

Abstract
Dysregulation of cell cycle machinery causes abnormal cell division, leading to cancer development. To drive cell cycle properly, expression levels of cell cycle regulators are tightly regulated through the cell cycle. Dual specificity tyrosine phosphorylation-regulated kinase 2 (DYRK2) is a Ser/Thr kinase, and its intracellular functions had not been elucidated for decades. Recent studies have shown that DYRK2 down-regulates key molecules on cell cycle control. This review mainly highlights the DYRK2 function during cell division. In addition, we summarize tumor suppressive role of DYRK2 in cancer cells and discuss future research directions for DYRK2 toward the novel cancer therapies.
Keywords: apoptosis, cell cycle regulation, DYRK2, EMT, proteasomal degradation
Introduction
Dual specificity tyrosine phosphorylation-regulated kinase 2 (DYRK2) is a member of the DYRK family that belongs to the CMGC group [(cyclin-dependent kinases (CDK), mitogen-activated protein kinases (MAPK), glycogen synthase kinase (GSK), and CDK-like kinases (CLKs)]. DYRKs have both tyrosine (Tyr) and serine/threonine (Ser/Thr) kinase activities. However, the Tyr kinase activity is only exerted for the autophosphorylation. All DYRKs phosphorylate their Tyr residues within the activation loop before completing translation, suggesting that they have a common basal activation machinery.1 Intriguingly, the Tyr kinase activity is lost after autophosphorylation. Therefore, DYRKs have only a Ser/Thr kinase activity in cells.
Members of the DYRK family are divided into 2 subclasses: class 1 (DYRK1A and DYRK1B) and class 2 (DYRK2, DYRK3, and DYRK4). Both subclasses share a central kinase domain and an adjacent N-terminal DYRK homology (DH) box. Class 1 DYRKs are mainly localized in the nucleus because they retain their nuclear localization sequence. Conversely, class 2 DYRKs are predominantly localized in the cytoplasm. The outside sequences of the central kinase domain are distinct between the subclasses and define their kinase characteristics including subcellular localization, substrate specificity, and tissue distribution.2 DYRKs are evolutionally conserved from yeast to mammals and regulate cell survival, differentiation, proliferation, and apoptosis. Since we revealed the apoptotic function of DYRK2 in 2007,3 further intriguing functions have been reported for DYRK2. In this review, we focus on the physiological functions of DYRK2 in human cancer cells.
Regulation of G1/S Transition as a Priming Kinase for GSK3
To prompt responses to intracellular events, molecules including transcription factors are required for rapid up- or down-regulation of genes and proteins. One of the downregulation machineries is the ubiquitin-proteasome system. In some cases, down regulation of proteins is initially triggered by priming phosphorylation. A phosphorylated protein is subsequently phosphorylated by glycogen synthase kinase 3 (GSK3) and thereafter the substrate protein is recognized and ubiquitinated by ubiquitin E3 ligase. Thus, the timing of the degradation might be determined by when and where the substrates are phosphorylated by a priming kinase and GSK3. The expression level, activity, and subcellular localization of the priming kinase and GSK3 are important to understanding the regulatory mechanism of substrates. During cell cycle progression, 2 major ubiquitin E3 ligases, the anaphase promoting complex/cyclosome (APC/C) and the Skp1-cullin-F-box-protein complex (SCF) regulate the expression levels of cell cycle regulators in a cell cycle phase-specific manner. Substrate specificity is determined by one of the components in E3 ligases. In the case of SCF, the F-box protein has an indispensable role in substrate recognition. Priming phosphorylation is essential for the recognition by F-box proteins that are a variable component in the SCF complex, suggesting that F-box proteins determine the substrate specificity of the SCF complex. F-box and WD-40 repeat domain containing protein 7 (Fbw7) and β-transduction repeat-containing protein (β-TrCP) are the most studied of the 69 known F-box proteins.4
One of the main functions of DYRK2 is acting as a priming kinase for GSK3. As direct substrates of DYRK2, we previously identified 2 transcription factors, c-Jun, and c-Myc.8 c-Jun and c-Myc play central roles in G1/S transition. These transcription factors are transiently expressed during the cell cycle. Expression levels of c-Jun and c-Myc rapidly decline as cells pass through S phase. Fbw7 recognizes phosphorylated c-Jun and c-Myc which is catalyzed by the priming kinase and GSK3, to initiate protein degradation via the proteasome. In the case of c-Jun and c-Myc, DYRK2 acts as a priming kinase for GSK3. Because c-Jun and c-Myc regulate G1/S transition, DYRK2 must play an important role in proper control of cell cycle progression (Fig. 1). Indeed, we found that DYRK2 phosphorylates c-Jun and c-Myc at Ser243 and Ser62, respectively.8 In DYRK2 knockdown cells, subsequent phosphorylation of c-Jun and c-Myc by GSK3 is absent. Because mutations of these phosphorylation sites have been reported, regulation of c-Jun and c-Myc expression is critical for cancer development. Taken together, a lack of DYRK2 activity accelerates cell proliferation with aberrant expression of c-Jun and c-Myc.
Figure 1.
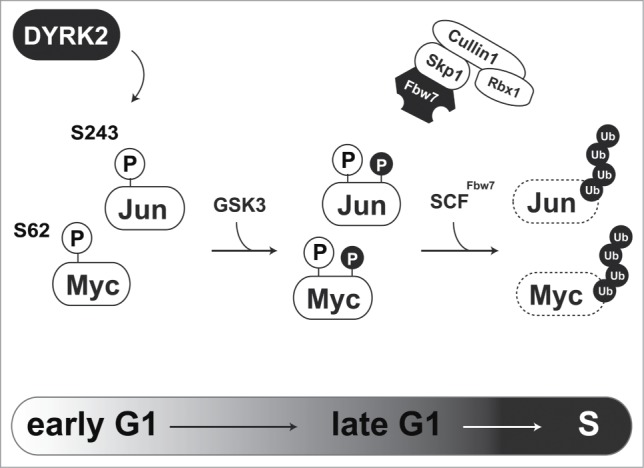
Regulation of G1/S transition via DYRK2-mediated c-Jun and c-Myc degradation. DYRK2-mediated phosphorylation of c-Jun and c-Myc triggers sequential phosphorylation by GSK3. Phosphorylated c-Jun and c-Myc are recognized and ubiquitinated by the SCFFbw7 complex.
Promoting G2/M Transition With EDVP E3 Ligase Complex
Some DYRK2 substrates are degraded independently of sequential phosphorylation by GSK3. Telomerase reverse transcriptase (TERT) and Katanin p60 are ubiquitinated by the EDD-DDB1-VprBP (EDVP) E3 ligase complex, which is triggered by DYRK2 phosphorylation (Fig. 2).9,10 TERT is a catalytic subunit of telomerase. Telomere repeats form the ends of chromosomes and are lost through cell division. However, telomere sequences are elongated by telomerase in certain cell types including cancer cells.11 DYRK2 phosphorylates TERT at Ser457 to induce TERT degradation via EDVP complex-mediated ubiquitination in G2/M phase. However, a DYRK2-TERT interaction has not been observed in S phase, suggesting that TERT is degraded in a cell cycle-specific manner. In DYRK2 knockdown cells, unphosphorylated TERT escapes from degradation, resulting in an increase of telomerase activity (Fig. 2, upper).10 Because high telomerase activity is a hallmark of cancer, TERT degradation is one of the tumor suppressive functions of DYRK2. Katanin p60 is a microtubule-severing enzyme that contributes to mitotic spindle formation. DYRK2 directly phosphorylates Katanin p60 at Ser42, Ser109, and Thr133 to induce its proteasomal degradation (Fig. 2, lower). Similar to TERT degradation, DYRK2 triggers Katanin p60 degradation as a scaffold protein for the EDVP complex. The interaction of DYRK2 with EDVP occurs specifically in G2/M phase because VprBP expression is altered during the cell cycle. These findings indicate that DYRK2 functions specifically as a component of the EDVP complex in G2/M phase. Knockdown of DYRK2 leads to dysregulation of cell division with aberrant expression of Katanin p60 and failure to complete cell division, resulting in an increased of the 4N population and polyploidy (>4N) cells.9 Interestingly, a GSK3 phosphorylation motif has not been found near DYRK2 phosphorylation sites, suggesting that DYRK2-mediated degradation of these molecules is independent of additional phosphorylation by GSK3. In these cases, DYRK2 functions as a scaffold protein for the EDVP complex (Fig. 2).
Figure 2.
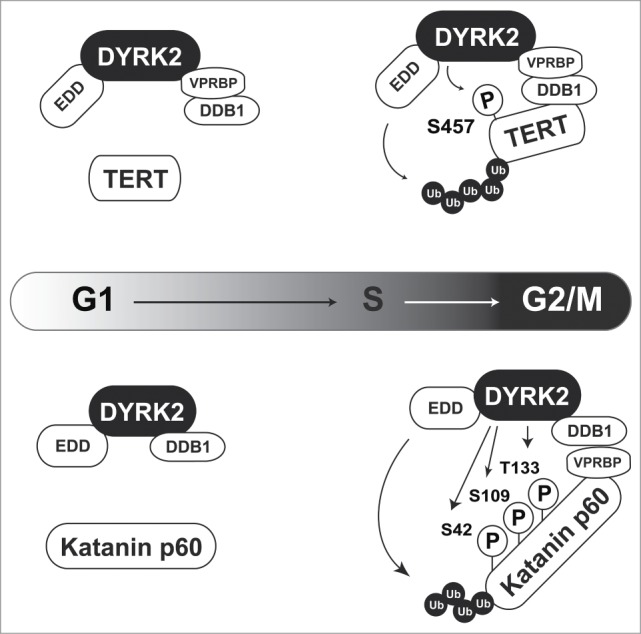
DYRK2 regulates TERT and Katanin stability as a scaffold protein for the EDVP complex. In G2/M phase, DYRK2 phosphorylates TERT (upper) and Katanin p60 (lower). These phosphorylations trigger ubiquitin ligase EDD-mediated ubiquitination.
Another Functions of DYRK2 in Tumor Cells
Apoptosis induction
DNA in eukaryotic cells is continuously damaged by ultraviolet light, genotoxic reagents, and reactive oxygen species. To prevent damaged DNA from being inherited by daughter cells during cell division, cells have checkpoint machinery. In cells bearing repairable DNA damage, the checkpoint is activated to arrest the cell cycle and repair the DNA lesion. However, upon exposure to severe DNA damage, cells undergo apoptosis to eliminate damaged cells. The transcription factor p53 functions as a master regulator of the checkpoint machinery.13 p53 binds to its consensus sequence, 5′-PuPuPuC(A/T)(T/A)GPyPyPy-3′, on the promoter region of target genes to promote transcription. In contrast, under normal conditions, p53 is constitutively degradated by murine double minute 2 (MDM2)-mediated ubiquitination. Under conditions of DNA damage, p53 escapes from degradation and transactivates its target genes involved in cell cycle arrest and DNA repair. p53 stabilization is elicited by rapid phosphorylation at Ser15 and Ser20, suggesting that these modifications inhibit degradation by MDM2. Notably, additional phosphorylation of p53 at Ser46 is indispensable for commitment to apoptotic induction. Two kinases, homeodomain-interacting protein kinase 2 (HIPK2) and DYRK2, have been identified as Ser46 kinases.3,14 Both kinases have mutual features in terms of structure and function. HIPK2 and DYRK2 belong to the CMGC group, and their position on the branch of CMGC kinases is adjacent to each other. These kinases are ubiquitinated by MDM2 under normal conditions and escape from the degradation following DNA damage.15,16 Stabilized HIPK2 and DYRK2 then phosphorylate p53 at Ser46 to irreversibly induce p53-dependent apoptosis. Phosphorylated p53 at Ser46 selectively transactivates apoptosis-related genes, indicating that the phosphorylation alters promoter selectivity among target genes. p53-regulated apoptosis inducing protein 1 (p53AIP1) was the first identified gene that is induced by Ser46-phosphorylated p53.17 p53AIP1 is induced by p53 and then localizes to the mitochondria after DNA damage. p53AIP1 triggers depolarization of the mitochondrial potential and cytochrome c release.17,18 Amphiregulin (AREG) has also been identified as a selective target gene of the Ser46-phosphorylated p53.19 AREG belongs to the epidermal growth factor (EGF) family. However, it has a bifunctional role in cell growth. Under genotoxic insult, AREG modulates tumor suppressive microRNAs biogenesis via RNA helicase DDX5. One of the microRNAs, miR-15, induces apoptosis by targeting the anti-apoptotic protein Bcl-2.20 In response to DNA damage, AREG modulates precursor miR-15 processing to inhibit Bcl-2 expression. Notably, Ser46-phosphorylated p53 by DYRK2 is selectively recruited to the promoter region of the AREG gene upon DNA damage.19 These findings suggest that DYRK2 induces apoptotic cell death via regulation of tumor suppressive microRNA biogenesis and that apoptosis induction is a major function of DYRK2 (Fig. 3). Additionally, DNA repair might be another function of DYRK2 under genotoxic stimuli. Smogorzewska and colleagues performed a genome-wide shRNA screening to identify the molecules involved in DNA repair.21 Through this screening, DYRK2 was ranked in the top 38 candidates. DYRK2 knockdown cells show slight sensitivity to DNA interstrand crosslinking agents, such as mitomycin C, indicating that DYRK2 might be associated with DNA repair. However, whether DYRK2 physiologically affects DNA repair has been elusive. Further study of DYRK2 functions under crosslinking damage is required to determine this possibility.
Figure 3.
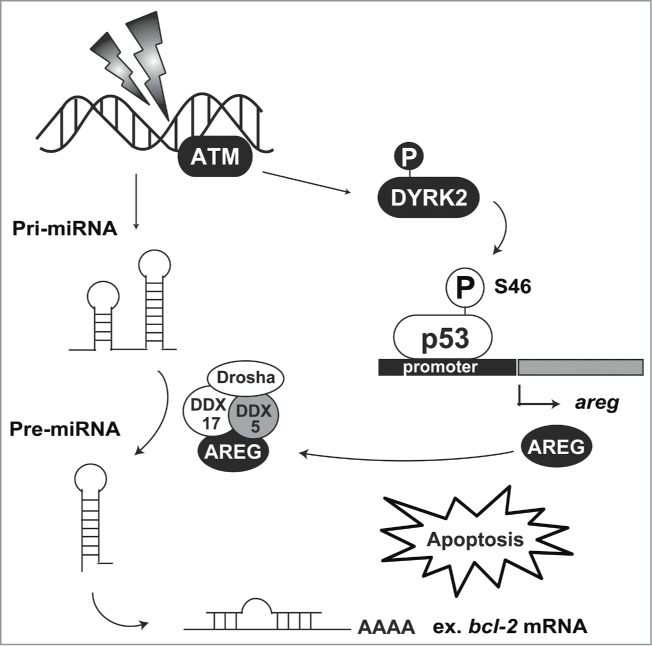
DYRK2-mediated apoptosis induction. Nuclear DYRK2 is ubiquitinated by MDM2 under normal conditions. Upon DNA damage, ATM phosphorylation stabilizes DYRK2 in the nucleus, and then DYRK2 phosphorylates p53 at Ser46. Phosphorylated p53 transactivates its target genes including areg. AREG interacts with the DDX5 complex and regulates precursor miR-15 processing. miR-15 attenuates bcl-2 mRNA translation, resulting in the acceleration of apoptosis induction.
Inhibition of metastasis
The epithelial-to-mesenchymal transition (EMT) is a hallmark of breast cancer invasion. Originally, EMT was defined by the morphological changes during early embryonic development. However, a similar phenomenon was observed in cancer metastasis. In the process of breast cancer metastasis, breast cancer cells lose expression of the adhesion protein E-cadherin, thereby acquiring a cell migration ability.22 Snail is the most studied transcription factor in EMT research. It is one of the members of the Snail superfamily of transcription factors. All Snail family members bind to the E-box domain, 5′-CACCTG-3′, to repress the expression of target genes including E-cadherin.23 Snail is an unstable protein because of constitutive ubiquitination by β-TrCP and FBXL14. β-TrCP-mediated ubiquitination of Snail requires priming phosphorylation at Ser104 and Ser107. However, this priming phosphorylations does not affect FBXL14-mediated ubiquitination.24 Snail, a zinc-finger transcription factor, has a pivotal role in EMT, a key step in cancer metastasis. In addition, Snail is also initially phosphorylated by DYRK2, and then additionally phosphorylated by GSK3. F-box protein β-TrCP recognizes these phosphorylations and triggers proteasomal degradation. In the absence of DYRK2, Snail escapes from β-TrCP-mediated degradation and then stabilizes (Fig. 4).25 As described above, DYRK2 phosphorylates Snail at Ser104 as a priming phosphorylation for GSK3 (Fig. 4). Knockdown of DYRK2 causes abnormal expression of Snail and inhibits E-cadherin expression, which eventually leads to bone metastasis. Notably, DYRK2 expression in invasive breast cancer tissues was declined.25 These findings indicate that DYRK2 negatively regulates EMT in human breast cancer tissues via Snail degradation.
Figure 4.
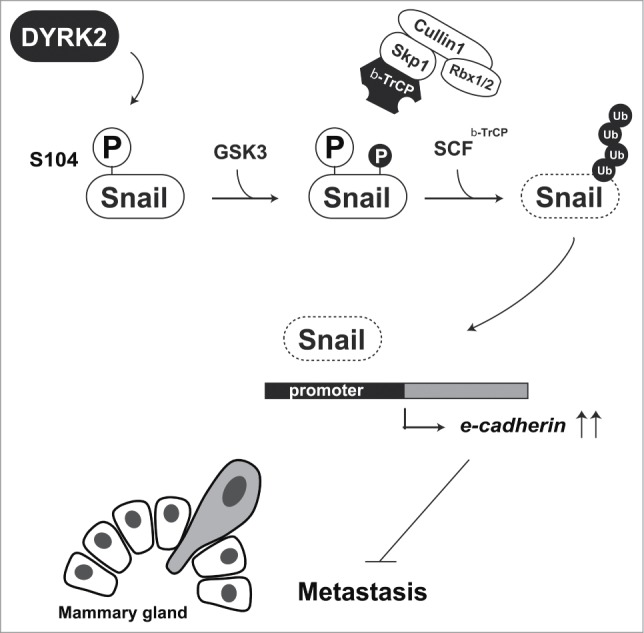
DYRK2-mediated Snail degradation protects against tumor cell metastasis. DYRK2 phosphorylation of Snail at Ser104 triggers sequential phosphorylation by GSK3. Phosphorylated Snail is recognized and ubiquitinated by the SCFβ-TrCP complex. Snail degradation leads to an induction of e-cadherin expression. Cells with highly expression of E-cadherin lose their cell migration ability.
As described previously, Katanin p60 degradation is triggered by DYRK2 phosphorylation (Fig. 2, lower). Ye and colleagues showed that Katanin p60 is associated with bone metastasis of prostate cancer cells. In this context, Katanin p60 expression may be elevated in prostate cancer tissues. Surprisingly, prostate cancer cells that highly express Katanin p60 showed a high migration ability, but also an inhibition of cell proliferation.26 Because DYRK2 expression declined in prostate cancer tissues,8 DYRK2 might inhibit cell migration via Katanin p60 degradation in prostatic adenocarcinomas.
DYRK2 Mutation and Expression in Human Cancer
As described previously, DYRK2 controls accurate cell cycle progression and inhibits cancer metastasis, suggesting that malfunction of DYRK2 might tumorigenesis. Somatic mutations of DYRK2 are found in breast, central nervous system, large intestine, ovarian, and prostate cancers. Almost all these mutations are missense mutations confined to the kinase domain. One of the somatic mutations, S471X, is a nonsense mutation that lacks kinase activity. As described above, DYRK2 down-regulates TERT expression, but the mutant loses the ability to interact with TERT and failes to de-stabilize its expression. Therefore, the S471X mutation contributes to tumor progression because of high telomerase activity.10
DYRK2 expression is down-regulated in various human cancer tissues including those in the kidney, colon, breast, anus, prostate, and esophagus.8 Furthermore, patients with non-small cell lung cancer that are positive for DYRK2 exhibit chemosensitivity, confirming that DYRK2 has a tumor suppressive function in vivo.27,28 In the early-stage of breast cancer, down-regulation of DYRK2 expression is correlated with lymph node metastasis.27,28 An aggressive breast cancer cell line, MDA-MB-231, has a high metastasis potential, and the expression level of DYRK2 in these cells is lower than that in other breast cancer cell lines such as MCF7. These data suggest that a lack of DYRK2 expression leads to breast cancer development. Intriguingly, mRNA levels of DYRK2 are equivalent in both MDA-MB-231 and MCF7 cells.25
DYRK2 accumulates in the presence of a proteasome inhibitor, MG-132, suggesting that DYRK2 expression is downregulated by a post-translational mechanism. Ubiquitin ligases DYRK2, SIAH2, and MDM2 have been identified in DYRK2 degradation.12,16 SIAH2 modulates DYRK2 polyubiquitination under hypoxic conditions. Hypoxic stress causes DYRK2 degradation, thereby failing to induce p53-dependent apoptosis. Perez and colleagues demonstrated that this machinery contributes to the acquisition of chemotherapy resistance in solid tumors.12 Our group found that MDM2 constitutively ubiquitinates nuclear DYRK2 under normal conditions. Nuclear stabilization of DYRK2 is required for p53-mediated induction of apoptosis.16 If nuclear DYRK2 escapes from proteasomal degradation, tumor cells would be eliminated by p53-mediated cell death. Considering the evidence showing that DYRK2 expression declines in invasive tumor tissues, the concept of escape from the proteasomal degradation would be a promising approach for cancer prevention.
Conclusions and Future Prospects
DYRK2 properly controls cell cycle progression through c-Jun, c-Myc, TERT, and Katanin p60 degradation.8-10,25 Most importantly, DYRK2 functions as a priming kinase for GSK3 and the SCF complex in G1 phase, because c-Jun and c-Myc degradation is essential for G1/S transition (Fig. 1). However, DYRK2 binds to the EDVP complex and triggers proteasomal degradation of TERT and Katanin p60 in G2/M phase (Fig. 2). Because dysregulation of cell cycle progression leads to tumorigenesis, DYRK2 might have a tumor suppressive role in cancer cells. In addition to cell cycle regulation, DYRK2 promotes p53-mediated apoptosis in response to severe DNA damage (Fig. 3). DYRK2 phosphorylation alters the promoter affinity of p53, suggesting that the phosphorylation changes its function but not its stability. Although DYRK2 has different functions under normal and stressed conditions, its differential functions serve to block tumorigenesis. Thus, DYRK2 maintains cellular homeostasis via these phosphorylations.
Dysregulation of c-Jun and c-Myc expression leads to infinite cell division. Furthermore, abnormal expression of Snail and TERT expression triggers bone metastasis, suggesting that the loss of DYRK2 strengthens the acquisition of cancer characteristics. To apply DYRK2 research to cancer prevention, we have to further investigate the underlying molecular mechanisms of DYRK2, which contribute to tumor inhibition. Several substrates have been identified thus far, but intracellular regulation of DYRK2 activity remains largely unclear. Chemical compounds that up-regulate DYRK2 activity might have the potential to inhibit cancer development. To discover activators of DYRK2, we have to elucidate the positive regulatory mechanisms of DYRK2 activity. Furthermore, up-regulation of DYRK2 expression might be a useful strategy for cancer prevention. Considering the DYRK2 expression declines in tumor tissues, and that DYRK2 is ubiquitinated by ubiquitin E3 ligases MDM2 and SIAH2, impeding the interaction of DYRK2 with these ubiquitin ligases might inhibit tumor development.
The tumorigenicity of DYRK2 in vivo has been examined by xenograft analyses.8,25 At present, there are no reports of DYRK2 knockout animals. Such knockout analysis might provide essential information regarding the physiological function of DYRK2 in vivo. Notably, the Catalog of Somatic Mutations in Cancer (COSMIC) database has revealed one nonsense mutation and 40 missense mutations in DYRK2. The pathological significance of these mutations remains poorly understood. To examine the physiological relevance of these mutations in cancer progression, knock-in mice bearing these mutations might be useful as disease model animals.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by grants from the Japan Society for the Promotion of Science, the Jikei University Graduate Research Fund, Uehara Memorial Foundation, the Nakajima Foundation, the Sagawa Foundation for Promotion of Cancer Research, Japan Foundation for Applied Enzymology, Project Mirai Cancer Research Grants, and the Vehicle Racing Commemorative Foundation.
References
- 1. Lochhead PA, Sibbet G, Morrice N, Cleghon V. Activation-loop autophosphorylation is mediated by a novel transitional intermediate form of DYRKs. Cell 2005; 121:925-36; PMID:15960979; http://dx.doi.org/ 10.1016/j.cell.2005.03.034 [DOI] [PubMed] [Google Scholar]
- 2. Becker W, Weber Y, Wetzel K, Eirmbter K, Tejedor FJ, Joost HG. Sequence characteristics, subcellular localization, and substrate specificity of DYRK-related kinases, a novel family of dual specificity protein kinases. J Biol Chem 1998; 273:25893-902; PMID:9748265; http://dx.doi.org/ 10.1074/jbc.273.40.25893 [DOI] [PubMed] [Google Scholar]
- 3. Taira N, Nihira K, Yamaguchi T, Miki Y, Yoshida K. DYRK2 is targeted to the nucleus and controls p53 via Ser46 phosphorylation in the apoptotic response to DNA damage. Mol Cell 2007; 25:725-38; PMID:17349958; http://dx.doi.org/ 10.1016/j.molcel.2007.02.007 [DOI] [PubMed] [Google Scholar]
- 4. Wang Z, Liu P, Inuzuka H, Wei W. Roles of F-box proteins in cancer. Nat Rev Cancer 2014; 14:233-47; PMID:24658274; http://dx.doi.org/ 10.1038/nrc3700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Varjosalo M, Bjorklund M, Cheng F, Syvanen H, Kivioja T, Kilpinen S, Sun Z, Kallioniemi O, Stunnenberg HG, He WW, et al. . Application of active and kinase-deficient kinome collection for identification of kinases regulating hedgehog signaling. Cell 2008; 133:537-48; PMID:18455992; http://dx.doi.org/ 10.1016/j.cell.2008.02.047 [DOI] [PubMed] [Google Scholar]
- 6. Weiss CS, Ochs MM, Hagenmueller M, Streit MR, Malekar P, Riffel JH, Buss SJ, Weiss KH, Sadoshima J, Katus HA, et al. . DYRK2 negatively regulates cardiomyocyte growth by mediating repressor function of GSK-3beta on eIF2Bepsilon. PLoS One 2013; 8:e70848; PMID:24023715; http://dx.doi.org/ 10.1371/journal.pone.0070848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Woods YL, Cohen P, Becker W, Jakes R, Goedert M, Wang X, Proud CG. The kinase DYRK phosphorylates protein-synthesis initiation factor eIF2Bepsilon at Ser539 and the microtubule-associated protein tau at Thr212: potential role for DYRK as a glycogen synthase kinase 3-priming kinase. Biochem J 2001; 355:609-15; PMID:11311121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Taira N, Mimoto R, Kurata M, Yamaguchi T, Kitagawa M, Miki Y, Yoshida K. DYRK2 priming phosphorylation of c-Jun and c-Myc modulates cell cycle progression in human cancer cells. J Clin Invest 2012; 122:859-72; PMID:22307329; http://dx.doi.org/ 10.1172/JCI60818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Maddika S, Chen J. Protein kinase DYRK2 is a scaffold that facilitates assembly of an E3 ligase. Nat Cell Biol 2009; 11:409-19; PMID:19287380; http://dx.doi.org/ 10.1038/ncb1848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jung HY, Wang X, Jun S, Park JI. Dyrk2-associated EDD-DDB1-VprBP E3 ligase inhibits telomerase by TERT degradation. J Biol Chem 2013; 288:7252-62; PMID:23362280; http://dx.doi.org/ 10.1074/jbc.M112.416792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Blasco MA. Telomeres and human disease: ageing, cancer and beyond. Nat Rev Genet 2005; 6:611-22; PMID:16136653; http://dx.doi.org/ 10.1038/nrg1656 [DOI] [PubMed] [Google Scholar]
- 12. Perez M, Garcia-Limones C, Zapico I, Marina A, Schmitz ML, Munoz E, Calzado MA. Mutual regulation between SIAH2 and DYRK2 controls hypoxic and genotoxic signaling pathways. J Mol Cell Biol 2012; 4:316-30; PMID:22878263; http://dx.doi.org/ 10.1093/jmcb/mjs047 [DOI] [PubMed] [Google Scholar]
- 13. Taira N, Yoshida K. Post-translational modifications of p53 tumor suppressor: determinants of its functional targets. Histol Histopathol 2012; 27:437-43; PMID:22374721 [DOI] [PubMed] [Google Scholar]
- 14. D'Orazi G, Cecchinelli B, Bruno T, Manni I, Higashimoto Y, Saito S, Gostissa M, Coen S, Marchetti A, Del Sal G, et al. . Homeodomain-interacting protein kinase-2 phosphorylates p53 at Ser 46 and mediates apoptosis. Nat Cell Biol 2002; 4:11-9; PMID:11780126; http://dx.doi.org/ 10.1038/ncb714 [DOI] [PubMed] [Google Scholar]
- 15. Rinaldo C, Prodosmo A, Mancini F, Iacovelli S, Sacchi A, Moretti F, Soddu S. MDM2-regulated degradation of HIPK2 prevents p53Ser46 phosphorylation and DNA damage-induced apoptosis. Mol Cell 2007; 25:739-50; PMID:17349959; http://dx.doi.org/ 10.1016/j.molcel.2007.02.008 [DOI] [PubMed] [Google Scholar]
- 16. Taira N, Yamamoto H, Yamaguchi T, Miki Y, Yoshida K. ATM augments nuclear stabilization of DYRK2 by inhibiting MDM2 in the apoptotic response to DNA damage. J Biol Chem 2010; 285:4909-19; PMID:19965871; http://dx.doi.org/ 10.1074/jbc.M109.042341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Oda K, Arakawa H, Tanaka T, Matsuda K, Tanikawa C, Mori T, Nishimori H, Tamai K, Tokino T, Nakamura Y, et al. . p53AIP1, a potential mediator of p53-dependent apoptosis, and its regulation by Ser-46-phosphorylated p53. Cell 2000; 102:849-62; PMID:11030628; http://dx.doi.org/ 10.1016/S0092-8674(00)00073-8 [DOI] [PubMed] [Google Scholar]
- 18. Wesierska-Gadek J, Gueorguieva M, Horky M. Roscovitine-induced up-regulation of p53AIP1 protein precedes the onset of apoptosis in human MCF-7 breast cancer cells. Mol Cancer Ther 2005; 4:113-24; PMID:15657359 [PubMed] [Google Scholar]
- 19. Taira N, Yamaguchi T, Kimura J, Lu ZG, Fukuda S, Higashiyama S, Ono M, Yoshida K. Induction of amphiregulin by p53 promotes apoptosis via control of microRNA biogenesis in response to DNA damage. Proc Natl Acad Sci U S A 2014; 111:717-22; PMID:24379358; http://dx.doi.org/ 10.1073/pnas.1313675111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cimmino A, Calin GA, Fabbri M, Iorio MV, Ferracin M, Shimizu M, Wojcik SE, Aqeilan RI, Zupo S, Dono M, et al. . miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci U S A 2005; 102:13944-9; PMID:16166262; http://dx.doi.org/ 10.1073/pnas.0506654102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Smogorzewska A, Desetty R, Saito TT, Schlabach M, Lach FP, Sowa ME, Clark AB, Kunkel TA, Harper JW, Colaiacovo MP, et al. . A genetic screen identifies FAN1, a Fanconi anemia-associated nuclease necessary for DNA interstrand crosslink repair. Mol Cell 2010; 39:36-47; PMID:20603073; http://dx.doi.org/ 10.1016/j.molcel.2010.06.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang Y, Shang Y. Epigenetic control of epithelial-to-mesenchymal transition and cancer metastasis. Exp Cell Res 2013; 319:160-9; PMID:22935683; http://dx.doi.org/ 10.1016/j.yexcr.2012.07.019 [DOI] [PubMed] [Google Scholar]
- 23. Batlle E, Sancho E, Franci C, Dominguez D, Monfar M, Baulida J, Garcia De Herreros A. The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat Cell Biol 2000; 2:84-9; PMID:10655587; http://dx.doi.org/ 10.1038/35000034 [DOI] [PubMed] [Google Scholar]
- 24. de Herreros AG, Peiro S, Nassour M, Savagner P. Snail family regulation and epithelial mesenchymal transitions in breast cancer progression. J Mammary Gland Biol Neoplasia 2010; 15:135-47; PMID:20455012; http://dx.doi.org/ 10.1007/s10911-010-9179-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mimoto R, Taira N, Takahashi H, Yamaguchi T, Okabe M, Uchida K, Miki Y, Yoshida K. DYRK2 controls the epithelial-mesenchymal transition in breast cancer by degrading Snail. Cancer Lett 2013; 339:214-25; PMID:23791882; http://dx.doi.org/ 10.1016/j.canlet.2013.06.005 [DOI] [PubMed] [Google Scholar]
- 26. Ye X, Lee YC, Choueiri M, Chu K, Huang CF, Tsai WW, Kobayashi R, Logothetis CJ, Yu-Lee LY, Lin SH. Aberrant expression of katanin p60 in prostate cancer bone metastasis. Prostate 2012; 72:291-300; PMID:21681775; http://dx.doi.org/ 10.1002/pros.21431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yamashita S, Chujo M, Moroga T, Anami K, Tokuishi K, Miyawaki M, Kawano Y, Takeno S, Yamamoto S, Kawahara K. DYRK2 expression may be a predictive marker for chemotherapy in non-small cell lung cancer. Anticancer Res 2009; 29:2753-7; PMID:19596956 [PubMed] [Google Scholar]
- 28. Enomoto Y, Yamashita SI, Yoshinaga Y, Fukami Y, Miyahara S, Nabeshima K, Iwasaki A. Downregulation of DYRK2 can be a predictor of recurrence in early stage breast cancer. Tumour Biol 2014; 35:11021-5; PMID:25095982; http://dx.doi.org/ 10.1007/s13277-014-2413-z [DOI] [PubMed] [Google Scholar]