

Abstract
Telomerase reactivation and immortalization are critical for human carcinoma progression. However, little is known about the mechanisms controlling this crucial step, due in part to the paucity of experimentally tractable model systems that can examine human epithelial cell immortalization as it might occur in vivo. We achieved efficient non-clonal immortalization of normal human mammary epithelial cells (HMEC) by directly targeting the 2 main senescence barriers encountered by cultured HMEC. The stress-associated stasis barrier was bypassed using shRNA to p16INK4; replicative senescence due to critically shortened telomeres was bypassed in post-stasis HMEC by c-MYC transduction. Thus, 2 pathologically relevant oncogenic agents are sufficient to immortally transform normal HMEC. The resultant non-clonal immortalized lines exhibited normal karyotypes. Most human carcinomas contain genomically unstable cells, with widespread instability first observed in vivo in pre-malignant stages; in vitro, instability is seen as finite cells with critically shortened telomeres approach replicative senescence. Our results support our hypotheses that: (1) telomere-dysfunction induced genomic instability in pre-malignant finite cells may generate the errors required for telomerase reactivation and immortalization, as well as many additional “passenger” errors carried forward into resulting carcinomas; (2) genomic instability during cancer progression is needed to generate errors that overcome tumor suppressive barriers, but not required per se; bypassing the senescence barriers by direct targeting eliminated a need for genomic errors to generate immortalization. Achieving efficient HMEC immortalization, in the absence of “passenger” genomic errors, should facilitate examination of telomerase regulation during human carcinoma progression, and exploration of agents that could prevent immortalization.
Keywords: carcinogenesis, c-Myc, genomic instability, human mammary epithelial cells, immortalization, p16INK4a, senescence, telomerase
Abbreviations
- BaP
benzo(a)pyrene
- CT
cholera toxin
- DDR
DNA damage response
- DMR
differentially methylated regions
- HMEC
human mammary epithelial cells
- OIS
oncogene-induced senescence
- p
passage
- p16sh
shRNA to p16INK4A
- PD
population doublings
- RB
retinoblastoma protein
- TTS
transcription start site
- X
oxytocin
Introduction
Acquisition of sufficient telomerase activity to maintain stable telomere lengths is necessary for immortalization of most human epithelial cells. In turn, immortalization appears essential for development and progression of malignant human carcinomas.1 Despite the crucial role of telomerase and immortalization in human carcinogenesis, the mechanisms that control telomerase expression, and the aberrations that allow telomerase reactivation during malignant progression, remain poorly understood. The lack of appropriate experimentally tractable model systems of human cancer–associated telomerase reactivation and immortalization has contributed to this knowledge gap. Murine cells have significant differences from human in regulation of telomerase, including less stringent telomerase repression.2,3 Consequently, telomerase activity is not limiting in most murine carcinoma model systems. On the other hand, there is a paucity of human epithelial cell immortalization models suitable for experimental examination of telomerase reactivation during carcinogenesis. Immortalization models employing transduction of ectopic hTERT, the catalytic subunit of telomerase, preclude identifying the errors responsible for telomerase reactivation during in vivo human carcinogenesis. Determining the errors responsible for driving cancer-associated immortalization using in vivo human tissues is difficult due to the many genomic errors and the genomic instability usually exhibited by carcinoma cells. Normal finite human epithelial cells contain intact genomes; short telomeres and widespread genomic instability can first be observed in many pre-malignant lesions, such as DCIS in breast.4,5 We have postulated that genomic instability caused by the critically shortened telomeres present in finite cells as they approach replicative senescence may give rise to rare errors permissive for telomerase reactivation, and underlie many of the passenger errors seen in carcinomas.1,6
Our previous studies have used pathologically relevant agents to transform normal finite lifespan human mammary epithelial cells (HMEC) to immortality.6-9 However, immortalization was clonal with multiple genomic errors present in immortalized lines,1 and the alterations specifically responsible for immortalization were not fully identified. The sporadic nature of the immortalization events has prevented examining the immortalization process as it occurs. We therefore sought to define a reproducible protocol, using agents that might recapitulate molecular alterations occurring during in vivo breast cancer progression, which could achieve non-clonal transformation of normal HMEC to immortality. Design of this protocol was based on our model of the tumor-suppressive senescence barriers normal HMEC need to bypass or overcome to attain immortality and malignancy6,10(see Fig. 1A). Further, we wanted to determine whether direct targeting of senescence barriers could generate immortal lines lacking gross genomic errors. Cultured HMEC can encounter at least 3 distinct tumor-suppressive senescence barriers.6,10,11 A first barrier, stasis, is stress-associated and mediated by the retinoblastoma protein (RB). HMEC at stasis express elevated levels of the cyclin-dependent kinase inhibitor CDKN2A/p16INK4A (p16), and do not show genomic instability or critically short telomeres.10,12,13 A second barrier, replicative senescence, is a consequence of critically shortened telomeres from ongoing replication in the absence of sufficient telomerase, and is associated with telomere dysfunction, genomic instability, and a DNA damage response (DDR).5,6,13,14 When functional p53 is present, this barrier has been called agonescence; cell populations remain mostly viable. If p53 function is abrogated, cells enter crisis and eventually die.6 Overcoming the third barrier, oncogene-induced senescence (OIS), is associated with acquiring telomerase activity and immortalization; thus a single additional oncogene can confer malignancy-associated properties once a cell is immortally transformed.11,15
Figure 1.
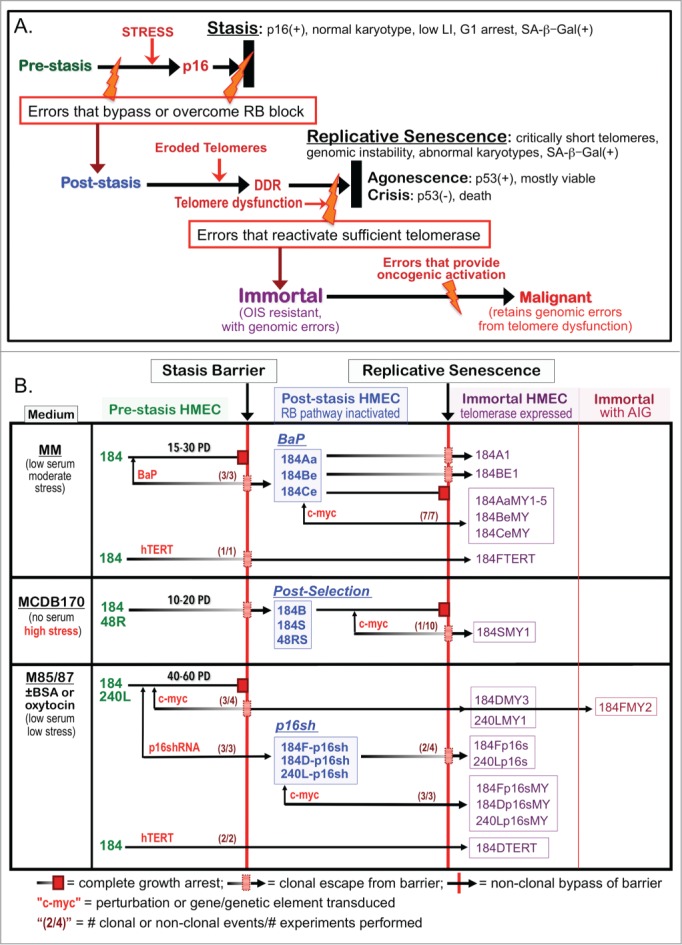
HMEC model system. (A) Schematic representation of cultured HMEC tumor-suppressive senescence barriers. Thick black bars represent the proliferation barriers of stasis and replicative senescence. Orange bolts represent genomic and/or epigenomic errors allowing these barriers to be bypassed or overcome. Red arrows indicate crucial changes occurring prior to a barrier. (B) Derivation of isogenic HMEC from specimens 184, 48R, and 240L at different stages of transformation ranging from normal pre-stasis to malignant. Cells were grown in media varying in stress induction, measured by increased p16 expression (left column), and exposed to various oncogenic agents (red). The distinct types of post-stasis HMEC are shown in the middle column; nomenclature for types is based on agent used for immortalization (e.g., BaP; p16sh) or historical naming (e.g., post-selection 19). Transduced finite cultures are indicated by specimen number and batch (e.g., 184F, 184D, 184B) followed by a “-“ and the agent transduced (e.g., -p16sh); the BaP post-stasis nomenclature is based on original publications, and includes specimen number and batch (e.g., 184A, 184B, 184C) 7, 16. New immortalized lines described in this paper are outlined in the right columns; nomenclature is based on the oncogenic agents employed (e.g., p16s for p16sh, MY for c-MYC, TERT). Numbers in parentheses before the barriers indicate how many time there was clonal or non-clonal escape from that barrier out of how many experiments performed (e.g., c-MYC-transduced pre-stasis HMEC were cultured to stasis 4 times; in 3 experiments there was clonal escape from stasis leading to 3 clonally immortalized lines).
By exposing normal pre-stasis HMEC to different culture conditions and oncogenic agents, we have generated numerous post-stasis and immortal HMEC with distinct phenotypes. HMEC grown in our original MM medium ceased growth at stasis after ∼15-30 population doublings (PD)(Fig. 1B, upper panel), but rare clonal outgrowths emerged after primary cultures were exposed to the chemical carcinogen benzo(a)pyrene (BaP), generating the BaP post-stasis populations (originally termed Extended Life).7,16 BaP post-stasis cultures examined lacked p16 expression, due to gene mutation or promoter silencing,12,17,18 and grew an additional 10–40 PD before agonescence. Rare immortal lines have emerged from BaP post-stasis populations at the telomere dysfunction barrier. Pre-stasis HMEC grown in serum-free MCDB170 medium showed more limited proliferative potential, with a rapid rise in p16 expression leading to stasis by ∼10–20 PD; cells at stasis exhibited abundant stress fibers.12,19 MCDB170 induces rare post-stasis cells, called post-selection, with silenced p16 as well as many other differentially methylated regions (DMR).12,18 Post-selection post-stasis HMEC proliferate for an additional 30-70 PD before the population ceases growth at agonescence. Immortal lines were produced by transducing BaP and post-selection post-stasis HMEC with the breast cancer-associated oncogenes c-MYC and/or ZNF217.1,8 In those studies, transduced c-MYC, a transactivator of hTERT, did not by itself immortalize post-selection post-stasis HMEC; however, when c-MYC was later transduced into the BaP post-stasis culture 184Aa, uniform immortalization was observed. Consequently, we tested the hypothesis that exposure to highly stressful (i.e., rapid p16-inducing) culture environments such as growth in serum-free MCDB170 produced post-stasis populations refractory to c-MYC induction of telomerase, whereas post-stasis cells that had not experienced high stress could be immortalized by c-MYC.
In the current studies, additional, independently derived BaP post-stasis cultures also showed induction of telomerase activity and uniform immortalization following c-MYC transduction. However, these BaP-exposed p16(-) cells harbor BaP-induced small genomic and epigenomic errors (18,20; Severson et al. in prep). We therefore generated and examined the effect of c-MYC transduction on HMEC populations made post-stasis by transduction of shRNA to p16 (p16sh) into unstressed pre-stasis cells. In addition to trying to achieve reproducible non-clonal immortalization, we wanted to examine whether direct targeting of the stasis and replicative senescence barriers could produce immortalized lines without gross genomic changes. We report that transduction of p16sh to bypass stasis, followed by transduced c-MYC to induce hTERT, efficiently immortalized pre-stasis HMEC populations grown in low stress-inducing media. Resultant immortalized lines possessed a normal karyotype at early passages, and none to few genomic copy number changes at higher passages. The failure of c-MYC to immortalize the p16(-) post-selection post-stasis HMEC was not due to differences in the hTERT gene locus DNA methylation state, or repressive (H3K27me3) or permissive (H3K4me3) histone modifications. These data indicate that just 2 oncogenic agents are sufficient to immortally transform unstressed normal HMEC, and support our hypothesis that the genomic instability commonly present in human carcinomas may not be required per se for transformation, but is needed to generate errors that can overcome tumor suppressive barriers.
Results
Immortalization of HMEC by p16sh and c-MYC
Figure 1A illustrates our model of the senescence barriers encountered by cultured primary HMEC and Fig. 1B shows the derivation and nomenclature of the finite and immortalized HMEC described in this study, and the agents employed to promote bypass or overcoming of the senescence barriers. In 10 independent experiments, c-MYC transduction of the post-selection post-stasis HMEC produced only one instance of clonal immortalization, generating the 184SMY1 line (Fig. 1B middle panel). Figure 2A shows the growth of post-selection 184B following transduction with c-MYC or control vector; net growth ceases at replicative senescence at passage (p) 15 in both conditions. Telomerase activity was examined using the TRAP assay in cells from this experiment, as well as one using the 184S post-selection batch, which ceases net growth at 22 p. In both cases, no significant TRAP activity could be detected in either control or c-MYC transduced populations, consistent with the failure to immortalize. Similar results were seen using post-selection post-stasis HMEC from another specimen, 48RS (Fig. S1A).
Figure 2.

Effect of c-MYC on post-stasis HMEC growth and TRAP activity. (A) Post-stasis post-selection 184B HMEC grown in MCDB170 were transduced with a c-MYC containing retrovirus (LXSN, red) or empty vector control at 7p (blue). Cultures ceased net growth at agonescence (15p). Post-selection 184S HMEC were transduced with c-MYC or control at 15p; net growth ceased at 22p (not shown). No significantly increased TRAP activity was seen following c-MYC transduction in either experiment. (B) BaP post-stasis 184Aa, 184Be, and 184Ce HMEC grown in MCDB170 were transduced with a c-MYC containing retrovirus (LXSN/ BH2), red) or empty vector (blue) at the indicated passages. Control cells ceased net growth at agonescence while c-MYC-transduced populations maintained proliferation indefinitely, associated with increased TRAP activity. The continuous exponential growth following c-MYC transduction reflects the visually observed non-clonal immortalization; growth was maintained throughout the dish with no areas of clonal growth. Proliferating control cultures of 184Ce expressed low TRAP activity. (C) Schematic representation of protocol to directly target senescence barriers to achieve non-clonal immortalization. (D) Pre-stasis 184D and 240L HMEC grown in M87A+CT+X were transduced at 3p with a p16sh-expressing retrovirus (MSCV, blue) or empty vector (black). At 4p cultures ±p16sh were transduced with c-MYC (BH2)(red +p16sh; purple -p16sh). c-MYC-transduced p16sh post-stasis HMEC maintained active growth indefinitely, associated with increased TRAP activity. The continuous exponential growth following c-myc transduction of the 4p p16sh-post-stasis populations reflects the observed non-clonal immortalization. Cells transduced with p16sh alone bypassed stasis and ceased net growth at agonescence, with rare clonal immortalization at agonescence. Cells transduced with c-MYC alone ceased growth at stasis, with rare clonal escape from stasis leading to immortalized lines. Control cultures transduced with empty vectors ceased growth at stasis. In some TRAP assays, heat-treated controls (+) were run next to unheated (-) samples. Positive TRAP control samples are indicted by “+” (E).
In contrast, c-MYC transduction into the BaP post-stasis culture, 184Aa, produced continuous cell growth with increasing TRAP activity (Fig. 2B). Similar results were seen in 5 independent experiments, generating the non-clonally immortalized lines 184AaMY1–5 (Fig. 1B upper panel). While both these post-stasis types lack p16 expression, this was due to mutation in 184Aa and promoter silencing in the post-selection HMEC.12,17 We therefore tested the effect of c-MYC transduction in 2 additional independent BaP post-stasis cultures that exhibited p16 promoter silencing, 184Be and 184Ce.12,18 Both populations showed continuous growth and increasing TRAP activity following c-MYC transduction, generating the non-clonally immortalized lines 184BeMY and 184CeMY (Figs. 1B upper panel, and 2B). These data indicate that these 2 different types of p16(-) post-stasis HMEC, BaP and post-selection, differ significantly in response to c-MYC transduction.
We then examined the effect of transduced c-MYC on HMEC made post-stasis by direct knockdown of p16 using p16sh (Figs. 1B lower panel, 2C, 2D). The non-clonal p16sh post-stasis populations would not harbor the BaP-induced errors present in the clonal BaP post-stasis cultures, and do not contain the extensive DMR present in the post-selection post-stasis HMEC.18 These studies used pre-stasis HMEC grown in media formulations (M87A or M85) that delay the onset of p16 expression and support up to ∼60 PD.10 Early passage pre-stasis HMEC from specimen 240 L and 2 batches from specimen 184 were transduced with p16sh-containing or control retrovirus, followed by c-MYC or control transduction at the next passage (Figs. 2C and D; Fig. S1B). At these early passages, <10% of the population expressed p16 protein.10 Control cultures ceased growth at stasis, and p16sh-transduced cultures ceased growth at replicative senescence, with rare exceptions. p16sh post-stasis cultures that received c-MYC showed uniform continuous growth and TRAP activity, generating the non-clonal immortal lines 184Dp16sMY, 184Fp16sMY and 240Lp16sMY (Fig. 1B lower panel). These studies indicate that the ability of c-MYC to induce rapid uniform immortalization in p16(-) post-stasis HMEC is not dependent upon pre-existing genomic errors.
Almost all pre-stasis HMEC receiving c-MYC alone ceased growth at stasis; however clonal outgrowths of rare cells that escaped stasis by unknown means produced clonal immortal lines (184DMY3, 184FMY2, 240LMY; Fig. 1B lower panel) with increased TRAP activity following the passages where most cells stopped at stasis (Fig. 2D). For example, pre-stasis 184D-myc initially stopped growth by 8–10 p. However, a culture reinitiated from 5 p frozen stocks exhibited 1-2 clonal outgrowths per dish at 9 p, against a background of senescing cells; these colonies maintained growth, generating the 184DMY3 line. We presume that once this population became post-stasis, the transduced c-MYC could immortalize it similar to the effect of c-MYC on the BaP and p16sh post-stasis populations. Although we have never observed spontaneous immortalization at the telomere dysfunction barrier in unperturbed post-selection post-stasis HMEC (cells that had experienced high pre-stasis culture stress), rare clonal outgrowths in a background of senescent cells were seen at this barrier in some p16sh post-stasis cultures (cells that had bypassed stasis prior to p16 elevation). These colonies maintained growth, generating the clonal immortal lines 184Fp16s and 240Lp16s (Fig. 1B lower panel). The immortalization-producing error in 184Fp16s must have occurred after 9 p, since re-initiation of frozen 9 p 184F-p16sh stock did not yield an immortal line (Fig. S1B). We hypothesize that the difference in spontaneous immortalization in the post-selection vs p16sh post-stasis HMEC, during the period of genomic instability, is related to the need for multiple errors for telomerase reactivation in the post-selection cells compared to the ability of just one error, such as transduced c-MYC, to immortalize the p16sh post-stasis HMEC. Altogether, these data suggest that a prior exposure to high culture stress may invoke alterations preventing c-MYC induction of hTERT in post-stasis populations.
Exposure of pre-stasis HMEC to high culture stress also influenced the ability of hTERT to produce efficient immortalization. Previous studies indicated that hTERT could not immortalize pre-stasis HMEC grown in high stress media such as MCDB170/MEGM,21 and yielded only one p16(-) clonal line (184FTERT) when transduced into 3 p HMEC grown in moderate stress MM medium.22 In contrast, hTERT transduced into 3 p HMEC grown in low stress M87A efficiently immortalized the population, with no growth slowdown at the stasis barrier (184DTERT, Figs. 1B lower panel; Fig. S1C). As expected, given hTERT's ability to immortalize post-selection post-stasis HMEC,21,22 transduction of hTERT into the p16sh post-stasis cells 240L-p16sh also produced efficient immortalization (240Lp16sTERT; Fig. S1D), with continuous growth similar to that seen following c-MYC transduction of 240L-p16sh (Fig. 1D).
We previously reported that proliferative pre-stasis HMEC grown in MM exhibit low levels of TRAP activity at 4 p.23 Pre-stasis HMEC from specimen 184 grown in M85/M87A also show low TRAP activity at early passages, but activity is not detectable when the cells approach stasis (Fig. 2D; Fig. S1E). Transduction with p16sh appeared to slightly increase TRAP activity compared to controls, with levels reduced by agonescence (Fig. 2D; Fig. S1B). The low TRAP activity in unperturbed 240L was increased by p16sh transduction, while the immortalized clonal line 240Lp16s emerged from replicative senescence with robust TRAP activity (Fig. 2D). c-MYC alone transiently increased TRAP activity in proliferative pre-stasis populations, with further increased activity seen in immortalized lines (184DMY3, 240LMY).
The effect of transduced p16sh in reducing p16 protein expression is shown by Western analysis in Fig. S2A for the finite and immortal cultures, and by immunochemistry for post-stasis 184D-p16sh and immortal 184DMY3 (Fig. S2C). Higher passage pre-stasis 184D and 240LB express significant p16; transduction of p16sh reduced most but not all p16 expression in both the p16sh post-stasis HMEC, and the immortal lines derived from them. p16 protein was seen in 2 of the MYC-alone transduced clonal immortal lines; the high expression in 240LMY suggests that an error elsewhere in the RB pathway enabled the cell giving rise to that line to overcome stasis, while the mixed p16 expression and 2 distinct morphologies present in 184DMY3 suggests it consists of 2 distinct clones, one of which retains p16 expression. HMEC lines containing transduced c-MYC showed variably increased MYC expression levels compared to normal pre-stasis HMEC (Fig. S2B). Of note, MYC levels in c-MYC-transduced normal pre-stasis HMEC were not significantly elevated, but were increased in abnormal post-selection 184B-myc, which did not immortalize. As has been suggested for cancer cells,24 dysregulation of MYC, as well as increased expression, may play a role in carcinogenesis, and in some circumstances, low level deregulated c-MYC may be more efficient at oncogenesis than overexpressed c-MYC.25,26
Altogether, these data indicate that post-selection post-stasis HMEC are refractory to c-MYC-induced telomerase induction and immortalization, while other p16(-) post-stasis types are readily immortalized by c-MYC, and are more vulnerable to immortalization from errors generated during telomere dysfunction. Most significantly, the data show that normal HMEC can be efficiently immortalized with endogenous telomerase reactivation by just 2 pathologically relevant oncogenic agents, p16sh and c-MYC.
Genomic profiles of immortally transformed HMEC lines
The studies described above have produced at least 12 new non-hTERT immortalized HMEC lines (Figure 1B, outlined in right columns). To examine the role of genomic errors in their generation, lines were assayed for karyotype and/or genome copy number by (a)array CGH. Karyotype at early passages following immortalization was determined for the non-clonally immortalized lines 184AaMY1 (17 p), 184BeMY (11 p), 184CeMY (12 p), 184Fp16sMY (16 p), 184Dp16sMY (16 p), and 240Lp16sMY (16 p)(Table 1, Figure 3A). aCGH was performed on these, and additional clonal lines, at higher passages (Fig. 3B; Fig. S3A). Clonal lines exhibited numerous copy number changes, consistent with a need to generate genomic errors to overcome stasis in the MYC-alone lines, and replicative senescence in the p16sh-alone lines. Some genomic errors, e.g., 1 q and 20 q amplification, are commonly seen in breast cancer.27
Table 1.
Karyology of non-clonally immortalized lines at early passage
| Cell line, passage | Karyotype and Aberrations [# cells examined] |
|---|---|
| 184Fp16sMY, 16p | 46,XX normal diploid [10] |
| 184Dp16sMY, 16p | 46,XX normal diploid [12] |
| 240Lp16sMY, 16p | 46,XX normal diploid [11] |
| 184AaMY1, 17p | 46,XX normal diploid [14] |
| 47,XX,+i(1)(q10) [6] | |
| 184BeMY, 11p | 45,X,add(X)(q28),-4,der(5)t(5;15)(q11.2;q11.2), der(12)t(5;12)(q11.2;q24.3),-15,+mar [cp16] |
| 184CeMY, 12p | 46,XX normal diploid [10] |
Figure 3.

Genomic analysis of newly developed lines from 184D and 240L. (A) Representative karyograms of newly derived immortalized lines at early passages; non-clonal 184Dp16sMY is show as an example of a normal karyotype: 46,XX. Individual abnormalities in 184AaMY1: 47,XX,+i(1)(q10), and 184BeMY: 46,X,add(X)(q28),-4,der(5)t(5;15)(q11.2;q11.2),der(12)t(5;12)(q11.2;q24.3),−15,+2mar, are shown by arrows. (B) aCGH analysis of lines at the indicated passage level using an Agilent human genome microarray with 44,000 probes per array.
The karyotype of all 3 p16sh-MYC-derived lines, and one of the 3 BaP-MYC lines (184CeMY), showed no abnormalities at early passage. At higher passages, 1-2 copy-number changes were observed in 184Dp16sMY (30p) and 240Lp16sMY (25 p). Both contained small deletions in the p16 locus on 9p21 that would not be obvious by karyology (Figure S3B), and a subpopulation of 240Lp16sMY showed a 1q amplification. MYC-induced genomic instability24 and/or retroviral-induced insertional mutagenesis9 could have produced a 1q error conferring preferential growth to a 240Lp16sMY cell. The origin of the 9p deletion in lines that had received both p16sh and c-MYC is currently unknown. The gross genomic errors in 184AaMY1 and 184BeMY are likely due to these post-stasis cultures being transduced by c-MYC close to the point of agonescence (Fig. 2B), when the populations would already contain cells with genomic errors due to telomere dysfunction,13 as these errors are not present in earlier passages of 184Aa or 184Be.20
In summary, by targeting the stasis and telomere dysfunction barriers with p16sh and c-MYC respectively, we could transform normal finite lifespan pre-stasis HMEC to immortality in the absence of gross genomic changes. These data are consistent with our hypothesis that cancer-associated genomic changes are needed to overcome tumor suppressive barriers and gain malignant properties, but gross genomic changes per se are not inherently necessary for cancer-associated immortalization.
Epigenetic state of the hTERT promoter in the cultured HMEC
The above data showed that c-MYC can induce telomerase activity and immortalization in p16(-) BaP and p16sh post-stasis, but not post-selection post-stasis HMEC. One possible basis for this difference could be distinct hTERT chromatin states that affect accessibility of c-MYC, an hTERT transactivator. To evaluate this possibility and to gain better understand of HMEC telomerase regulation, the hTERT gene locus was examined for DNA methylation and permissive (H3K4me3) or repressive (H3K27me3) histone modifications using 5-methylcytosine and chromatin immunoprecipitations (ChIP) coupled to custom tiling microarray hybridization. Post-stasis BaP, post-selection, and p16sh cultures were examined along with other HMEC with different levels of TRAP activity, ranging from normal pre-stasis 184D (low activity, Fig. 2D; Fig. S1C), isogenic 184 mammary fibroblasts (no activity, not shown), immortal 184A1 (moderate activity; Fig. S1C), and several breast tumor lines. The DNA methylation microarray results for the ∼6kb region that brackets the hTERT transcriptional start site are shown in Figure 4A, lower panel. The TERT locus was extensively methylated in all samples analyzed, with no differences detected or correlated to the level of basal or MYC-inducible TRAP activity. To increase resolution and sensitivity of the DNA methylation analysis, 2 regions were analyzed in greater detail using MassARRAY (Figs. 4B, C). One region that extended from 400 bp upstream to 200 bp downstream of the transcription start site (TSS) was unmethylated in the pre-stasis, post-stasis, and in vitro immortalized HMEC assayed, but partially methylated in some breast cancer cell lines. A second region located 850 to 1400 bp upstream of the TSS was extensively DNA methylated in all HMEC cultures, with lower levels in 2 of the 4 cancer cell lines. Therefore, there was no obvious correlation between DNA methylation state and TRAP activity among the cell types analyzed.
Figure 4.
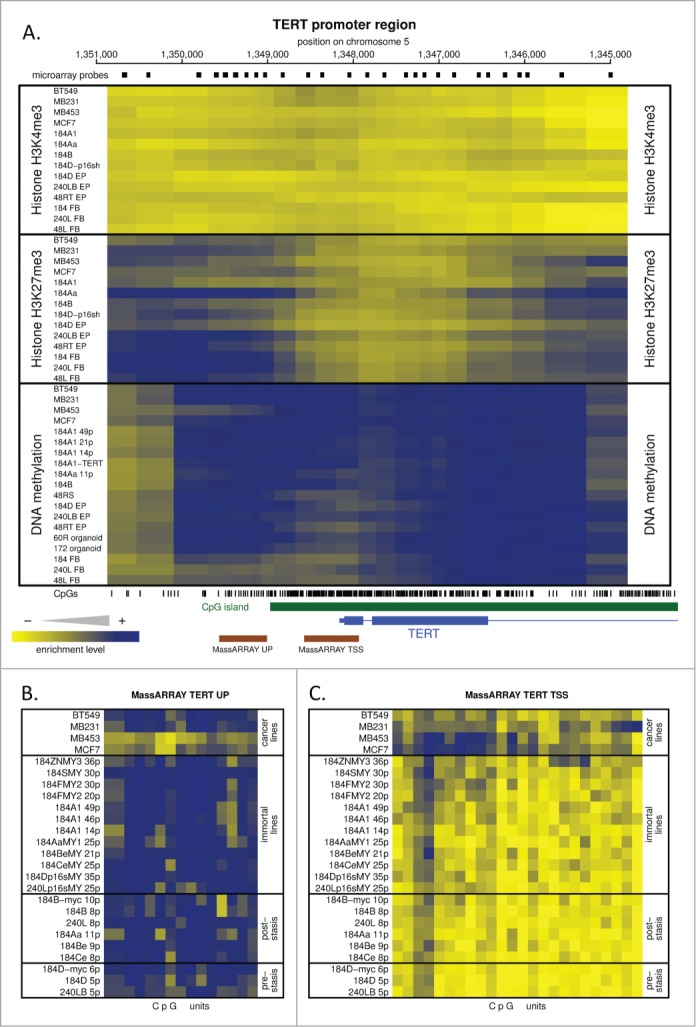
Epigenetic analysis of the hTERT gene promoter. (A) Shows the tiling microarray data from the TERT promoter region displayed as a heatmap, with blue indicating high enrichment of particular epigenetic mark and yellow indicating no enrichment. This region includes the areas bound by H3K4me3 and transcription factors including c-MYC according to online data (http://genome.ucsc.edu). Upper and middle sections of the heatmap show permissive H3K4me3 and repressive H3K27me3 histone marks, respectively; the bottom section shows DNA methylation data. Two regions (UP and TSS) indicated by brown bars at the bottom were analyzed for DNA methylation at higher resolution by MassARRAY analysis. The small black rectangles above the heatmap indicate positions of individual microarray probes. The vertical bars below the heatmap indicate positions of individual CpG dinucleotides. The CpG island is marked in green. The 5’ part of the hTERT gene is in blue. The genomic coordinates at the top are hg18. (B and C) MassARRAY analysis data for regions UP and TSS indicated in (A). The data are presented as a heatmap with methylated CpG units in blue and unmethylated CpG units in yellow.
The unmethylated region immediately surrounding the TSS of the hTERT gene suggests a state permissive to transcription, so the absence of TRAP activity in some of these cultures might be due to other epigenetic marks. Using ChIP linked microarray, we analyzed the HMEC for 2 histone modifications at the hTERT gene region - H3K27me3, a polycomb-mediated repressive modification,28 and H3K4me3, a permissive modification present on all active and even some inactive promoters.29 Figure 4A (middle panel) shows that all the cultures have repressive H3K27me3 near the hTERT promoter, with no detectable correlation to TRAP activity. Surprisingly, the permissive H3K4me3 mark was not detected in the hTERT promoter region in any of the analyzed samples (Fig. 4A, top panel), including the in vitro immortalized and cancer lines, known to possess sufficient telomerase activity to maintain stable telomeres. The genomic region displayed in Figure 4 includes the area occupied by H3K4me3 in TERT-expressing human embryonic stem cells according to the online data (http://neomorph.salk.edu/human_methylome/) and we detected the permissive H3K4me3 at the GAPDH promoter (Fig. S4) and other active genes covered by the microarray.
Overall, the data show that the epigenetic states of the hTERT locus in the analyzed HMEC samples, with respect to DNA methylation, H3K4me3, and H3K27me3, are indistinguishable from one another and therefore do not appear to play a role in the differential response of post-stasis types to c-MYC transduction.
Characterization of immortally transformed HMEC lines
The newly developed lines were characterized for lineage markers by FACS and immunofluorescence, and for AIG. Most of the lines did not display the malignancy-associated property of AIG (Fig. 1B); the one exception, 184FMY2, has other properties associated with more aggressive breast cancer cells (see below).
FACS analyses using the cell surface markers CD227 (Muc-1) and CD10 (Calla) can distinguish CD227+/CD10- luminal from CD227-/CD10+ myoepithelial lineages in normal pre-stasis HMEC (Fig. S5A). While normal pre-stasis 240L HMEC exhibit distinct luminal and myoepithelial populations, all the cell lines exhibited a predominantly basal/myoepithelial-like phenotype, showing expression of CD10, along with minor to significant expression of CD227 (Figs. S5B and C). All lines examined showed expression of the basal-associated intermediate filament protein keratin 14 and little or no expression of luminal-associated keratin 19 (Fig. S6).
Antibodies recognizing the surface antigens CD44 and CD24 have been widely used in the putative identification of carcinoma cells with tumor-initiating properties.30,31 Normal pre-stasis 240 L HMEC are predominantly CD44hi/CD24hi, with a small CD44lo/CD24hi subpopulation. Almost all the lines exhibited co-expression of CD44 and CD24 at varying levels in all cells, but some had separate subpopulations with increased CD44 and decreased CD24, e.g., 184CeMY and 240Lp16sMY. Interestingly, the 184FMY2 cell line with AIG exhibited a very prominent CD44hi/CD24low population and evidence of EMT (Vrba, Garbe, Stampfer, Futscher unpublished), but no tumor-forming ability when injected subcutaneously in immune-compromised mice (data not shown). In general, these immortalized lines derived from young reduction mammoplasty specimens displayed basal-like phenotypes compared to the heterogeneous composition of their normal pre-stasis populations.
Discussion
Immortalization of normal cultured HMEC using agents associated with breast cancer pathogenesis in vivo has been difficult to achieve. We report here that reproducible non-clonal immortalization was attained by targeting 2 tumor suppressive senescence barriers, stasis and replicative senescence, and that resultant immortalized lines exhibit normal karyotypes at early passage. Our prior studies have indicated that stasis is enforced in cultured HMEC by elevated p16 levels maintaining RB in an active state. Unlike some other human epithelial cell types, e.g., keratinocytes,32 p53-dependent p21 is not upregulated in cultured HMEC at stasis;10,12,13 consequently, transduction of shRNA to p16 can be sufficient to bypass stasis. Overcoming the telomere dysfunction barrier at replicative senescence requires, at minimum, sufficient levels of telomerase activity to maintain stable telomere lengths. Transduction of c-MYC could induce telomerase activity and immortalization in some, but not all types of p16(-) post-stasis HMEC. These results demonstrate that bypassing these 2 barriers is sufficient to transform normal finite HMEC to immortality; genomic instability and gross genomic errors are not required. The data also validate our model of the functionally and molecularly distinct tumor suppressive senescence barriers encountered by cultured HMEC: stasis, a stress-associated arrest independent of telomere length and extent of replication, and replicative senescence due to ongoing replication in the absence of sufficient telomerase producing critically short telomeres and telomere dysfunction.6,10
Expression of sufficient telomerase activity is crucial for human carcinoma progression. Almost all human breast cancer cell lines and tissues have detectable telomerase;33,34 the ALT method for telomere maintenance is very rare.35 The presence of short telomeres and genomic instability in most DCIS, as well as in pre-malignant lesions from other human organ systems, indicates that these lesions did not develop from cells expressing sufficient telomerase for telomere maintenance.4,5,36,37 While malignancy requires immortality to support ongoing tumor cell proliferation, telomerase can also provide significant additional malignancy-promoting properties.38 Telomerase reactivation has been associated with gaining resistance to OIS,11,15,39 and expression of hTERT can confer resistance to TGF-b growth inhibition22 and affect other signaling pathways.38,40 Given the importance of telomerase and immortalization for human carcinogenesis, it is surprising that so little is known about the regulation of hTERT as normal cells transform to cancer. The lack of appropriate experimentally tractable model systems has contributed to this knowledge gap. Unlike humans, small short-lived animals such as mice do not exert stringent repression of telomerase activity in adult cells, which can spontaneously immortalize in culture.2,3 Comparison of the human and mouse TERT gene shows significant differences in regulatory regions.41 The importance of telomerase in murine carcinogenesis has been demonstrated using animals engineered to lack telomerase activity,42 however such models do not address the mechanisms that allow endogenous hTERT to become reactivated during human carcinogenesis. There has also been a lack of human epithelial cell systems that model immortalization as it might occur during in vivo tumorigenesis. The use of ectopic hTERT to achieve immortalization precludes study of the factors that regulate endogenous hTERT in vivo, while viral oncogenes such as HPVE6E7 or SV40T are not etiologic agents for most human carcinomas, including breast, and have many characterized and uncharacterized effects.
We have employed reduction mammoplasty-derived primary HMEC grown under different culture conditions and exposed to a number of oncogenic agents, to generate cell types that may represent the different stages and heterogeneity of in vivo malignant progression.6-9,11 Prior studies revealed divergence in transformation pathways at the earliest stage, becoming post-stasis. Post-selection post-stasis HMEC exhibited ∼200 DMR, most of which are also found in breast cancer cells, compared to ∼10 in BaP and ∼5 in p16sh post-stasis HMEC.18 Of note, it has been suggested that post-selection post-stasis HMEC (also referred to as vHMEC,43 and sold commercially as “normal” primary HMEC (Lonza CC-2551; Life Technologies A10565)) may be on a pathway to metaplastic cancer.44 Here we show an additional difference among post-stasis types: the inability of post-stasis post-selection HMEC to become immortalized by transduced c-MYC. While the molecular processes underlying this difference remain unknown, we note an association with prior exposure to culture stress. Post-selection HMEC overcame stasis following growth in medium that rapidly induces p16, whereas p16sh post-stasis HMEC bypassed stasis prior to p16 induction. The distinct properties of the post-selection HMEC may result from their prior experience of p16-inducing stresses. Current studies are addressing the hypothesis that mechanical stressors may influence telomerase expression. Functionally, our results suggest that neither post-selection HMEC, nor pre-stasis HMEC cultured in MCDB170-type media, would be suitable substrates for the immortalization protocol presented here.
The molecular phenotype of cancer cells likely varies depending upon initial target cell as well as the specific errors that promote transformation. Progenitor cell types have been suggested to be the initial target in some situations.45-47 Our M87A/85 media support proliferation of pre-stasis HMEC with progenitor lineage markers, and allow robust proliferation prior to p16 upregulation.10,48 Such lower stress/p16-inducing conditions may be reflective of early stage carcinogenesis in vivo, if unstressed progenitor cells are initial targets.
Our results support the hypothesis that genomic errors are needed to overcome tumor suppressive barriers, but instability and aneuploidy per se may not be required for transformation.6,10 While all our clonally derived lines exhibit multiple genomic alterations,1,8,9 non-clonal lines without gross genomic errors could be generated by directly targeting the 2 main barriers to immortality, stasis and replicative senescence. Most human carcinomas contain many genomic changes, however, only a small number of these are estimated to play a driving role in carcinogenesis.49 Several hypotheses have addressed the causes of genomic instability and aneuploidy in carcinomas, including mutator phenotype,50 DNA damage,51 and altered genomic copy number models.52,53 We, and others, have proposed that the inherent genomic instability during telomere dysfunction at replicative senescence may be responsible for initiating most of the genomic errors seen in primary breast cancers.4,6,10,54,55 This instability will render most cells non-proliferative or dead, but rare cells that generate errors allowing telomerase reactivation may immortalize, carrying with them all the other errors accumulated to that point. Consequently, genomic instability in pre-malignant cells may be the source of many of the “passenger” mutations present in carcinomas, as well as of “driver” mutations that influence prognosis. If bridge-fusion-breakage cycles have begun, immortalized cells will maintain some ongoing instability.9 This hypothesis is consistent with DCIS cells possessing short telomeres, genomic instability, and many breast cancer-associated properties, including specific genomic errors and aggressiveness,56-59 as well as detection of telomerase activity in some DCIS tissues. Further, our results suggest that once a cell acquires the errors that allow stasis bypass, and then maintains proliferation to telomere dysfunction, no external agents may be needed to support rare progression to immortality. Although gross genomic changes were not required for immortalization of post-stasis HMEC by transduced c-MYC, epigenetic changes might be needed: changes have been observed associated with immortalization, even in non-clonally immortalized lines with no gross karyotypic abnormalities (18 and unpublished). Our genomically normal non-clonal immortalized lines lack malignancy-associated properties; however, we and others have seen that these OIS-resistant populations can be readily further transformed to AIG and/or tumorigenicity by transduction of individual oncogenes.1,11,60 Genomic analysis of non-clonal lines malignantly transformed at early passage will be needed to determine whether a malignant phenotype can be achieved without gross genomic errors.
Our DNA methylation and histone modification analysis of the TERT locus provides an overview of the hTERT epigenetic state in normal to malignant cells, with varying expression of telomerase activity, from one organ system. We did not find any changes in DNA methylation or histone modification state that could explain the distinct responses to transduced c-MYC by post-selection post-stasis HMEC compared to the BaP and p16sh post-stasis types. Overall, we did not find a correlation between DNA methylation or histone modification and TRAP activity in all the HMEC examined. Specifically, the CpG-rich region that immediately surrounds the TERT TSS is DNA unmethylated in pre-stasis, post-stasis, and TRAP(+) immortal HMEC cultures. These results using isogenic HMEC indicate that the lack of DNA methylation in this region may be permissive for, but is not by itself indicative of telomerase activity.61 This DNA methylation state is similar to what is seen in TERT-expressing human embryonic stem cells (hESC) or induced pluripotent stem cells (http://neomorph.salk.edu/human_methylome/). Outside of the TERT TSS region, the rest of the TERT promoter is densely DNA methylated in most of the examined HMEC, consistent with previous reports for human cancer cells,61,62 as is the large CpG island that extends from the promoter to approximately 5 kb into the gene itself, similar to hESC and iPSC (http://neomorph.salk.edu/human_methylome/). Our histone modification analysis did not detect the H3K4me3 mark at the TERT promoter/TSS in HMEC with and without telomerase activity. The polycomb-specific H3K27me3 mark was detected both upstream and downstream of the TSS region, but similar to DNA methylation, the H3K27me3 levels decreased near the TSS. These results are in contrast to hESC cells, where the TERT promoter exists in a bivalent state, occupied by both H3K4me3 and H3K27me3 (http://neomorph.salk.edu/human_methylome/). Altogether, these analyses highlight some unusual qualities of the hTERT locus, in addition to the absence of any obvious epigenetic regulation correlated with TRAP activity. The absence of permissive H3K4me3 mark and the presence of 2 distinct repressive epigenetic marks at the HMEC TERT promoter suggests it exists in a repressed or inactive chromatin state, regardless of TRAP activity or finite vs immortal status. This type of redundant chromatin repression may reflect human cells general need, as part of tumor suppression, to limit TERT induction to prevent sustained aberrant overexpression and cell immortalization. Further support of this possibility is the presence of very high DNA methylation levels in the unusually large CpG island at the 5’end of the hTERT gene, a structure usually associated with transcriptional repression and heterochromatic state. Additionally, since TERT expression is usually very low and dynamic, being predominant during S-phase, at a given moment promoters permissive for transcription may be present only in a small proportion of the cells, making it difficult to detect active chromatin.
The process of telomerase reactivation during human carcinogenesis may present a valuable target for clinical intervention. While breast cancers are known to be heterogeneous, both among and within a given tumor, the requirement for immortalization is common to almost all human carcinomas. Further, unlike the signaling pathways involved in cell growth and survival, there are no commonly used alternative pathways to telomerase reactivation during HMEC immortalization, thus decreasing the possibility for emergence of therapeutic resistance. However, development of potential therapeutics has been limited by the lack of information on the mechanisms underlying human epithelial cell immortalization, and by the absence of a significant immortalization barrier in murine carcinogenesis, precluding usage of murine models for testing pharmacologic interventions in immortalization. The reproducible immortalization of HMEC in the absence of “passenger” errors that is achievable with our system can facilitate further examination of the mechanisms involved in hTERT regulation during carcinogenesis. Better understanding of hTERT regulation may offer new clinical opportunities that involve not just targeting telomerase activity but the reactivation process itself.
Material and Methods
Cell culture
Finite lifespan HMEC from specimens 184, 240L, and 48R were obtained from reduction mammoplasty tissue of women aged 21, 19, and 16 respectively. Pre-stasis 184 (batch D), 240 L (batch B), 48R (batch T) HMEC were grown in M87A supplemented with 0.5 ng/ml cholera toxin (CT), and 0.1 nM oxytocin (X) (Bachem); pre-stasis 184 (batch F) were grown in M85+CT, as described.10 Post-selection post-stasis HMEC 184 (batch B, agonescence at ∼passage (p) 15; batch S, agonescence at ∼22 p), and 48R batch S, agonescence at ∼22 p, as well as BaP post-stasis 184Aa, 184Be, and 184Ce HMEC (agonescence at ∼16 p, 10 p, 15 p respectively) were grown in serum-free MCDB170 medium (commercially available versions MEGM, Lonza, or M171, Life Technologies) plus supplements.19 Total PD level was calculated as described.10 Anchorage-independent growth (AIG) was assayed as described9 using 1.5% methylcellulose solution made up in M87A+CT+X. Details on the derivation and culture of these HMEC can be found at http://hmec.lbl.gov. Research was conducted under LBNL Human Subjects Committee IRB protocols 259H001 and 108H004.
Retroviral transduction
The p16 shRNA vector (MSCV) was obtained from Greg Hannon Narita,63 Four different c-Myc vectors were used: LXSN for 184B, 184S, 184Aa, 184F; pBabe–hygro (BH2) for 184Be, 184Ce, 184D, 240LB; LNCX2-MYC-ires-GFP for 48RS;60 Myc:ER for 184S, 184B.64 The hTERT vector pBabe-hygro-TERT was obtained from Bob Weinberg.65 The p16-containing construct was pLenti-p16-neo vector, plasmid 22260, Addgene.Retroviral stocks were generated, supernatants collected in MCDB170 medium containing 0.1% bovine serum albumin or M87A medium, and infections performed as described.9
TRAP assays
Telomerase activity assays employed the TRAPeze Telomerase detection kit (Millipore) using 0.2 μg of protein extract per reaction. Reaction products were separated on a 10% polyacrylamide gel and visualized using a Storm 860 imaging system (Molecular Dynamics).
DNA isolation
Genomic DNA was extracted using the DNeasy Blood and Tissue Kit (Qiagen) according to manufacturer protocol and quantified spectrophotometrically.
Comparative genomic hybridizations (CGH) and karyology
CGH was performed at the Genomics Shared Service of the Arizona Cancer Center using the Agilent human genome CGH microarray with 44,000 probes per array, and analyzed using Bioconductor in an R environment.66 Low passage isogenic pre-stasis HMEC were used as a reference. CGH for the 184F lines was performed as described.5 Karyology was performed as described.67
Epigenetic analysis of the hTERT gene
Methyl cytosine DNA immunoprecipitation (MeDIP), chromatin immunoprecipitation (ChIP), sample labeling and microarray hybridization were performed as described.68 Microarray data were analyzed in R66 as described68(GEO Accession number GSE48504). DNA methylation analysis by MassARRAY was performed as described.18 Primer sequences are listed in Table S1; oligonucleotides were obtained from Integrated DNA Technologies.
Western and ELISA analysis
Protein lysates for p16 were collected and processed as described23 and 50 μg samples were resolved on a 4–12% Novex Bis/Tris gel (Invitrogen). Protein lysates for c-MYC were prepared using cell extraction buffer (Invitrogen cat# FNN0011) with protease inhibitors (Sigma cat# P2714). For detection of c-MYC by western blot, 25 μg of extracts were separated on a 4–12% Criterion TGX gel (Biorad). Separated proteins were transferred to Immobilon PVDF membrane (Millipore) and blocked in PBS 0.05% Tween20 with 1% nonfat milk for 1 hour. Binding of mAb Y69 to c-MYC (Abcam) and mAb G175–405 to p16 (BD Biosciences) was detected by chemiluminescence using the VersaDoc MP imaging system and quantified using Quanity-One software (Biorad). The total c-MYC ELISA assay (Invitrogen cat# KH02041) was performed following manufacturer's directions.
Immunohistochemistry and immunofluorescence
Immunohistochemical analysis for p16 was performed as described using the JC822 or MAB G175-405 antibody (BD Bioscience). Immunofluorescence was performed as described23 using anti-K14 (1:500, Thermo, polyclonal) and anti-K19 (1:500, Sigma, clone A53-B/A2). Cells were counterstained with DAPI (Sigma) and imaged with an epifluorescence Axioplan microscope (Carl Zeiss).
FACS
Cells were trypsinized and resuspended in ice-cold M87A media. Cells were stained for surface antigens using anti-CD227-FITC (Becton Dickinson, clone HMPV), anti-CD10-PE or –APC (BioLegend, clone HI10a), anti-CD24-Alexa488 (Biolegend, clone ML5), or anti-CD44-PE (BioLegend, clone IM7). Results were obtained on a FACS Calibur (Becton Dickenson) analysis platform as described.48
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Gerri Levine, Batul Merchant, and Annie Pang for technical assistance.
Supplemental Materials
Supplemental materials for this article can be found on the publisher's website.
Funding
This work was supported by DOD BCRP BC060444 (JCG, MRS), NIH CA24844 (JCG, MRS), NIH NIA R00AG033176 and R01AG040081 (JCG, KS, MAL), SWEHSC NIEHS ES06694 and NIH CA23074 (LV, PN, GW, BWF), Margaret E. and Fenton L. Maynard Endowment for Breast Cancer Research (BWF), RVO:60077344 (PN), University of Arizona Cytogenomics Laboratory (LF, AB), ACS RSG CCG 122517 (MJ), and the Office of Energy Research, Office of Health and Biological Research, US. Department of Energy under Contract No. DE-AC02-05CH11231. (JCG, KS, MAL, MRS).
References
- 1. Stampfer MR, Garbe JC, Labarge MA. An Integrated Human Mammary Epithelial Cell Culture System for Studying Carcinogenesis and Aging. In: Schatten H, ed. Cell and Molecular Biology of Breast Cancer. New York: Springer, 2013:323–61. [Google Scholar]
- 2. Greider CW. Telomeres, telomerase and senescence. BioEssays : news and reviews in molecular, Cell Dev Biol 1990; 12:363–9. [DOI] [PubMed] [Google Scholar]
- 3. Seluanov A, Hine C, Bozzella M, Hall A, Sasahara TH, Ribeiro AA, Catania KC, Presgraves DC, Gorbunova V. Distinct tumor suppressor mechanisms evolve in rodent species that differ in size and lifespan. Aging Cell 2008; 7:813-23; PMID:18778411; http://dx.doi.org/ 10.1111/j.1474-9726.2008.00431.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Meeker AK, Argani P. Telomere shortening occurs early during breast tumorigenesis: a cause of chromosome destabilization underlying malignant transformation? J Mammary Gland Biol Neo 2004; 9:285-96; PMID:15557801; http://dx.doi.org/ 10.1023/B:JOMG.0000048775.04140.92 [DOI] [PubMed] [Google Scholar]
- 5. Chin K, de Solorzano CO, Knowles D, Jones A, Chou W, Rodriguez EG, Kuo WL, Ljung BM, Chew K, Myambo K, et al. In situ analyses of genome instability in breast cancer. Nat Genet 2004; 36:984-8; PMID:15300252; http://dx.doi.org/ 10.1038/ng1409 [DOI] [PubMed] [Google Scholar]
- 6. Garbe JC, Holst CR, Bassett E, Tlsty T, Stampfer MR. Inactivation of p53 function in cultured human mammary epithelial cells turns the telomere-length dependent senescence barrier from agonescence into crisis. Cell Cycle 2007; 6:1927-36; PMID:17671422; http://dx.doi.org/ 10.4161/cc.6.15.4519 [DOI] [PubMed] [Google Scholar]
- 7. Stampfer MR, Bartley JC. Induction of transformation and continuous cell lines from normal human mammary epithelial cells after exposure to benzo; a.pyrene. ProcNat Acad Sci U S A 1985; 82:2394-8; PMID:3857588; http://dx.doi.org/ 10.1073/pnas.82.8.2394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nonet G, Stampfer MR, Chin K, Gray JW, Collins CC, Yaswen P. The ZNF217 gene amplified in breast cancers promotes immortalization of human mammary epithelial cells. Cancer Res 2001; 61:1250-4; PMID:11245413 [PubMed] [Google Scholar]
- 9. Stampfer MR, Garbe J, Nijjar T, Wigington D, Swisshelm K, Yaswen P. Loss of p53 function accelerates acquisition of telomerase activity in indefinite lifespan human mammary epithelial cell lines. Oncogene 2003; 22:5238-51; PMID:12917625; http://dx.doi.org/ 10.1038/sj.onc.1206667 [DOI] [PubMed] [Google Scholar]
- 10. Garbe JC, Bhattacharya S, Merchant B, Bassett E, Swisshelm K, Feiler HS, Wyrobek AJ, Stampfer MR. Molecular distinctions between stasis and telomere attrition senescence barriers shown by long-term culture of normal human mammary epithelial cells. Cancer Res 2009; 69:7557-68; PMID:19773443; http://dx.doi.org/ 10.1158/0008-5472.CAN-09-0270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Olsen CL, Gardie B, Yaswen P, Stampfer MR. Raf-1-induced growth arrest in human mammary epithelial cells is p16-independent and is overcome in immortal cells during conversion. Oncogene 2002; 21:6328-39; PMID:12214273; http://dx.doi.org/ 10.1038/sj.onc.1205780 [DOI] [PubMed] [Google Scholar]
- 12. Brenner AJ, Stampfer MR, Aldaz CM. Increased p16INK4a expression with onset of senescence of human mammary epithelial cells and extended growth capacity with inactivation. Oncogene 1998; 17:199-205; PMID:9674704; http://dx.doi.org/ 10.1038/sj.onc.1201919 [DOI] [PubMed] [Google Scholar]
- 13. Romanov SR, Kozakiewicz BK, Holst CR, Stampfer MR, Haupt LM, Tlsty TD. Normal human mammary epithelial cells spontaneously escape senescence and acquire genomic changes. Nature 2001; 409:633-7; PMID:11214324; http://dx.doi.org/ 10.1038/35054579 [DOI] [PubMed] [Google Scholar]
- 14. Stampfer MR, Bodnar A, Garbe J, Wong M, Pan A, Villeponteau B, Yaswen P. Gradual phenotypic conversion associated with immortalization of cultured human mammary epithelial cells. Mol Biol Cell 1997; 8:2391-405; PMID:9398663; http://dx.doi.org/ 10.1091/mbc.8.12.2391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sherman MY, Meng L, Stampfer M, Gabai VL, Yaglom JA. Oncogenes induce senescence with incomplete growth arrest and suppress the DNA damage response in immortalized cells. Aging Cell 2011; 10:949-61; PMID:21824272; http://dx.doi.org/ 10.1111/j.1474-9726.2011.00736.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Stampfer MR, Bartley JC. Human mammary epithelial cells in culture: differentiation and transformation. Cancer Treat Res 1988; 40:1-24; PMID:2908646; http://dx.doi.org/ 10.1007/978-1-4613-1733-3_1. [DOI] [PubMed] [Google Scholar]
- 17. Brenner AJ, Aldaz CM. Chromosome 9p allelic loss and p16/CDKN2 in breast cancer and evidence of p16 inactivation in immortal breast epithelial cells. Cancer Res 1995; 55:2892-5; PMID:7796417 [PubMed] [Google Scholar]
- 18. Novak P, Jensen TJ, Garbe JC, Stampfer MR, Futscher BW. Step-wise DNA methylation changes are linked to escape from defined proliferation barriers and mammary epithelial cell immortalization. Cancer research 2009; 67:5251-8; http://dx.doi.org/ 10.1158/0008-5472.CAN-08-4977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hammond SL, Ham RG, Stampfer MR. Serum-free growth of human mammary epithelial cells: Rapid clonal growth in defined medium and extended serial passage with pituitary extract. Proc Natl Acad Sci USA 1984; 81:5435-9; PMID:6591199; http://dx.doi.org/ 10.1073/pnas.81.17.5435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Severson PL, Vrba L, Stampfer MR, Futscher BW. Exome-wide Mutation Profile in Benzo; a.pyrene-derived Post-stasis and Immortal Human Mammary Epithelial Cells. Mutation Research submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kiyono T, Foster SA, Koop JI, McDougall JK, Galloway DA, Klingelhutz AJ. Both Rb/p16INK4a inactivation and telomerase activity are required to immortalize human epithelial cells. Nature 1998; 396:84-8; PMID:9817205; http://dx.doi.org/ 10.1038/23962 [DOI] [PubMed] [Google Scholar]
- 22. Stampfer MR, Garbe J, Levine G, Lichtsteiner S, Vasserot AP, Yaswen P. Expression of the telomerase catalytic subunit, hTERT, induces resistance to transforming growth factor beta growth inhibition in p16INK4A(-) human mammary epithelial cells. Proc Nat Acad Sci U S A 2001; 98:4498-503; PMID:11287649; http://dx.doi.org/ 10.1073/pnas.071483998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Garbe J, Wong M, Wigington D, Yaswen P, Stampfer MR. Viral oncogenes accelerate conversion to immortality of cultured human mammary epithelial cells. Oncogene 1999; 18:2169-80; PMID:10327063; http://dx.doi.org/ 10.1038/sj.onc.1202523 [DOI] [PubMed] [Google Scholar]
- 24. Dang CV. MYC on the path to cancer. Cell 2012; 149:22-35; PMID:22464321; http://dx.doi.org/ 10.1016/j.cell.2012.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chen D, Kon N, Zhong J, Zhang P, Yu L, Gu W. Differential effects on ARF stability by normal versus oncogenic levels of c-Myc expression. Molecular cell 2013; 51:46-56; PMID:23747016; http://dx.doi.org/ 10.1016/j.molcel.2013.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Murphy DJ, Junttila MR, Pouyet L, Karnezis A, Shchors K, Bui DA, Brown-Swigart L, Johnson L, Evan GI. Distinct thresholds govern Myc's biological output in vivo. Cancer cell 2008; 14:447-57; PMID:19061836; http://dx.doi.org/ 10.1016/j.ccr.2008.10.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chin K, DeVries S, Fridlyand J, Spellman PT, Roydasgupta R, Kuo WL, Lapuk A, Neve RM, Qian Z, Ryder T, et al. Genomic and transcriptional aberrations linked to breast cancer pathophysiologies. Cancer Cell 2006; 10:529-41; PMID:17157792; http://dx.doi.org/ 10.1016/j.ccr.2006.10.009 [DOI] [PubMed] [Google Scholar]
- 28. Simon JA, Kingston RE. Mechanisms of polycomb gene silencing: knowns and unknowns. Nat Rev Mol Cell Biol 2009; 10:697-708; PMID:19738629 [DOI] [PubMed] [Google Scholar]
- 29. Guenther MG, Levine SS, Boyer LA, Jaenisch R, Young RA. A chromatin landmark and transcription initiation at most promoters in human cells. Cell 2007; 130:77-88; PMID:17632057; http://dx.doi.org/ 10.1016/j.cell.2007.05.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bloushtain-Qimron N, Yao J, Snyder EL, Shipitsin M, Campbell LL, Mani SA, Hu M, Chen H, Ustyansky V, Antosiewicz JE, et al. Cell type-specific DNA methylation patterns in the human breast. Proc Nat Acad Sci U S A 2008; 105:14076-81; PMID:18780791; http://dx.doi.org/ 10.1073/pnas.0805206105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Park SY, Gonen M, Kim HJ, Michor F, Polyak K. Cellular and genetic diversity in the progression of in situ human breast carcinomas to an invasive phenotype. J Clin Invest 2010; 120:636-44; PMID:20101094; http://dx.doi.org/ 10.1172/JCI40724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rheinwald JG, Hahn WC, Ramsey MR, Wu JY, Guo Z, Tsao H, De Luca M, Catricala C, O'Toole KM. A two-stage, p16INK4a-and p53-dependent keratinocyte senescence mechanism that limits replicative potential independent of telomere status. Mol Cell Biol 2002; 22:5157-72; PMID:12077343; http://dx.doi.org/ 10.1128/MCB.22.14.5157-5172.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Carey LA, Hedican CA, Henderson GS, Umbricht CB, Dome JS, Varon D, Sukumar S. Careful histological confirmation and microdissection reveal telomerase activity in otherwise telomerase-negative breast cancers. Clin Cancer Res: an official J Am Assoc Cancer Res 1998; 4:435-40; PMID:9516933 [PubMed] [Google Scholar]
- 34. Yashima K, Milchgrub S, Gollahon LS, Maitra A, Saboorian MH, Shay JW, Gazdar AF. Telomerase enzyme activity and RNA expression during the multistage pathogenesis of breast carcinoma. Clin Cancer Res: an official J Am Assoc Cancer Res 1998; 4:229-34; PMID:9516976 [PubMed] [Google Scholar]
- 35. Subhawong AP, Heaphy CM, Argani P, Konishi Y, Kouprina N, Nassar H, Vang R, Meeker AK. The alternative lengthening of telomeres phenotype in breast carcinoma is associated with HER-2 overexpression. Modern pathology: an official journal of the United States and Canadian Academy of Pathology, Inc; 2009; 22:1423-31; PMID:19734843 [DOI] [PubMed] [Google Scholar]
- 36. Meeker AK, Hicks JL, E.A. P, March GE, Bennett CJ, Delannoy MJ, De Marzo AM. Telomere shortening is an early somatic DNA alteration in human prostate tumorigenesis. Cancer Res 2002; 62:6405-9; PMID:12438224 [PubMed] [Google Scholar]
- 37. Chene G, Tchirkov A, Pierre-Eymard E, Dauplat J, Raoelfils I, Cayre A, Watkin E, Vago P, Penault-Llorca F. Early telomere shortening and genomic instability in tubo-ovarian preneoplastic lesions. Clin Cancer Res : an official J Am Assoc Cancer Res 2013; 19:2873-82; PMID:23589176; http://dx.doi.org/ 10.1158/1078-0432.CCR-12-3947 [DOI] [PubMed] [Google Scholar]
- 38. Blasco MA. Telomerase beyond telomeres. Nature reviews Cancer 2002; 2:627-32; PMID:12154355; http://dx.doi.org/ 10.1038/nrc862 [DOI] [PubMed] [Google Scholar]
- 39. Suram A, Herbig U. The replicometer is broken: telomeres activate cellular senescence in response to genotoxic stresses. Aging Cell 2014; PMID:25040628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mukherjee S, Firpo EJ, Wang Y, Roberts JM. Separation of telomerase functions by reverse genetics. Proc Nat Acad Sci U S A 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Horikawa I, Chiang YJ, Patterson T, Feigenbaum L, Leem SH, Michishita E, Larionov V, Hodes RJ, Barrett JC. Differential cis-regulation of human versus mouse TERT gene expression in vivo: Identification of a human-specific repressive element. Proc Nat Acad Sci U S A 2005; 102:18437-42; PMID:16344462; http://dx.doi.org/ 10.1073/pnas.0508964102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. O'Hagan RC, Chang S, Maser RS, Mohan R, Artandi SE, Chin L, DePinho RA. Telomere dysfunction provokes regional amplification and deletion in cancer genomes. Cancer Cell 2002; 2:149-55; PMID:12204535; http://dx.doi.org/ 10.1016/S1535-6108(02)00094-6 [DOI] [PubMed] [Google Scholar]
- 43. Zhang J, Pickering CR, Holst CR, Gauthier ML, Tlsty TD. p16INK4a modulates p53 in primary human mammary epithelial cells. Cancer Res 2006; 66:10325-31; PMID:17079452; http://dx.doi.org/ 10.1158/0008-5472.CAN-06-1594 [DOI] [PubMed] [Google Scholar]
- 44. Keller PJ, Arendt LM, Skibinski A, Logvinenko T, Klebba I, Dong S, Smith AE, Prat A, Perou CM, Gilmore H, et al. Defining the cellular precursors to human breast cancer. Proc Nat Acad Sci U S A 2012; 109:2772-7; PMID:21940501; http://dx.doi.org/ 10.1073/pnas.1017626108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lim E, Vaillant F, Wu D, Forrest NC, Pal B, Hart AH, Asselin-Labat ML, Gyorki DE, Ward T, Partanen A, et al. Aberrant luminal progenitors as the candidate target population for basal tumor development in BRCA1 mutation carriers. Nat Med 2009; 15:907-13; PMID:19648928; http://dx.doi.org/ 10.1038/nm.2000 [DOI] [PubMed] [Google Scholar]
- 46. Molyneux G, Geyer FC, Magnay FA, McCarthy A, Kendrick H, Natrajan R, Mackay A, Grigoriadis A, Tutt A, Ashworth A, et al. BRCA1 basal-like breast cancers originate from luminal epithelial progenitors and not from basal stem cells. Cell Stem Cell 2010; 7:403-17; PMID:20804975; http://dx.doi.org/ 10.1016/j.stem.2010.07.010 [DOI] [PubMed] [Google Scholar]
- 47. Proia TA, Keller PJ, Gupta PB, Klebba I, Jones AD, Sedic M, Gilmore H, Tung N, Naber SP, Schnitt S, et al. Genetic predisposition directs breast cancer phenotype by dictating progenitor cell fate. Cell Stem Cell 2011; 8:149-63; PMID:21295272; http://dx.doi.org/ 10.1016/j.stem.2010.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Garbe JC, Pepin F, Pelissier FA, Sputova K, Fridriksdottir AJ, Guo DE, Villadsen R, Park M, Petersen OW, Borowsky AD, et al. Accumulation of multipotent progenitors with a basal differentiation bias during aging of human mammary epithelia. Cancer Res 2012; 72:3687-701; PMID:22552289; http://dx.doi.org/ 10.1158/0008-5472.CAN-12-0157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wood LD, Parsons DW, Jones S, Lin J, Sjoblom T, Leary RJ, Shen D, Boca SM, Barber T, Ptak J, et al. The genomic landscapes of human breast and colorectal cancers. Science 2007; 318:1108-13; PMID:17932254; http://dx.doi.org/ 10.1126/science.1145720 [DOI] [PubMed] [Google Scholar]
- 50. Loeb LA. Human cancers express mutator phenotypes: origin, consequences and targeting. Nat Rev Cancer 2011; 11:450-7; PMID:21593786; http://dx.doi.org/ 10.1038/nrc3063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Negrini S, Gorgoulis VG, Halazonetis TD. Genomic instability–an evolving hallmark of cancer. Nat Rev Mol Cell Biol 2010; 11:220-8; PMID:20177397; http://dx.doi.org/ 10.1038/nrm2858 [DOI] [PubMed] [Google Scholar]
- 52. Kolodner RD, Cleveland DW, Putnam CD. Cancer. Aneuploidy drives a mutator phenotype in cancer. Science 2011; 333:942-3; PMID:21852477; http://dx.doi.org/ 10.1126/science.1211154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Storchova Z, Pellman D. From polyploidy to aneuploidy, genome instability and cancer. Nat Rev Mol Cell Biol 2004; 5:45-54; PMID:14708009; http://dx.doi.org/ 10.1038/nrm1276 [DOI] [PubMed] [Google Scholar]
- 54. Soler D, Genesca A, Arnedo G, Egozcue J, Tusell L. Telomere dysfunction drives chromosomal instability in human mammary epithelial cells. Genes, Chrom Cancer 2005; 44:339-50; http://dx.doi.org/ 10.1002/gcc.20244 [DOI] [PubMed] [Google Scholar]
- 55. Pampalona J, Frias C, Genesca A, Tusell L. Progressive telomere dysfunction causes cytokinesis failure and leads to the accumulation of polyploid cells. PLoS Genet 2012; 8:e1002679; PMID:22570622; http://dx.doi.org/ 10.1371/journal.pgen.1002679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Warnberg F, Nordgren H, Bergkvist L, Holmberg L. Tumour markers in breast carcinoma correlate with grade rather than with invasiveness. British J Cancer 2001; 85:869-74; PMID:11556839; http://dx.doi.org/ 10.1054/bjoc.2001.1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Miron A, Varadi M, Carrasco D, Li H, Luongo L, Kim HJ, Park SY, Cho EY, Lewis G, Kehoe S, et al. PIK3CA mutations in in situ and invasive breast carcinomas. Cancer Res 2010; 70:5674-8; PMID:20551053; http://dx.doi.org/ 10.1158/0008-5472.CAN-08-2660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ma XJ, Salunga R, Tuggle JT, Gaudet J, Enright E, McQuary P, Payette T, Pistone M, Stecker K, Zhang BM, et al. Gene expression profiles of human breast cancer progression. Proc Nat Acad Sci U S A 2003; 100:5974-9; PMID:12714683; http://dx.doi.org/ 10.1073/pnas.0931261100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Espina V, Liotta LA. What is the malignant nature of human ductal carcinoma in situ? Nat Rev Cancer 2011; 11:68-75; PMID:21150936; http://dx.doi.org/ 10.1038/nrc2950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Cipriano R, Kan CE, Graham J, Danielpour D, Stampfer M, Jackson MW. TGF-beta signaling engages an ATM-CHK2-p53-independent RAS-induced senescence and prevents malignant transformation in human mammary epithelial cells. Proc Nat Acad Sci U S A 2011; 108:8668-73; PMID:21555587; http://dx.doi.org/ 10.1073/pnas.1015022108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Zinn RL, Pruitt K, Eguchi S, Baylin SB, Herman JG. hTERT is expressed in cancer cell lines despite promoter DNA methylation by preservation of unmethylated DNA and active chromatin around the transcription start site. Cancer Res 2007; 67:194-201; PMID:17210699; http://dx.doi.org/ 10.1158/0008-5472.CAN-06-3396 [DOI] [PubMed] [Google Scholar]
- 62. Renaud S, Loukinov D, Alberti L, Vostrov A, Kwon YW, Bosman FT, Lobanenkov V, Benhattar J. BORIS/CTCFL-mediated transcriptional regulation of the hTERT telomerase gene in testicular and ovarian tumor cells. Nucl Acids Res 2011; 39:862-73; PMID:20876690; http://dx.doi.org/ 10.1093/nar/gkq827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Narita M, Nunez S, Heard E, Lin AW, Hearn SA, Spector DL, Hannon GJ, Lowe SW. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell 2003; 113:703-16; PMID:12809602; http://dx.doi.org/ 10.1016/S0092-8674(03)00401-X [DOI] [PubMed] [Google Scholar]
- 64. Eilers M, Picard D, Yamamoto KR, Bishop JM. Chimaeras of myc oncoprotein and steroid receptors cause hormone-dependent transformation of cells. Nature 1989; 340:66-8; PMID:2662015; http://dx.doi.org/ 10.1038/340066a0 [DOI] [PubMed] [Google Scholar]
- 65. Counter CM, Hahn WC, Wei W, Caddle SD, Beijersbergen RL, Lansdorp PM, Sedivy JM, Weinberg RA. Dissociation among in vitro telomerase activity, telomere maintenance, and cellular immortalization. Proc Natl Acad Sci USA 1998; 95:14723-8; PMID:9843956; http://dx.doi.org/ 10.1073/pnas.95.25.14723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. R_Development_Core_Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing. Vienna, Austria, 2011. [Google Scholar]
- 67. Barch MJ. The AGT Cytogenetics Laboratory Manual, 3rd edition New York: Lippincott-Raven, 1997. [Google Scholar]
- 68. Vrba L, Garbe JC, Stampfer MR, Futscher BW. Epigenetic regulation of normal human mammary cell type-specific miRNAs. Genome Res 2011; 21:2026-37; PMID:21873453; http://dx.doi.org/ 10.1101/gr.123935.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.