19:17–24; PMID:22139015; http://dx.doi.org/ 10.1038/nsmb.2177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ying S, Hamdy FC, Helleday T. Mre11-dependent degradation of stalled DNA replication forks is prevented by BRCA2 and PARP1. Cancer Res 2012; 72:2814–21; PMID:22447567; http://dx.doi.org/ 10.1158/0008-5472.CAN-11-3417 [DOI] [PubMed] [Google Scholar]
- 18.Schlacher K, Wu H, Jasin M. A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell 2012; 22:106–16; PMID:22789542; http://dx.doi.org/ 10.1016/j.ccr.2012.05.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Petermann E, Orta ML, Issaeva N, Schultz N, Helleday T. Hydroxyurea-stalled replication forks become progressively inactivated and require two different RAD51-mediated pathways for restart and repair. Mol Cell 2010; 37:492–502; PMID:20188668; http://dx.doi.org/ 10.1016/j.molcel.2010.01.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu L, Hickson ID. The Bloom's syndrome helicase suppresses crossing over during homologous recombination. Nature 2003; 426:870–4; PMID:14685245; http://dx.doi.org/ 10.1038/nature02253 [DOI] [PubMed] [Google Scholar]
- 21.Chaudhury I, Sareen A, Raghunandan M, Sobeck A. FANCD2 regulates BLM complex functions independently of FANCI to promote replication fork recovery. Nucleic Acids Res 2013; 41:6444–59; PMID:23658231; http://dx.doi.org/ 10.1093/nar/gkt348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sidorova JM, Li N, Folch A, Monnat RJ Jr. The RecQ helicase WRN is required for normal replication fork progression after DNA damage or replication fork arrest. Cell Cycle 2008; 7:796–807; PMID:18250621; http://dx.doi.org/ 10.4161/cc.7.6.5566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brown ET, Holt JT. Rad51 overexpression rescues radiation resistance in BRCA2-defective cancer cells. Mol Carcinog 2009; 48:105–9; PMID:18618591; http://dx.doi.org/ 10.1002/mc.20463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bryant HE, Petermann E, Schultz N, Jemth AS, Loseva O, Issaeva N, Johansson F, Fernandez S, McGlynn P, Helleday T. PARP is activated at stalled forks to mediate Mre11-dependent replication restart and recombination. EMBO J 2009; 28:2601–15; PMID:19629035; http://dx.doi.org/ 10.1038/emboj.2009.206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Davies AA, Masson JY, McIlwraith MJ, Stasiak AZ, Stasiak A, Venkitaraman AR, West SC. Role of BRCA2 in control of the RAD51 recombination and DNA repair protein. Mol Cell 2001; 7:273–82; PMID:11239456; http://dx.doi.org/ 10.1016/S1097-2765(01)00175-7 [DOI] [PubMed] [Google Scholar]
- 26.Davies OR, Pellegrini L. Interaction with the BRCA2 C terminus protects RAD51-DNA filaments from disassembly by BRC repeats. Nat Struct Mol Biol 2007; 14:475–83; PMID:17515903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Esashi F, Galkin VE, Yu X, Egelman EH, West SC. Stabilization of RAD51 nucleoprotein filaments by the C-terminal region of BRCA2. Nat Struct Mol Biol 2007; 14:468–74; PMID:17515904; http://dx.doi.org/ 10.1038/nsmb1245 [DOI] [PubMed] [Google Scholar]
- 28.Esashi F, Christ N, Gannon J, Liu Y, Hunt T, Jasin M, West SC. CDK-dependent phosphorylation of BRCA2 as a regulatory mechanism for recombinational repair. Nature 2005; 434:598–604; PMID:15800615; http://dx.doi.org/ 10.1038/nature03404 [DOI] [PubMed] [Google Scholar]
- 29.Hao Y, Chun A, Cheung K, Rashidi B, Yang X. Tumor suppressor LATS1 is a negative regulator of oncogene YAP. J Biol Chem 2008; 283:5496–509; PMID:18158288; http://dx.doi.org/ 10.1074/jbc.M709037200 [DOI] [PubMed] [Google Scholar]
- 30.Hamilton G, Yee KS, Scrace S, O'Neill E. ATM regulates a RASSF1A-dependent DNA damage response. Curr Biol: CB 2009; 19:2020–5; PMID:19962312; http://dx.doi.org/ 10.1016/j.cub.2009.10.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.van der Weyden L, Papaspyropoulos A, Poulogiannis G, Rust AG, Rashid M, Adams DJ, Arends MJ, O'Neill E. Loss of RASSF1A synergizes with deregulated RUNX2 signaling in tumorigenesis. Cancer Res 2012; 72:3817–27; PMID:22710434; http://dx.doi.org/ 10.1158/0008-5472.CAN-11-3343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhao B, Wei X, Li W, Udan RS, Yang Q, Kim J, Xie J, Ikenoue T, Yu J, Li L, et al.. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev 2007; 21:2747–61; PMID:17974916; http://dx.doi.org/ 10.1101/gad.1602907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chan EH, Nousiainen M, Chalamalasetty RB, Schafer A, Nigg EA, Sillje HH. The Ste20-like kinase Mst2 activates the human large tumor suppressor kinase Lats1. Oncogene 2005; 24:2076–86; PMID:15688006; http://dx.doi.org/ 10.1038/sj.onc.1208445 [DOI] [PubMed] [Google Scholar]
- 34.Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER 3rd, Hurov KE, Luo J, Bakalarski CE, Zhao Z, Solimini N, Lerenthal Y, et al.. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 2007; 316:1160–6; PMID:17525332; http://dx.doi.org/ 10.1126/science.1140321 [DOI] [PubMed] [Google Scholar]
- 35.Tao W, Zhang S, Turenchalk GS, Stewart RA, St John MA, Chen W, Xu T. Human homologue of the Drosophila melanogaster lats tumour suppressor modulates CDC2 activity. Nat Genet 1999; 21:177–81; PMID:9988268; http://dx.doi.org/ 10.1038/5960 [DOI] [PubMed] [Google Scholar]
- 36.Yabuta N, Mukai S, Okamoto A, Okuzaki D, Suzuki H, Torigata K, Yoshida K, Okada N, Miura D, Ito A, et al.. N-terminal truncation of Lats1 causes abnormal cell growth control and chromosomal instability. J Cell Sci 2013; 126:508–20; PMID:23230145; http://dx.doi.org/ 10.1242/jcs.113431 [DOI] [PubMed] [Google Scholar]
- 37.Li Y, Pei J, Xia H, Ke H, Wang H, Tao W. Lats2, a putative tumor suppressor, inhibits G1/S transition. Oncogene 2003; 22:4398–405; PMID:12853976; http://dx.doi.org/ 10.1038/sj.onc.1206603 [DOI] [PubMed] [Google Scholar]
- 38.Xu T, Wang W, Zhang S, Stewart RA, Yu W. Identifying tumor suppressors in genetic mosaics: the Drosophila lats gene encodes a putative protein kinase. Development 1995; 121:1053–63; PMID:7743921 [DOI] [PubMed] [Google Scholar]
- 39.John MA, Tao W, Fei X, Fukumoto R, Carcangiu ML, Brownstein DG, Parlow AF, McGrath J, Xu T. Mice deficient of Lats1 develop soft-tissue sarcomas, ovarian tumours and pituitary dysfunction. Nat Genet 1999; 21:182–6; PMID:9988269; http://dx.doi.org/ 10.1038/7741 [DOI] [PubMed] [Google Scholar]
- 40.Yu T, Bachman J, Lai ZC. Mutation analysis of large tumor suppressor genes LATS1 and LATS2 supports a tumor suppressor role in human cancer. Protein Cell 2015; 6:6–11; PMID:25482410; http://dx.doi.org/ 10.1007/s13238-014-0122-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hesson LB, Cooper WN, Latif F. Evaluation of the 3p21.3 tumour-suppressor gene cluster. Oncogene 2007; 26:7283–301; PMID:17533367; http://dx.doi.org/ 10.1038/sj.onc.1210547 [DOI] [PubMed] [Google Scholar]
- 42.Dammann R, Takahashi T, Pfeifer GP. The CpG island of the novel tumor suppressor gene RASSF1A is intensely methylated in primary small cell lung carcinomas. Oncogene 2001; 20:3563–7; PMID:11429703; http://dx.doi.org/ 10.1038/sj.onc.1204469 [DOI] [PubMed] [Google Scholar]
- 43.Dammann R, Yang G, Pfeifer GP. Hypermethylation of the cpG island of Ras association domain family 1A (RASSF1A), a putative tumor suppressor gene from the 3p21.3 locus, occurs in a large percentage of human breast cancers. Cancer Res 2001; 61:3105–9; PMID:11306494 [PubMed] [Google Scholar]
- 44.Tommasi S, Dammann R, Zhang Z, Wang Y, Liu L, Tsark WM, Wilczynski SP, Li J, You M, Pfeifer GP. Tumor susceptibility of Rassf1a knockout mice. Cancer Res 2005; 65:92–8; PMID:15665283 [PubMed] [Google Scholar]
- 45.Tommasi S, Besaratinia A, Wilczynski SP, Pfeifer GP. Loss of Rassf1a enhances p53-mediated tumor predisposition and accelerates progression to aneuploidy. Oncogene 2011; 30:690–700; PMID:20890300; http://dx.doi.org/ 10.1038/onc.2010.440 [DOI] [PubMed] [Google Scholar]
- 46.van der Weyden L, Arends MJ, Dovey OM, Harrison HL, Lefebvre G, Conte N, Gergely FV, Bradley A, Adams DJ. Loss of Rassf1a cooperates with Apc(Min) to accelerate intestinal tumourigenesis. Oncogene 2008; 27:4503–8; PMID:18391979; http://dx.doi.org/ 10.1038/onc.2008.94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Matallanas D, Romano D, Yee K, Meissl K, Kucerova L, Piazzolla D, Baccarini M, Vass JK, Kolch W, O'Neill E. RASSF1A elicits apoptosis through an MST2 pathway directing proapoptotic transcription by the p73 tumor suppressor protein. Mol Cell 2007; 27:962–75; PMID:17889669; http://dx.doi.org/ 10.1016/j.molcel.2007.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.O'Neill E, Rushworth L, Baccarini M, Kolch W. Role of the kinase MST2 in suppression of apoptosis by the proto-oncogene product Raf-1. Science 2004; 306:2267–70; PMID:15618521; http://dx.doi.org/ 10.1126/science.1103233 [DOI] [PubMed] [Google Scholar]
- 49.Honorio S, Agathanggelou A, Schuermann M, Pankow W, Viacava P, Maher ER, Latif F. Detection of RASSF1A aberrant promoter hypermethylation in sputum from chronic smokers and ductal carcinoma in situ from breast cancer patients. Oncogene 2003; 22:147; PMID:12527916; http://dx.doi.org/ 10.1038/sj.onc.1206057 [DOI] [PubMed] [Google Scholar]
- 50.Hoque MO, Begum S, Topaloglu O, Jeronimo C, Mambo E, Westra WH, Califano JA, Sidransky D. Quantitative detection of promoter hypermethylation of multiple genes in the tumor, urine, and serum DNA of patients with renal cancer. Cancer Res 2004; 64:5511–7; PMID:15289362; http://dx.doi.org/ 10.1158/0008-5472.CAN-04-0799 [DOI] [PubMed] [Google Scholar]
- 51.Vaissiere T, Hung RJ, Zaridze D, Moukeria A, Cuenin C, Fasolo V, Ferro G, Paliwal A, Hainaut P, Brennan P, et al.. Quantitative analysis of DNA methylation profiles in lung cancer identifies aberrant DNA methylation of specific genes and its association with gender and cancer risk factors. Cancer Res 2009; 69:243–52; PMID:19118009; http://dx.doi.org/ 10.1158/0008-5472.CAN-08-2489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Buhmeida A, Merdad A, Al-Maghrabi J, Al-Thobaiti F, Ata M, Bugis A, Syrjanen K, Abuzenadah A, Chaudhary A, Gari M, et al.. RASSF1A methylation is predictive of poor prognosis in female breast cancer in a background of overall low methylation frequency. Anticancer Res 2011; 31:2975–81; PMID:21868547 [PubMed] [Google Scholar]
- 53.Ko E, Lee BB, Kim Y, Lee EJ, Cho EY, Han J, Shim YM, Park J, Kim DH. Association of RASSF1A and p63 with poor recurrence-free survival in node-negative stage I-II non-small cell lung cancer. Clin Cancer Res: Off J Am Assoc Cancer Res 2013; 19:1204–12; PMID:23319821; http://dx.doi.org/ 10.1158/1078-0432.CCR-12-2848 [DOI] [PubMed] [Google Scholar]
- 54.Wang J, Wang B, Chen X, Bi J. The prognostic value of RASSF1A promoter hypermethylation in non-small cell lung carcinoma: a systematic review and meta-analysis. Carcinogenesis 2011; 32:411–6; PMID:21156971; http://dx.doi.org/ 10.1093/carcin/bgq266 [DOI] [PubMed] [Google Scholar]
- 55.Murria R, Palanca S, de Juan I, Egoavil C, Alenda C, Garcia-Casado Z, Juan MJ, Sanchez AB, Santaballa A, Chirivella I, et al.. Methylation of tumor suppressor genes is related with copy number aberrations in breast cancer. Am J Cancer Res 2015; 5:375–85; PMID:25628946 [PMC free article] [PubMed] [Google Scholar]
- 56.Fischer JR, Ohnmacht U, Rieger N, Zemaitis M, Stoffregen C, Manegold C, Lahm H. Prognostic significance of RASSF1A promoter methylation on survival of non-small cell lung cancer patients treated with gemcitabine. Lung Cancer 2007; 56:115–23; PMID:17196704; http://dx.doi.org/ 10.1016/j.lungcan.2006.11.016 [DOI] [PubMed] [Google Scholar]
- 57.Hogg RP, Honorio S, Martinez A, Agathanggelou A, Dallol A, Fullwood P, Weichselbaum R, Kuo MJ, Maher ER, Latif F. Frequent 3p allele loss and epigenetic inactivation of the RASSF1A tumour suppressor gene from region 3p21.3 in head and neck squamous cell carcinoma. Eur J Cancer 2002; 38:1585–92; PMID:12142046; http://dx.doi.org/ 10.1016/S0959-8049(01)00422-1 [DOI] [PubMed] [Google Scholar]
- 58.Yee KS, Grochola L, Hamilton G, Grawenda A, Bond EE, Taubert H, Wurl P, Bond GL, O'Neill E. A RASSF1A polymorphism restricts p53/p73 activation and associates with poor survival and accelerated age of onset of soft tissue sarcoma. Cancer Res 2012; 72:2206–17; PMID:22389451; http://dx.doi.org/ 10.1158/0008-5472.CAN-11-2906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gao B, Xie XJ, Huang C, Shames DS, Chen TT, Lewis CM, Bian A, Zhang B, Olopade OI, Garber JE, et al.. RASSF1A polymorphism A133S is associated with early onset breast cancer in BRCA1/2 mutation carriers. Cancer Res 2008; 68:22–5; PMID:18172292; http://dx.doi.org/ 10.1158/0008-5472.CAN-07-5183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kanzaki H, Hanafusa H, Yamamoto H, Yasuda Y, Imai K, Yano M, Aoe M, Shimizu N, Nakachi K, Ouchida M, et al.. Single nucleotide polymorphism at codon 133 of the RASSF1 gene is preferentially associated with human lung adenocarcinoma risk. Cancer Lett 2006; 238:128–34; PMID:16125301; http://dx.doi.org/ 10.1016/j.canlet.2005.07.006 [DOI] [PubMed] [Google Scholar]
- 61.Schagdarsurengin U, Seidel C, Ulbrich EJ, Kolbl H, Dittmer J, Dammann R. A polymorphism at codon 133 of the tumor suppressor RASSF1A is associated with tumorous alteration of the breast. Int J Oncol 2005; 27:185–91; PMID:15942659 [PubMed] [Google Scholar]
- 62.Bergqvist J, Latif A, Roberts SA, Hadfield KD, Lalloo F, Howell A, Evans DG, Newman WG. RASSF1A polymorphism in familial breast cancer. Familial Cancer 2010; 9:263–5; PMID:20361264; http://dx.doi.org/ 10.1007/s10689-010-9335-8 [DOI] [PubMed] [Google Scholar]
- 63.Pan D. The hippo signaling pathway in development and cancer. Dev Cell 2010; 19:491–505; PMID:20951342; http://dx.doi.org/ 10.1016/j.devcel.2010.09.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Donninger H, Clark J, Rinaldo F, Nelson N, Barnoud T, Schmidt ML, Hobbing KR, Vos MD, Sils B, Clark GJ. The RASSF1A tumor suppressor regulates XPA-mediated DNA repair. Mol Cell Biol 2015; 35:277–87; PMID:25368379; http://dx.doi.org/ 10.1128/MCB.00202-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Heller RC, Marians KJ. Replisome assembly and the direct restart of stalled replication forks. Nat Rev Mol Cell Biol 2006; 7:932–43; PMID:17139333; http://dx.doi.org/ 10.1038/nrm2058 [DOI] [PubMed] [Google Scholar]