

Abstract
The Fe(II)- and 2-oxoglutarate (2-OG)-dependent dioxygenases comprise a large and diverse enzyme superfamily the members of which have multiple physiological roles. Despite this diversity, these enzymes share a common chemical mechanism and a core structural fold, a double-stranded β-helix (DSBH), as well as conserved active site residues. The prolyl hydroxylases are members of this large superfamily. Prolyl hydroxylases are involved in collagen biosynthesis and oxygen sensing in mammalian cells. Structural–mechanistic studies with prolyl hydroxylases have broader implications for understanding mechanisms in the Fe(II)- and 2-OG-dependent dioxygenase superfamily. Here, we describe crystal structures of an N-terminally truncated viral collagen prolyl hydroxylase (vCPH). The crystal structure shows that vCPH contains the conserved DSBH motif and iron binding active site residues of 2-OG oxygenases. Molecular dynamics simulations are used to delineate structural changes in vCPH upon binding its substrate. Kinetic investigations are used to report on reaction cycle intermediates and compare them to the closest homologues of vCPH. The study highlights the utility of vCPH as a model enzyme for broader mechanistic analysis of Fe(II)- and 2-OG-dependent dioxygenases, including those of biomedical interest.
Prolyl hydroxylases are members of the large and diverse superfamily of enzymes, the Fe(II)- and 2-oxoglutarate (2-OG)-dependent dioxygenases.1 Enzymes from the 2-OG oxygenase superfamily catalyze a large and diverse range of reactions and have a variety of physiological roles.2−4 Examples include antibiotic synthesis in microorganisms,5 histone modifications,6 and DNA repair/RNA modification.7,8 Despite this diversity of roles, these enzymes share a common reaction mechanism (Figure 1).1 They utilize Fe(II) and the cosubstrate 2-OG in the active site to activate oxygen, resulting in the two-electron oxidation of the substrate. This is coupled to the decarboxylation of 2-OG to produce carbon dioxide and succinate (Figure 1). This oxidative decarboxylation of 2-OG yields a highly reactive Fe(IV) intermediate, which is responsible for abstraction of hydrogen from the substrate.9,10 As well as a conserved reaction mechanism, these enzymes have a highly conserved double-stranded β-helix (DSBH) domain that supports a highly conserved HXD/E···H iron binding motif.11,12 The selectivity of individual enzymes is dictated by additional motifs that surround the DSBH core.
Figure 1.
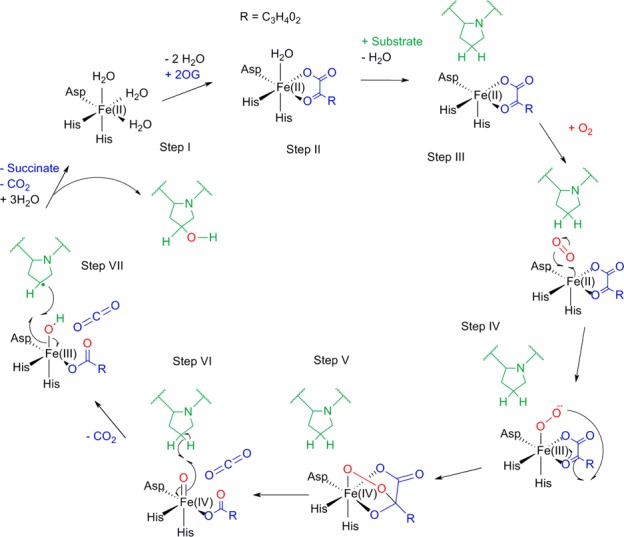
Mechanism of the Fe(II)- and 2-OG-dependent dioxygenases. In its resting position, the enzyme active site contains a ferrous iron ion coordinated by two His residues and one Asp residue (step I). The binding of 2-OG displaces two water molecules (step II); binding of a third water molecule is displaced/weakened upon substrate binding (step III). Substrate binding allows oxygen binding, which subsequently forms an anionic intermediate (step IV) that attacks the ketone of 2-OG to form a cyclic peroxide molecule (step V). The collapse of this intermediate causes formation of the Fe(IV)oxo species, which abstracts a hydrogen from the substrate (step VI). The substrate radical then reacts with the Fe(III)–OH complex to form the hydroxylated substrate (step VII) and restores the enzyme to the resting position.
Prolyl hydroxylases catalyze the hydroxylation of proline residues in proteins and peptides. This post-translational modification is important in cellular oxygen sensing, collagen biosynthesis, and ribosomal protein synthesis by the action of prolyl hydroxylase domain proteins (PHDs), collagen prolyl hydroxylases, and the ribosomal prolyl-3-hydroxylase OGFOD1,13 respectively. In humans, there are three PHD enzymes (PHD1–3) that modulate the stability of isoforms of the hypoxia inducible factor (HIF), a transcription factor responsible for regulation of genes involved in the hypoxic response.14,15 Of the three PHD enzymes, PHD2 is the most important in oxygen sensing,16,17 and studies of PHD2 have characterized its unusually slow reaction with oxygen, relative to other Fe(II)- and 2-OG-dependent dioxygenases, which is thought to be important in its role as an oxygen sensor.18
The mammalian tetrameric collagen prolyl-4-hydroxylases (C-P4H) target X-Pro-Gly repeats in procollagen polypeptides, leading to the increased stability of the collagen triple helix.19 Hydroxylation of collagen-like domains and proline-rich proteins also occurs in plants and algae, and various prolyl hydroxylases have been identified. These enzymes are generally smaller and monomeric compared to the mammalian collagen prolyl hydroxylases. They also differ in terms of substrate specificity and can hydroxylate different sequences of proline-rich peptides.19 Many of the plant prolyl hydroxylases are thought to be involved in the production of hydroxyproline-rich glycoproteins (HRGPs), major components of plant cell walls as well as other cellular functions.19
A viral collagen prolyl hydroxylase from Paramecium bursaria Chlorella Virus-1 (PBCV-1) was first described by Eriksson and co-workers.20 The enzyme has a distinct sequence similarity (between 15 and 23% identity) with the C-terminal half of the α-subunit of animal collagen prolyl hydroxylases. Substrates for the enzyme were determined after sequence analysis of the PBCV-1 genome highlighted many open reading frames for proteins containing regions of proline-rich repeats, similar to the collagen prolyl hydroxylase substrates.20 Hoffart et al. have shown that during the reaction cycle a high-spin Fe(IV) intermediate forms, which is responsible for the abstraction of hydrogen from substrate (Figure 1).9 High-spin Fe(IV) intermediates are often transient in nature and therefore difficult to characterize. That this intermediate can be observed makes mechanistic analysis of the enzymatic reaction cycle accessible. The enzyme is relatively small for a 2-OG oxygenase (25 kDa) and is a monomeric protein. It is soluble at high concentrations and can be tagged with a polyhistidine (His6) extension. This facilitates rapid purification in quantities suitable for (time-resolved) spectroscopy studies for which a relatively high concentration of protein is required.
Here, crystal structures of the viral collagen prolyl hydroxylase from P. bursaria Chlorella Virus-1 (PBCV-1) in both manganese-bound and zinc- and 2-OG-bound conformations are reported. Crystallographic determination of co-complexes with a peptide substrate was not possible despite considerable effort, so molecular dynamics simulations were used to simulate the mode of binding of a proline-rich peptide substrate. Stopped-flow spectroscopy was used to monitor the formation and/or decay of a metal–ligand charge-transfer (MLCT) complex and formation of a hydroxylated product, providing information about the kinetics of the reaction cycle and information about the spectroscopic properties of chemical reaction intermediates (Figure 1).
Experimental Procedures
Protein Expression and Purification
A gene encoding an N-terminally truncated variant of prolyl hydroxylase from P. bursaria Chlorella Virus-1 (PBCV-1) (residues 36–242) (NCBI GI number 9631654) was synthesized with codon optimization for Escherichia coli and cloned into pET-28a with an N-terminal His6 tag using the NdeI/XhoI sites. The variant was produced to remove a predicted N-terminal transmembrane helix. The truncated protein (hereafter termed vCPH) was produced in E. coli Rosetta 2(DE3)pLysS cells (Novagen) and purified using a three-step process. The initial nickel affinity chromatography step used a 5 mL HisTrap HP (GE Healthcare) column with a protocol involving a wash step in 0.5 M NaCl, 20 mM sodium phosphate buffer, and 20 mM imidazole (pH 7.4) followed by an elution gradient from 50 to 250 mM imidazole. After elution, vCPH was exhaustively dialyzed in 20 mM Tris (pH 8) before being loaded onto a ResourceQ (GE Healthcare) anionic exchange column. An elution gradient was applied to the column with the NaCl concentration increasing from 0 to 375 mM over 20 column volumes. After elution, vCPH was concentrated and diluted into 0.2 M EDTA and 12 mM ammonium acetate to remove metal impurities and left overnight at 4 °C. vCPH was then further concentrated before being loaded onto a HiLoad 26/600 Superdex 200 (GE Healthcare) gel filtration column, equilibrated with 100 mM HEPES (pH 7.5).
Peptide Synthesis
Peptides were synthesized as C-terminal amides as described previously21 using a CS Bio CS336 multichannel solid phase peptide synthesizer with Fmoc-protected amino acids, PL-AMS resin (Polymer Laboratories) with Rink amide linker and DIC/HOBT coupling, deprotected (88:5:5:2 CF3CO2H:phenol:H2O:triisopropylsilane), and purified by reversed phase high-performance liquid chromatography using a Vydac 218TP C18 10-15u column (Grace Davison Discovery Sciences). Fmoc-trans-4-hydroxyproline (Fmoc-Hyp) was from Bachem UK.
Substrate Hydroxylation Assay
Hydroxylase activity was measured by mass spectrometry. Reactions were performed in a total volume of 50 μL containing 50 mM Tris-HCl buffer (pH 7.5), with 4 mM ascorbate, 300 mM 2-OG, a 50 μM Fe(NH4)2(SO4)2·6H2O stock solution (prepared freshly in 200 μM HCl to prevent oxidation), 4 μM enzyme, and 100 μM substrate. Controls used buffer instead of enzyme; all assay tubes were shaken in an incubator at 37 °C for the appropriate amount of time and then quenched with an equal volume of methanol (50 μL).
Mass spectrometry measurements were performed after spotting 2 μL of a sample mixture {50% peptide solution and 50% CHCA matrix [recrystallized α-cyano-4-hydroxy-cinnamic acid (Laser Bio Laboratories)]} onto a MALDI 96-well sample plate. Peptide hydroxylation was measured in positive ion mode, with a laser voltage of 12000 and a reflectron voltage of 5200. Spectra were analyzed using MassLynx version 4.0, and the percentage peptide hydroxylation was calculated using eq 1:
| 1 |
where IOH and Inon-OH correspond to peak intensities of hydroxylated and nonhydroxylated peptides, respectively.22
Crystallization
vCPH was concentrated to 30 mg/mL in 100 mM HEPES (pH 7.5) before being supplemented with 1.1 mM MnCl2. Crystals of vCPH were grown by sitting drop vapor diffusion and incubated at 4 °C and arose from a reservoir solution containing 0.1 M imidazole, a MES monohydrate (acid) buffer system, 0.1 M amino acids [0.2 M sodium l-glutamate, 0.2 M alanine (racemic), 0.2 M glycine, 0.2 M lysine HCl (racemic), and 0.2 M serine (racemic)], and 30% PEGMME 550 PEG 20K (pH 6.5) [Morpheus HT96 condition H1, Molecular Dimensions]. The complexed 2-OG structure was obtained by soaking crystals in mother liquor supplemented with 250 mM ZnSO4 and 100 mM 2-OG for 16 h. All crystals were subsequently cryofrozen by being plunged into liquid nitrogen.
Data Collection and Phase Determination
X-ray diffraction data were collected from single cryocooled crystals at Diamond Light Source and subsequently integrated and scaled using XDS.23
Model Building and Refinement
All structures were determined by molecular replacement in Phaser24 using a search model derived from a related prolyl hydroxylase from Chlamydomonas reinhardtii [Cr-P4H; Protein Data Bank (PDB) entry 2JIG]. Xtriage25 was used to analyze the data sets and indicated significant twinning was present in both the Mn(II) and 2-OG data. Twinning was detected using Britton analyses, an H-test, and the maximum likelihood methodology as implemented in Xtriage. A pseudomerohedral twin operator (h, −k, −l) was found and used to automatically account for the twinning during subsequent refinement. The resulting models were completed through iterative rounds of rebuilding in COOT26 and refinement in Phenix.25 Structure validation with MOLPROBITY27 and PDB_REDO28 was integrated as part of the iterative rebuild and refinement procedure.
Molecular Modeling of a Peptide-Bound Structure
In the absence of a peptide-bound crystal structure, a model was generated by first superimposing the structure of the 2-OG-bound vCPH with that of Cr-P4H in complex with a peptide (PDB entry 3GZE) using Secondary Structure Matching (SSM) in Coot.29 The peptide substrate from superimposed model 3GZE was then substituted for the sequence of the peptide substrate of vCPH, PKPAPK, and merged into the PDB file of the vCPH 2-OG structure (vCPH-2OG-Pep). The resultant model was subsequently superimposed onto the vCPH·Mn(II) structure and the peptide backbone real space refined into the electron density observed in this structure from a symmetry-related monomer’s N-terminal His tail that was ordered within the peptide binding site (Figure 3A). The side chain orientations were used to inform initial rotamer choices of the mutated peptide; however, these were not constrained in the molecular dynamics (MD) simulations. The resulting model was then exported to the Rosetta FlexPepDock web server30 and used as the starting model for molecular dynamics and energy minimization. The peptide altered its position to a conformation similar to that of the peptide bound in the 3GZE structure of Cr-P4H. MD simulations were used to explore the conformation of the βII−βIII loop in the absence and presence of the peptide substrate (with 2-OG and Mn bound in both cases). In the Mn(II)-bound structure, the βII−βIII loop adopts a folded back conformation, similar to that of Cr-P4H in the absence of ligand.31 The α1−β2 loop was first modeled using SWISS-MODEL,32,33 and MD simulations were then conducted using Gromacs34,35 version 4.6.1 with the Gromos 53a6 force field.36 To ensure maximal sampling, an initial 10 ns of simulation at 300 K was followed by periodic annealing with the temperature alternating between 300 and 350 K every 1 ns, with heating and cooling over a 10 ps window for a total of 50 ns. This method did not require constraints on the protein or peptide to avoid unfolding during the high-temperature dynamics, but for the peptide-bound protein, a center-of-mass distance restraint of 10 kJ mol–1 Å–2 was applied between the peptide and the protein to prevent unbinding.
Figure 3.
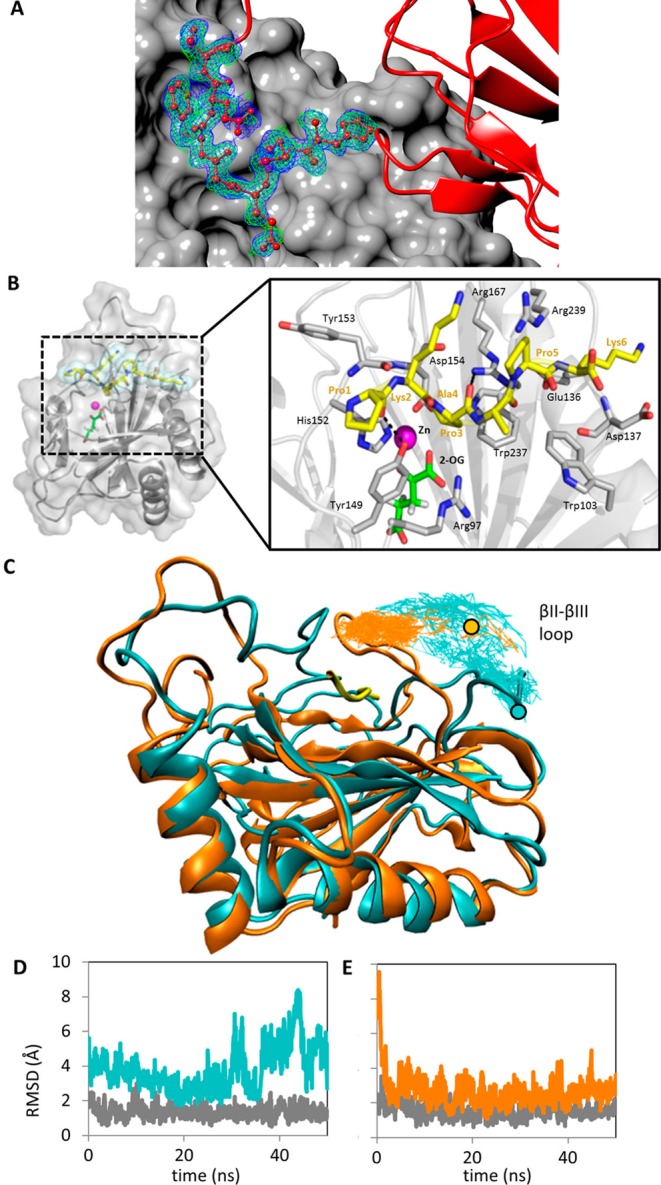
Model of peptide-bound vCPH. (A) Electron density of a symmetry-related monomer’s N-terminal His tail that was ordered within the peptide binding site of the Mn(II)-bound structure. The green map shows the Fo – Fc omit map contoured at 2σ. The blue map shows the 2Fo – Fc final refinement map contoured at 1σ. Residues shown as balls and sticks are residues 34–40 of chain A. The red ribbon is the backbone of chain A. The gray surface rendering is the adjacent chain A from within the crystallographic lattice highlighting the clear interaction of this region of chain. (B) Location of the peptide substrate PKPAPK modeled into the structure of vCPH. vCPH is colored gray; the Zn(II) ion is shown as a magenta sphere, and the 2-OG molecule is colored green. The peptide is shown as yellow sticks surrounded by cyan spheres. Residues of the peptide substrate are labeled in yellow, and residues from the crystal structure are labeled in black. Hydrogen bonds are shown as black dashed lines. (C) Conformation of the βII−βIII loop with and without a peptide bound. Peptide-free protein (cyan) and peptide-bound protein (orange, with peptide colored yellow). The traces follow the center of mass of the α-carbons of residues 125–128 during 50 ns of periodic annealing initiated after 10 ns of 300 K simulation, with the circle indicating the starting position. (D and E) rmsd of the βII−βIII loop relative to the average following alignment to the protein backbone. For comparison, the rmsd for the periodic annealing simulations (cyan for peptide-free, orange for peptide-bound) is shown as well as that for 300 K simulations (gray).
Dissociation Constant Determination
The dissociation constant (Kd) of the 2-OG–vCPH complex was determined by measuring absorbance changes associated with the MLCT complex after titration of 2-OG into a solution of vCPH·Fe(II) using a Cary 50Bio UV–vis spectrophotometer (Agilent technologies). After addition of a small volume of a 2-OG solution, spectra were normalized to account for the addition of small volumes of ligand and the absorbance was set to zero at 700 nm. The absorbance values at 520 nm were plotted against ligand concentration, and the data were then fitted to determine the Kd for the enzyme–2-OG complex (eq 2):
| 2 |
where Y is the relative change in fluorescence, Y0 is fluorescence in the absence of ligand, dY is the change in Y, [E] is the enzyme concentration, [L] is the ligand concentration, and Kd is the vCPH–2-OG complex dissociation constant.
The dissociation constants for peptide–vCPH complexes were determined by tryptophan fluorescence quenching using a Cary Eclipse fluorescence spectrophotometer (Agilent technologies). The crystal structure shows Trp237 present in the peptide and 2-OG binding site, indicating binding of ligands would affect tryptophan fluorescence. For tryptophan fluorescence experiments, samples were excited at 295 nm and the integrals of the area under the emission spectra (peak at ≈350 nm) were first measured and adjusted to account for volume changes upon the addition of peptide. The initial fluorescence intensity was adjusted to 1, and fluorescence intensities with ligand bound were calculated relative to the initial fluorescence and described as a factor of 1. The relative fluorescence was then plotted against ligand concentration and fitted to eq 2 to calculate the dissociation constant.
Transient State Kinetics
Transient state kinetic studies of the reaction cycle were investigated using a stopped-flow spectrophotometer (TgK Scientific). The stopped-flow equipment used a 75 W xenon Arc lamp and a photomultiplier tube as the absorbance and fluorescence detectors. Prior to measurement of absorbance changes, a baseline for protein absorbance of protein and buffer was taken. For fluorescence measurements, the initial fluorescence intensity was set to 100%, and subsequent fluorescence measurements were relative to this intensity. Changes in fluorescence emission were measured as a percentage of signal change.
Single-wavelength stopped-flow data were analyzed using OriginPro software (version 8.5) to give an observed rate constant for the reaction. The data were fitted to an exponential decay function:
| 3 |
where A*i is the amplitude, t is the time, ki is the observed rate constant of the ith exponential component, y is the change in absorbance or fluorescence, and b is the offset value to take into account the non-zero baseline. Transients were fitted from the dead time of the instrument (approximately 2 ms), and each transient was independently fitted to the moving average (nine consecutive points were averaged) of the change in absorbance or fluorescence. Typically, three to five transients were averaged to give the data shown.
The dependence of the observed rate constants on the concentration of ligand was used to gain more information about the spectral features being measured. When the kobs was plotted against ligand/substrate concentration and showed a linear relationship, the data were fitted to the following equation for second-order kinetics:
| 4 |
where kobs is the observed rate constant, kon is the on-rate constant, [L] is the ligand or substrate concentration, and koff is the off-rate constant.
Rapid Quench Mass Spectrometry Experiments
Rapid quench-flow experiments were used to measure levels of cosubstrate and product on a millisecond time scale during a reaction. A rapid quench-flow apparatus (RQF-63, TgK Scientific) was used in an anaerobic glovebox (Belle Technologies). Samples were quenched after the reaction solution had been mixed with 1% CF3CO2H (TFA) and analyzed using MALDI-TOF-MS (peptide hydroxylation) or LC–MS (succinate formation).10,18 For LC–MS, chromatographic separation was performed at 50 °C using a Waters ACQUITY BEH Amide 1.7 μm, 2.1 mm × 100 mm column on a Waters ACQUITYTM ultraperformance liquid chromatography (UPLC) system. The following eluents were used: mobile phase A, 10% H2O, 90% (v/v) acetonitrile, and 10 mM ammonium formate; mobile phase B, 50% H2O, 50% (v/v) acetonitrile, and 10 mM ammonium formate. The elution gradient was 0 to 5.0 min linear from 10 to 50% B and 5.0 to 7.0 min at 10% B for re-equilibration of the column. A constant flow rate of 0.4 mL/min was used. Analytes were detected in negative ionization mode using single-reaction monitoring (SRM) on a Quattro triple-quadruple mass spectrometer (Waters) with a cone voltage of 15 V and a capillary voltage of 3.0 kV. The desolvation temperature was set to 250 °C and the source temperature to 120 °C. For MALDI-MS, peptide hydroxylation was measured as described above.
Results
Description of Mn(II)-Bound vCPH and vCPH in Complex with 2-OG
Two structures of vCPH were obtained, one in complex with Mn(II) and another in complex with Zn(II) and a 2-OG molecule (Table 1). Mn(II) and Zn(II) were used in crystal screens, rather than Fe(II), to make an inactive enzyme complex. The Mn(II)-bound structure contains two molecules per asymmetric unit, and these superimpose with a root-mean-square deviation (rmsd) of 0.34 Å. Several electron density features remain within the active site of the Mn(II)-complexed structure; these proved to be insufficient to accurately model and have been highlighted as UNK in the final structure. The metal ions were modeled on the basis of the known metals present during protein production and crystallogenesis and validated using the CheckMyMetal (CMM) server.37 The crystal packing of the vCPH·Mn(II) structure shows a symmetry-related molecule from a neighboring monomer within the crystal lattice has inserted a portion of its N-terminus and His tag linker into the peptide binding site of each monomer.
Table 1. Data Collection and Refinement Statistics of the Mn- and 2-OG-Bound Structures of vCPH.
| Mn-bound vCPH | 2-OG-bound vCPH | |
|---|---|---|
| wavelength (Å) | 1.0 | 1.0 |
| resolution range (Å) | 28.42–1.7 (1.761–1.7) | 28.43–1.598 (1.655–1.598) |
| space group | P1211 | P1211 |
| unit cell dimensions | 33.77 Å, 157.67 Å, 41.01 Å, 90°, 90.02°, 90° | 33.79 Å, 156.95 Å, 41.24 Å, 90°, 90.02°, 90° |
| total no. of reflections | 137720 (8139) | 187145 (17646) |
| no. of unique reflections | 44328 (3627) | 55206 (5361) |
| multiplicity | 3.1 (2.2) | 3.4 (3.3) |
| completeness (%) | 94.56 (77.65) | 97.68 (94.94) |
| mean I/σ(I) | 10.40 (1.92) | 13.55 (2.38) |
| Rmerge | 0.080 (0.405) | 0.0576 (0.416) |
| Rmeas | 0.096 | 0.069 |
| CC1/2 | 0.996 (0.715) | 0.998 (0.781) |
| CC* | 0.999 (0.913) | 0.999 (0.937) |
| Rwork | 0.156 | 0.178 |
| Rfree | 0.202 | 0.211 |
| no. of non-hydrogen atoms | 3577 | 3326 |
| no. of macromolecules | 3045 | 2907 |
| no. of ligands | 6 | 23 |
| no. of waters | 767 | 551 |
| no. of protein residues | 378 | 360 |
| rmsd for bonds (Å) | 0.005 | 0.014 |
| rmsd for angles (deg) | 0.88 | 1.46 |
| Ramachandran favored (%) | 98 | 98 |
| Ramachandran outliers (%) | 0 | 0.57 |
| Clash score | 1.49 | 2.92 |
| average B factor (Å2) | 15.69 | 21.52 |
| average B factor for macromolecules (Å2) | 12.29 | 19.96 |
| average B factor for ligands (Å2) | 22.67 | 16.08 |
| average B factor for solvent (Å2) | 35.30 | 33.28 |
The vCPH·Mn(II) structure is comprised of a total of 185 of a possible 206 residues spanning residues 34–78 and 100–242 (numbering is maintained from the full length gene; residues 34 and 35 are the start of the His tag linker) (Figure 2A). The C-terminal Lys242 is disordered in chain A. The monomeric fold is comprised of two β-sheets and two α-helices (Figure 2A). Starting at the N-terminus, the β1 strand extends the major sheet of the DSBH (βI, VIII, III, and VI) by hydrogen bonding to βVI. This is followed by α1, which packs across the major β-sheet, and a disordered loop region, 21 residues long. After this, β2 extends the major β-sheet by hydrogen bonding to βI, followed by α2, which packs across the entire major β-sheet (Figure 2A). The region following the βI strand (Tyr148–Asp157) is present as a β-strand in other members of this enzyme family. However, in the Mn-bound vCPH structure, this region does not appear to maintain regular hydrogen bonding associated with β-sheets. Despite this, this region is still labeled βII to conserve the nomenclature of this highly conserved structural motif. The βVII strand also appears not to form uniform antiparallel β-sheet hydrogen bonds with the adjacent βIV strand. Two pairs of cysteine residues form disulfide bonds in vCPH. One pair, Cys47 and Cys129, links a region N-terminal to β1 to the loop between α2 and βI (Figure 2A). It is possible that this linkage may stabilize the α2−βI loop and allow the α2 helix to pack along the major β-sheet. The second pair consists of Cys158 and Cys162, which are part of a βII−βIII loop region near the active site.
Figure 2.

Structure of a viral collagen prolyl hydroxylase (vCPH). (A) Overall topology of the Mn-bound vCPH (PDB entry 5C5U). β-Strands (labeled I–VIII) of the DSBH motif are colored red, additional β-strands gray, and α-helices and loop regions blue. The Mn(II) ion is shown as a dark green sphere. A peptide found in a neighboring molecule in the crystal lattice is colored green. A disordered loop region is shown as a dashed line. (B) Changes in the βII loop upon binding of 2-OG to vCPH. Residues from the Mn-bound structure are colored red, and residues from the 2-OG-bound structure (PDB entry 5C5T) are colored cyan. The metal binding residues are colored yellow; the Zn(II) ion is shown as a magenta sphere, and the 2-OG molecule is colored green. The inset shows the interactions of Tyr149 with residues in the 2-OG-bound structure. (C) Position of 2-OG in the active site of vCPH. Residues from the Mn-bound structure are colored red. Residues from the 2-OG-bound structure are colored cyan, and hydrogen bonds are shown as dashed lines. The Zn(II) ion is shown as a magenta sphere, and waters are shown as blue spheres (Mn-bound structure only). 2-OG is colored green along with the 2Fo – Fc density, contoured at the 2σ level. Tyr149 from the 2-OG-bound structure has been omitted for the sake of clarity.
The overall topology of the 2-OG-bound structure is similar to that of the Mn-bound structure. However, significant changes in the βII strand active site residues are observed upon 2-OG binding (Figure 2B) (βII supports two of the three metal binding residues). In the Mn-bound structure, Tyr149 is flipped out away from the active site, but when 2-OG binds to vCPH, Tyr149 is flipped into the active site such that it is positioned to hydrogen bond to a water molecule that hydrogen bonds to the C-1 carboxylate moiety of 2-OG. Tyr148 and Tyr150 also appear in a different orientation in the 2-OG-bound structure compared to that in the Mn-bound structure. These changes allow the βII and βVII strands to form regular hydrogen bonds to their adjacent β-strands. They also mean certain residues are more ordered in the 2-OG-bound structure as 16 residues of the α1−β2 loop region are disordered (Lys78–Asp93) in the 2-OG-bound structure, compared to the 21 residues (Ser79–Ser99) in the Mn-bound structure. The change in orientation of the Tyr149 side chain allows interactions with both His152 and Arg97 (Figure 2B, inset); Arg97 in the Mn(II) structure, however, forms part of the disordered region.
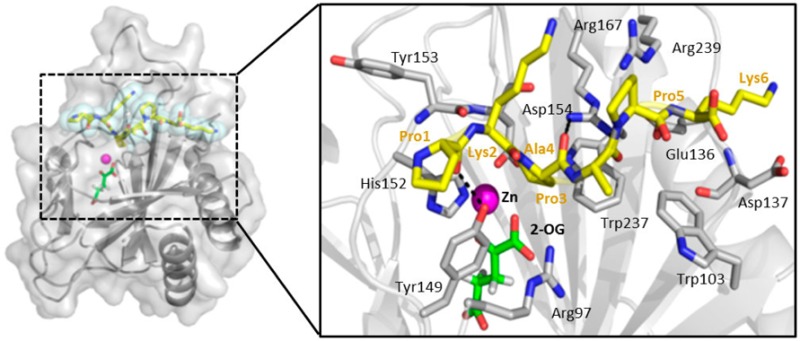
The metals bound in the active sites of our structures are bound by the conserved iron-coordinating residues His152, Asp154, and His221.12 The proximal His152 and Asp154 residues originate from the βII strand, and the distal His221 originates from the βVII strand (Figure 2A). Three water molecules are also coordinated to the Mn(II) ion and occupy places trans to both the equatorial and axial histidines (His152 and His221, respectively) and trans to Asp154 (Figure 2C). In the 2-OG-bound structure, the 2-OG molecule is bound to the Zn(II) ion in a bidentate manner where one oxygen of the C-1 carboxylate is ligated trans to the axial His221 and the C-2 carbonyl is trans to Asp154. The position trans to His152, which contained a water molecule in the Mn-bound structure, is now vacant when 2-OG is bound. It is likely that this position is the site of oxygen binding according to the consensus mechanism (Figure 1). This would mean that the active site might be required to undergo a rearrangement to position the reactive oxygen adjacent to the oxidized bond of the substrate, as observed for other prolyl hydroxylases.38 Several hydrogen bonds, electrostatic contacts, and hydrophobic interactions are made with the 2-OG molecule within the 2-OG binding pocket. These include the conserved basic Lys231 residue (Figure 2C).
Peptide Substrate Interactions with vCPH
In the absence of a peptide-bound crystal structure, a model of vCPH was generated with peptide PKPAPK bound in the active site (see Experimental Procedures). This is a shorter sequence of (PAPK)n, a substrate described by Hoffart et al.20 This was used as the starting model for molecular dynamics and energy minimization studies (Figure 3). PKPAPK adopts a PPII helical structure in the model and binds vCPH across the entrance to the active site between the βII−βIII loop and the loop region between α1 and β2 (Figure 3B). This is a conformation similar to that of the peptide bound in the structure of Cr-P4H.39 The peptide predominantly interacts with vCPH via hydrophobic residues from the β2 and βII strands (Figure 3B). Hydrogen bonds are formed from Tyr149 to the main chain atoms of Pro1, and Arg167 forms hydrogen bonds to the main chain atoms of Ala4 and Pro3. Pro3 of the peptide is pointed toward the Zn(II) ion in the active site, placing the C4 atom in the proximity of the metal ion, indicating that the peptide is positioned optimally for hydroxylation (Figure 3B).
The conformations of the two loops near the peptide binding site were simulated using MD calculations after the disordered α1−β2 loop was modeled using SWISS-MODEL. The simulations showed that in the absence of peptide the βII−βIII loop adopted a more open conformation (Figure 3C), whereas with peptide bound, this loop quickly adopts a conformation closed around the peptide. The fact that the loop in the peptide-bound protein remains in this closed conformation during the periodic annealing reveals that this is a metastable state. On the other hand, as can be seen from the rmsd (Figure 3D,E) and the traces following the loop motion (Figure 3C), the βII−βIII loop is likely to be more disordered without the peptide bound.
Rapid Quench-Flow Experiments Observing Product Formation
The PBCV-1 genome contains many predicted proteins with proline-rich repeat sequences, including proteins with (PAPK)n repeats in which n is >20.40 Recombinant vCPH is able to hydroxylate various proline-rich peptide sequences.20 Different peptide sequences were assayed to determine how the sequence affects the rate of product formation. These were variants of the (PAPK)n sequence determined to be preferable substrates for vCPH by Eriksson et al.20 (Figure 4). Initially, we analyzed (PAPK)n, where n = 2 and 5, by MALDI mass spectrometry. We found that with n = 2, two hydroxylations were observed (a mass shift from 806.7 to 838.7 Da), whereas for n = 5, up to eight hydroxylations were observed (a mass shift from 1984.1 to 2111.5 Da). Under the same conditions, (PAPP)5 underwent up to four hydroxylations (a mass shift from 1851.6 to 1915.2 Da), whereas (PEPV)5 was not hydroxylated (Figure 4). Eriksson et al.20 previously showed that only “internal” prolines (i.e., not those at the N- and C-terminal positions) were being hydroxylated. This is likely the case for the (PAPK)n and (PAPP)5 substrates. However, as (PEPV)5 was not hydroxylated, this suggests specificity for only certain proline-rich peptides.
Figure 4.

MALDI-TOF spectra showing peptide hydroxylation by vCPH. The plots show the change in mass of peptides after they were incubated with vCPH. The top plots show the masses of the peptides after overnight incubations, and the bottom plots show the initial masses of the peptides. The number of oxygen molecules added to each peptide is indicated. Assay conditions: 4 μM vCPH, 160 μM ascorbate, 15 mM 2-OG, 1 μM FeSO4, and 100 μM peptide in 50 mM Tris-HCl buffer (pH 7.5). Reaction mixtures were incubated at 37 °C and reactions quenched with methanol.
Other potential substrates were then tested with vCPH, with a view of identifying a good substrate undergoing only a single hydroxylation suitable for kinetic analyses. One of the hydroxylation sites in (PAPK)2 was blocked by introduction of a trans-4-hydroxyproline residue (Hyp) to give PAPKHypAPK; this peptide was found to undergo only a single hydroxylation (a mass shift from 822.8 to 838.8 Da). Subsequent studies employed longer peptides to allow adequate binding, but also undergoing a single hydroxylation as shown by mass spectrometry. These were (GAGK)2PAGK(GAGK)2 and (HypAHypK)2PAHypK(HypAHypK)2 (Figure 5).
Figure 5.

Product formation of the reaction of vCPH with three peptide substrates. Plots of the reaction of vCPH in complex with Fe(II), 2-OG, and peptide with oxygen are shown. Three peptide substrates were assayed and the samples chemically quenched with 1% TFA at different time points. Quenched samples were analyzed to determine the percentage of peptide hydroxylation using eq 1 (A) and levels of succinate (B) during the reaction. The data were fitted to a single-exponential function shown as a red line eq 3. Final conditions: 800 μM vCPH, 800 μM FeSO4, 1 mM 2-OG, and 1 mM peptide in 100 mM HEPES (pH 7.5) under anaerobic conditions at 5 °C, reacted with O2-saturated buffer.
The dissociation constants of the vCPH–peptide complexes were determined by monitoring tryptophan fluorescence quenching upon addition of peptide substrate to a solution of vCPH in complex with Fe(II). The peptides studied in detail were all found to bind tightly to vCPH as they had dissociation constants between 1.6 and 2.9 μM (Table 2). This suggests that only stretches of hydrophobic residues rather than prolines are necessary as the glycine-rich sequence, (GAGK)2PAGK(GAGK)2, had a dissociation constant similar to those of proline-rich sequences.
Table 2. Kinetic Parameters of Product Formation in vCPH.
| (PAPK)3 | (GAGK)2PAGK(GAGK)2 | (HypAHypK)2PAHypK(HypAHypK)2 | |
|---|---|---|---|
| Rapid Quench MS Measurements | |||
| rate of peptide hydroxylation (s–1) | ND | 0.102 ± 0.022 | 0.84 ± 0.11 |
| rate of succinate formation (s–1) | 0.052 ± 0.060 | 0.047 ± 0.005 | 0.77 ± 0.26 |
| Tryptophan Fluorescence Quenching Measurements | |||
| dissociation constant (μM) | 1.60 ± 0.26 | 1.65 ± 0.33 | 2.86 ± 0.27 |
The rate of product formation in the reaction catalyzed by vCPH using three different peptides [(PAPK)3, (HypAHypK)2PAHypK(HypAHypK)2, and (GAGK)2PAGK(GAGK)2] was determined by rapid quench-flow experiments. Samples were collected when the reaction of vCPH·Fe(II)·2-OG with peptide and oxygen was chemically quenched with 1% trifluoroacetic acid (TFA) at various time points. Hydroxylated peptide formed was quantified by MALDI-TOF MS and succinate generated by LC–MS (Figure 5). The rate constants of peptide hydroxylation and succinate formation are listed in Table 2. Rate constants for the (PAPK)3 peptide were not measured because of the multiple hydroxylation sites on the peptide, but rates were obtained for succinate formation.
The rate constant for hydroxylation of (HypAHypK)2PAHypK(HypAHypK)2 was determined to be faster than that for the (GAGK)2PAGK(GAGK)2 peptide (Figure 5A), suggesting that (hydroxylated) proline-rich peptides are preferable to glycine-rich peptides as vCPH substrates. The rate of peptide hydroxylation and the rate of succinate formation are similar for both peptides (GAGK)2PAGK(GAGK)2 and (HypAHypK)2PAHypK(HypAHypK)2 (Table 2). This is consistent with the consensus mechanism, which implies that hydroxylation is coupled to 2-OG decarboxylation (Figure 1).
Characterization of the MLCT Complex in vCPH
The dissociation constant for the vCPH–2OG complex in the presence of Fe(II) was determined by measuring absorbance changes at 520 nm. These absorbance changes are associated with the MLCT complex (ε520 = 250 M–1 cm–19). A broad spectral feature was observed over 400–700 nm after 2-OG was added to vCPH with an absorbance maximum at approximately 520 nm (Figure 6A). The values at 520 nm were plotted against 2-OG concentration and fitted to eq 2 to determine a Kd for the 2-OG–vCPH complex (746 ± 128 μM). This value differs from that reported by Hoffart et al. (27 ± 6 μM). However, the agreement of our value with the value determined by tryptophan fluorescence (681 ± 128 μM) (Table 3) indicates a weaker binding mode may be at play.
Figure 6.

Kinetic studies of MLCT complex formation. (A) Titration of 2-OG into a solution of vCPH in complex with Fe(II). The left-hand plot shows the difference spectra of vCPH after increasing concentrations of 2-OG are added to a vCPH Fe(II) solution. Spectra shown are samples with 0–5 mM 2-OG present. The right-hand plot shows the absorbance values at 520 nm vs 2-OG concentration. The data were fitted to the Morrison equation (eq 2), which is shown as a black line. Conditions: 150 μM vCPH, 200 μM FeSO4 in 0.5 M NaCl, 50 mM Tris, 10% (v/v) glycerol buffer (pH 7.6) at 25 °C under anaerobic conditions. (B) Dependence of the observed rate constant on the concentration of 2-OG during MLCT complex formation. The left-hand plot shows single-wavelength transients at 520 nm for the formation of the MLCT complex after vCPH, in complex with Fe(II), is mixed with 2-OG in a stopped flow. The data were fitted to an exponential equation (eq 3). The right-hand plot shows the observed rate constants of formation at 520 nm of the first faster phase vs 2-OG concentration. Final conditions: 200 μM vCPH, 250 μM FeSO4 in 50 mM Tris, and 0.5 M NaCl (pH 7.6) at 10 °C mixed with varying concentrations of 2-OG under anaerobic conditions. (C) 2-OG concentration dependence of the transient state kinetics of vCPH tryptophan quenching upon MLCT complex formation. The top left-hand plot is an example of transients measured when vCPH in complex with Fe(II) was mixed with 2-OG in the stopped flow. The data were fitted to an exponential equation (eq 3). The top right-hand plot shows the observed rate constants of tryptophan quenching for the first faster phase vs 2-OG concentration, and the bottom plot shows their respective amplitudes. Final conditions: 5 μM vCPH, 20 μM FeSO4 mixed with varying concentrations of 2-OG in a 50 mM Tris, 0.5 mM NaCl buffer (pH 7.6) under anaerobic conditions at 10 °C. Samples were excited at 295 nm, and fluorescence at 350 nm was measured.
Table 3. Kinetic Parameters of MLCT Complex Formation in vCPH.
| Kd (μM) | kon (mM–1 s–1) | koff (s–1) | |
|---|---|---|---|
| 520 nm absorbance measurements | 746 ± 128 | 17.8 ± 5.0 | 65.8 ± 30.9 |
| tryptophan quenching measurements | 681 ± 128 | 19.8 ± 0.9 | 31.6 ± 1.4 |
The transient state kinetics of MLCT complex formation were investigated by stopped-flow spectroscopy. Absorbance changes at 520 nm were measured after vCPH·Fe(II) was mixed with varying concentrations of 2-OG (Figure 6B). A single- or double-exponential reaction trace was observed (Figure 6B). Figure 6B shows the observed rate constants calculated from the single-wavelength traces of the first (faster) phase plotted against 2-OG concentration. The data show a linear relationship between 2-OG concentration and the observed rate constant, confirming that this first phase is a second-order process. The data were fitted to a linear equation (eq 4) to calculate the kon and koff values, which were 17.8 ± 5.0 mM–1 s–1 and 65.8 ± 30.9 s–1, respectively (Table 3). The rates of the slower phase were found to be independent of 2-OG concentration and were in the range of 2–20 s–1 (data not shown). Experimental difficulties associated with absorption measurements {poor signal-to-noise ratio due to protein precipitation of high concentrations of the vCPH·Fe(II)·2OG [or vCPH·Fe(II)] complex, combined with a low extinction coefficient} meant absorbance amplitude values could not be determined and multiple measurements per 2-OG concentration were not possible, resulting in the lack of error bars in Figure 6B. However, these difficulties were not experienced in the rapid quench experiments, potentially because of the presence of substrate, allowing higher protein concentrations to be used. As an alternative method, changes in tryptophan fluorescence were measured to determine whether they report on 2-OG binding. A single- or double-exponential trace of fluorescence quenching was observed when the vCPH·Fe(II) complex was mixed with 2-OG (Figure 6C). The first exponential rate constants had a linear dependence on 2-OG concentration, similar to absorbance measurements. The kon and koff rates of this first exponential phase (19.8 ± 0.9 mM–1 s–1 and 31.6 ± 1.4 s–1, respectively) were comparable to those determined by absorbance measurements (Table 3), indicating that tryptophan quenching also reports on 2-OG binding. Also, the dissociation constant measured by tryptophan quenching (680 ± 130 μM) (Table 3) was comparable to the value presented in Figure 6A. The amplitudes of the stopped-flow transients also did not appear to saturate until ≈2 mM 2-OG (Figure 6C), which would indicate incomplete binding at lower concentrations, consistent with 2-OG binding weakly to vCPH. Despite difficulties in measuring absorbance changes associated with the MLCT complex, these data suggest a multiple-step mechanism for 2-OG binding.
Discussion
Comparison between the Structure of vCPH and Those of Structural Homologues
The crystal structure of a viral collagen prolyl hydroxylase has been determined for the first time (Figure 2). vCPH contains many of the structural features conserved in other Fe(II)- and 2-OG-dependent dioxygenases, including the DSBH motif and iron binding residues12 (Figure 2A). To date, the crystal structures of five prolyl-4-hydroxylases, including vCPH, have been determined. These are human HIF prolyl hydroxylase 2 (PHD2),41C. reinhardtii prolyl hydroxylase (Cr-P4H),31 a prolyl hydroxylase from Bacillus anthracis (anthrax P4H),42 and a Pseudomonas aeruginosa prolyl hydroxylase domain-containing protein (PPHD).43 The structure of a human ribosomal prolyl-3-hydroxylase, OGFOD1 (Tpa1p in Saccharomyces cerevisiae), has also been reported, but this is more distantly related to the prolyl-4-hydroxylases.44,45 vCPH superimposes well with the prolyl-4-hydroxylase proteins, and the DSBH fold, α1, β1, β2, and α2 of vCPH are in positions similar to those of the topologically similar regions in these three enzymes (Figure 7A). These other prolyl hydroxylases often have N-terminal extensions and differences in loops from the core DSBH motif.
Figure 7.

Structural comparison of the prolyl-4-hydroxylases. (A) Superimposition of four available crystal structures of prolyl-4-hydroxylases. The core DSBH is colored gray, and the differing structural motifs are colored. vCPH is colored cyan, Cr-P4H magenta (PDB entry 2JIG), anthrax P4H green (PDB entry 3ITQ), and PHD2 yellow (PDB entry 2G19). Colored text refers to structures defined by the colors. (B) Comparison of vCPH (cyan) and Cr-P4H (magenta) (PDB entry 2JIG). Tyr149 from vCPH has been omitted for the sake of clarity. 2-OG from vCPH is colored blue and 2-OG from Cr-P4H red. (C) Comparison of vCPH (cyan) and PHD2 (yellow) (PDB entry 3OUJ). 2-OG from vCPH is colored blue and 2-OG from PHD2 brown. Water molecules are shown as small cyan, magenta, and yellow spheres for vCPH, Cr-P4H, and PHD2, respectively. (D) Structural alignment of the sequences of the prolyl-4-hydroxylases. Sequences were aligned by their secondary structures, using the Promal3D server. The secondary structure of vCPH is labeled above the sequence. β-Sheets are shown as red arrows and α-helices as blue squares. Residues involved in iron, 2-OG, and peptide binding are labeled as triangles, circles, and squares, respectively. Underlined residues in the βII−βIII loop indicate the conserved D/E-X-X-N/D motif. Key: α-helix, h; β-strand, e; conserved amino acids in bold and uppercase letters; aliphatic, l; aromatic, @; hydrophobic, h; alcohol, o; polar residues, p; tiny, t; small, s; bulky residues, b; positively charged, +; negatively charged, −; charged, c.
Unique to vCPH, in 2-OG oxygenase structures determined to date, is a disulfide bond formed between a region N-terminal to β1 and the α2−βI loop (Cys47 and Cys129) (Figure 2A). In Cr-P4H and anthrax P4H, this N-terminal to β1 region is shorter and contains an α-helix, and in PHD2, this is a longer loop that interacts with the C-terminal helix of PHD2. It is possible that this disulfide bond is present in vCPH to give stability to this region, which is unnecessary in the shorter loop regions of Cr-P4H and anthrax P4H. Stability of this equivalent region in PHD2 is provided by interactions of the extended loop with the C-terminal helix. Another disulfide bond is present in the βII−βIII loop (Cys158 and Cys162). This bond should stabilize the loop region. In Cr-P4H, this loop is stabilized by hydrogen bonds between Asp149 and Asn152, which are conserved in other prolyl hydroxylases (Figure 7D).39 In anthrax P4H, this region contains an α-helix and hydrogen bonds between residues of the loop, which would stabilize the region.
Comparison of the 2-OG Binding Site with Homologues
The 2-OG binding site in vCPH is similar to that of other prolyl C-4 hydroxylases. The majority of residues that form hydrogen bonds to 2-OG in vCPH are conserved in Cr-P4H (Thr186, Lys231, Tyr143, Gln139, and Tyr149 in vCPH) (Figure 7B,D). Exceptions include Asn235 in vCPH, which is Thr241 in Cr-P4H. However, regardless of the residue in this position, it still forms hydrogen bonds with the C1 carboxylate of 2-OG [2,4-pyridine-dicarboxylate (PDCA) in Cr-P4H]. Gln130 in Cr-P4H is hydrogen bonded to the main chain atoms of Ala153 in the βII−βIII loop, which is folded over the active site, whereas the equivalent residue Gln139 in vCPH forms an interaction with 2-OG via a water molecule (Figure 7B). It is possible that this hydrogen bond in vCPH will resemble the equivalent residue in Cr-P4H if the topologically similar loop region in vCPH folds over the active site. The active site of vCPH also shares many conserved residues with PHD2 (Figure 7C). However, they have significantly different Kd values for the 2-OG–enzyme complex under comparable conditions. The value calculated for vCPH was 746 ± 128 μM (Table 3), which is much higher than that of PHD2 (≤2 μM).18,46
Upon comparison of the structures of vCPH and PHD2, the structural origin of the variation in the dissociation constant for the 2-OG–enzyme is not clear. However, one difference is a different basic residue coordinating C5 of 2-OG (Lys231 in vCPH, Arg383 in PHD2) (Figure 7C), and studies of a PHD2 R383K variant have shown weaker binding of 2-OG to the PHD2–Fe(II) complex.47 This may explain the weaker binding of 2-OG in vCPH compared to that in the structurally similar PHD2. The active site of PHD2 is proposed to be specifically tailored for the tight binding of 2-OG and slow reactivity with oxygen.47 Further comparative studies of vCPH and PHD2 might shed light on why PHD2 is so slow to react with oxygen.
Tyr149 in the Mn-bound structure of vCPH is flipped out from the active site (Figure 2B). When 2-OG is bound, this residue flips toward the active site where it forms hydrogen bonds with 2-OG via a water molecule. The adjacent Tyr150 residue also has a similar flipping movement. This is similar to that observed for the equivalent residues in Cr-P4H.31 This movement has been suggested to modulate loop movements upon substrate binding. Tyr150 in Mn-bound vCPH is in the position where the α1−β2 loop would be expected when the peptide substrate is bound. When this residue flips out, upon 2-OG binding, it may allow the loop movement to occur and facilitate peptide substrate binding. This tyrosine residue is conserved in other prolyl hydroxylases (Figure 7D), and this structure shows this residue is a potential “conformational switch” as also suggested by Koski and colleagues.39
2-OG is bound to the Zn (substituting for Fe) with the 1-carboxylate trans to the distal histidine (His221) and the 2-oxo group trans to the aspartate residue (Asp154) (Figure 2C). This mode of binding is observed in crystal structures of other family members, including PHD2, carbapenem synthase (CarC),48 and anthocyanidin synthase (ANS).49 The positioning of 2-OG in this manner means that the unoccupied coordination site on iron, where oxygen could bind, is oriented away from the peptide substrate. Several suggestions of how the oxygen becomes oriented toward the substrate during catalysis have been discussed.1,12,50 One of these possibilities includes a rearrangement of the C-1 carboxylate of the 2-OG molecule. It has been discussed that in Cr-P4H a change in position of the 2-OG molecule to a position where oxygen binds trans to the axial histidine would require the movement of the side chain of Tyr140. It is possible that this might apply to vCPH as Tyr149 is in a position topologically similar to that of Tyr140.
Significance of the Kinetics of MLCT Complex Formation in vCPH
Aspects of the catalytic cycle of vCPH were analyzed using stopped-flow spectroscopy, including using a peptide that undergoes a single hydroxylation. Specifically, absorbance changes at 520 nm upon mixing of vCPH with 2-OG report on 2-OG binding (Figure 6B). Absorbance data were difficult to obtain because of enzyme precipitation at the required high enzyme concentrations (>200 μM) for observation of the relatively weak absorption signals. Consequently, tryptophan fluorescence was used to measure 2-OG binding at a substantially reduced vCPH concentration (5 μM) (Figure 6C). Tryptophan fluorescence changes mirrored data obtained by absorbance measurements suggesting that both signals observed in stopped-flow studies report on 2-OG binding (i.e., MLCT complex formation) (Table 3). This is further supported by the presence of Trp237 in the 2-OG binding pocket, which would be affected by ligand binding (Figure 2C). A two-step binding mechanism in which a faster transient developed at the higher concentrations of 2-OG assayed was identified (Figure 6B,C). The faster kinetic phase was determined to have second-order kinetics (kon and koff values 17.8 ± 5.0 mM–1 s–1 and 65.8 ± 30.9 s–1, respectively, for absorbance changes and 19.8 ± 0.9 mM–1 s–1 and 31.6 ± 1.4 s–1, respectively, for tryptophan fluorescence measurements). However, the slower phase obeyed first-order kinetics as the observed rate constants were independent of 2-OG concentration. These data are consistent with a model in which 2-OG binds vCPH initially in a 2-OG concentration-dependent manner. A slower first-order active site rearrangement occurs after this initial binding phase (Scheme 1), where k1 and k2 describe the kon and koff values, respectively.
Scheme 1.
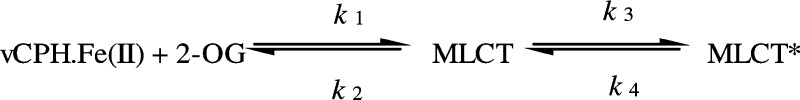
Rate constant k3 can be described by the observed rate constants measured from the second (slower) phase that was independent of 2-OG concentration. A similar experiment conducted on TauD determined that binding of 2-OG to the enzyme·Fe(II) complex was also multiphasic.51 Price et al. showed evidence that this complex kinetic scheme arises from multiple conformational states of the enzyme.51 Other studies of Fe(II)- and 2-OG-dependent dioxygenases have shown that the DSBH core is more conformationally flexible, in particular in the apo state (i.e., without 2OG or metal) of DNA demethylase AlkB52,53 and PHD2.53,54 It seems likely that similar conformational flexibility is a feature of the multistep mechanism for binding of 2-OG to vCPH.
Mode of Peptide Binding in vCPH
Loop regions close to the active site are important in substrate binding in other prolyl-4-hydroxylases.38,39 Molecular dynamics simulations demonstrate that peptide binding facilitates formation of a “closed” conformation of vCPH (Figure 3). This closed conformation has also been observed in Cr-P4H and PHD2. However, the βII−βIII loop in PHD2 is shorter than the equivalent in vCPH and does not directly participate in substrate binding at the active site.
The βII−βIII loop contains a conserved D/E-X-X-N/D motif present in other collagen prolyl hydroxylases39 (Figure 7D), which has been shown to maintain the structure of the loop region via a hydrogen bonding network.39 This motif is also present in vCPH (Figure 7D), suggesting a similar role for these residues. However, these simulations do not show this hydrogen bonding network is present. Another conserved sequence of residues, the D/N-X-X-S/T-G motif, present in the equivalent α1−β2 loop region in various other collagen prolyl hydroxylases is not present in vCPH (Figure 7D).39 This sequence was shown to be important in peptide binding in Cr-P4H, and its absence in vCPH suggests the mode of peptide binding differs from those of other collagen prolyl hydroxylases.
Hydrophobic residues surrounding the hydroxylated proline residue in the Cr-P4H peptide-bound structure are conserved in vCPH (Figure 7D). These residues are Val81 (Val79 in Cr-P4H) and Gly82 (Val80 in Cr-P4H), which may serve the same function in vCPH. The peptide binding groove is also similar to that of Cr-P4H, as most residues in vCPH are conserved in Cr-P4H apart from Asp157 in vCPH, which is Gly128 in Cr-P4H, and Arg239, which is His245 in Cr-P4H (Figure 7D).39 This indicates that the prolyl hydroxylases may have a conserved binding groove that is able to accommodate proline-rich peptides via hydrophobic interactions.
Rapid quench mass spectrometry indicates that, of the peptides tested, (GAGK)2PAGK(GAGK)2 was found to have a rate of hydroxylation lower than that of (HypAHypK)2PAHypK(HypAHypK)2. This suggests a preference for proline residues in the sequences rather than glycine residues. However, significant differences in the dissociation constants of these peptides are not observed. Longer peptides have been shown to be better substrates with lower Km and higher Vmax values.20 As there are likely several hydrophobic interactions with the peptide and the peptide binding groove, it is likely that longer peptides form additional hydrophobic interactions leading to tighter binding. Rate constants for peptide hydroxylation are similar to those for succinate formation for peptides (GAGK)2PAGK(GAGK)2 and (HypAHypK)2PAHypK(HypAHypK)2. This agrees with the proposed mechanism (Figure 1) for the vCPH reaction cycle, in which these two processes are tightly coupled, and the fact that in the case of vCPH there is minimal accumulation of intermediate species between succinate formation and peptide hydroxylation.1,9,10 The rates of substrate hydroxylation and succinate formation for vCPH are much faster than those of its homologue, PHD218 [approximately 43- and 65-fold increases, respectively for (HypAHypK)2PAHypK(HypAHypK)2 in vCPH]. The rearrangement of the 2-OG molecule during catalysis and a metal-coordinated water molecule stabilized by hydrogen bonding with the metal binding Asp residue have both been proposed as factors contributing to the slow reaction of PHD2 with oxygen.18,47,55 Relatively fast activation by oxygen in vCPH may be caused by differences in the structure. Asp315 of PHD2 interacts with both the active site Fe(II) and a Fe(II)-coordinated water molecule.47 This water is strongly ligated in PHD2, and a D315E variant with a weaker interaction with this water molecule, showed more rapid kinetics with respect to oxygen, possibly as a result of more facile H2O release to allow oxygen binding. Whether such a strongly ligated water molecule is present in vCPH is not certain at present, but the crystal structure reveals no such water molecule present in the Zn(II)- and 2-OG-bound structure. The lack of a strongly bound water molecule in a Zn(II)-bound vCPH could be another potential factor responsible for the faster peptide hydroxylation. It is, however, unclear whether this is the case in a Fe(II)-bound structure. Further studies of vCPH could elucidate why the activation of oxygen in PHD2 is slow relative to other prolyl hydroxylases.
Concluding Remarks
The crystal structure of vCPH and analysis of key aspects of the enzyme reaction cycle add to the growing body of structural understanding of the Fe(II)- and 2-OG-dependent prolyl hydroxylases. The study reveals that vCPH is an excellent and accessible model for mechanistic analysis of the growing family of prolyl hydroxylase enzymes, and more broadly the Fe(II)- and 2-OG-dependent dioxygenase superfamily of enzymes.
Acknowledgments
We thank Diamond Light Source for access to beamline i04-1 (Proposal MX7146) that contributed to the results presented here.
Glossary
Abbreviations
- 2-OG
2-oxoglutarate
- DSBH
double-stranded β-helix
- LC–MS
liquid chromatography–mass spectrometry
- MALDI-TOF MS
matrix-assisted laser desorption ionization time-of-flight mass spectrometry
- MLCT
metal to ligand charge transfer
- PBCV-1
P. bursaria Chlorella Virus-1
- PHD
prolyl hydroxylase domain proteins
- vCPH
truncated viral collagen prolyl hydroxylase.
Accession Codes
All coordinates and data have been deposited as Protein Data Bank entries 5C5T and 5C5U.
Author Contributions
J.E.L. purified and crystallized vCPH, performed the majority of kinetic experiments, analyzed the crystal structures, and wrote the paper. C. Levy collected and processed the crystallography data and generated the crystal structure models. L.O.J. performed molecular modeling simulations of vCPH. H.T. and J.E.L. performed the rapid quench assays and MS product detection. E.F. coordinated this aspect of the study and contributed to manuscript preparation. C. Loenarz purified vCPH, designed, synthesized, and purified peptides, and performed peptide hydroxylation assays. S.J. synthesized peptides and performed peptide hydroxylation assays. S.H. assisted in analyzing the MLCT complex binding model. C.J.S. and N.S.S. conceived and coordinated the study and wrote the paper. All authors analyzed the results and approved the final version of the manuscript.
This work was supported by the Biotechnology and Biological Sciences Research Council (BBSRC) (Grant BB/M017702/1), a Royal Society Dorothy Hodgkin Fellowship (E.F.), a L’Oreal-UNESCO For Women In Science Fellowship (E.F.), a Clarendon-St Hugh’s College-W. Louey Scholarship (H.T.), a Leverhulme Fellowship (C.L.), and the Wellcome Trust. N.S.S. is an Engineering and Physical Sciences Research Council (EPSRC) Established Career Fellow (EP/J02019211).
The authors declare no competing financial interest.
References
- Hausinger R. P. (2004) FeII/alpha-ketoglutarate-dependent hydroxylases and related enzymes. Crit. Rev. Biochem. Mol. Biol. 39, 21–68. 10.1080/10409230490440541. [DOI] [PubMed] [Google Scholar]
- Loenarz C.; Schofield C. J. (2008) Expanding chemical biology of 2-oxoglutarate oxygenases. Nat. Chem. Biol. 4, 152–156. 10.1038/nchembio0308-152. [DOI] [PubMed] [Google Scholar]
- Loenarz C.; Schofield C. J. (2011) Physiological and biochemical aspects of hydroxylations and demethylations catalyzed by human 2-oxoglutarate oxygenases. Trends Biochem. Sci. 36, 7–18. 10.1016/j.tibs.2010.07.002. [DOI] [PubMed] [Google Scholar]
- Hewitson K. S.; Granatino N.; Welford R. W. D.; McDonough M. A.; Schofield C. J. (2005) Oxidation by 2-oxoglutarate oxygenases: non-haem iron systems in catalysis and signalling. Philos. Trans. R. Soc., A 363, 807–828. 10.1098/rsta.2004.1540. [DOI] [PubMed] [Google Scholar]
- Neary J. M.; Powell A.; Gordon L.; Milne C.; Flett F.; Wilkinson B.; Smith C. P.; Micklefield J. (2007) An asparagine oxygenase (AsnO) and a 3-hydroxyasparaginyl phosphotransferase (HasP) are involved in the biosynthesis of calcium-dependent lipopeptide antibiotics. Microbiology 153, 768–776. 10.1099/mic.0.2006/002725-0. [DOI] [PubMed] [Google Scholar]
- Tsukada Y.; Fang J.; Erdjument-Bromage H.; Warren M. E.; Borchers C. H.; Tempst P.; Zhang Y. (2005) Histone demethylation by a family of JmjC domain-containing proteins. Nature 439, 811–816. 10.1038/nature04433. [DOI] [PubMed] [Google Scholar]
- Trewick S. C.; Henshaw T. F.; Hausinger R. P.; Lindahl T.; Sedgwick B. (2002) Oxidative demethylation by Escherichia coli AlkB directly reverts DNA base damage. Nature 419, 174–178. 10.1038/nature00908. [DOI] [PubMed] [Google Scholar]
- Mishina Y.; He C. (2006) Oxidative dealkylation DNA repair mediated by the mononuclear non-heme iron AlkB proteins. J. Inorg. Biochem. 100, 670–678. 10.1016/j.jinorgbio.2005.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffart L. M.; Barr E. W.; Guyer R. B.; Bollinger J. M.; Krebs C. (2006) Direct spectroscopic detection of a C-H-cleaving high-spin Fe(IV) complex in a prolyl-4-hydroxylase. Proc. Natl. Acad. Sci. U. S. A. 103, 14738–14743. 10.1073/pnas.0604005103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price J. C.; Barr E. W.; Tirupati B.; Bollinger J. M.; Krebs C. (2003) The first direct characterization of a high-valent iron intermediate in the reaction of an alpha-ketoglutarate-dependent dioxygenase: a high-spin FeIV complex in taurine/alpha-ketoglutarate dioxygenase (TauD) from Escherichia coli. Biochemistry 42, 7497–7508. 10.1021/bi030011f. [DOI] [PubMed] [Google Scholar]
- Aik W.; McDonough M. A.; Thalhammer A.; Chowdhury R.; Schofield C. J. (2012) Role of the jelly-roll fold in substrate binding by 2-oxoglutarate oxygenases. Curr. Opin. Struct. Biol. 22, 691–700. 10.1016/j.sbi.2012.10.001. [DOI] [PubMed] [Google Scholar]
- Clifton I. J.; McDonough M. A.; Ehrismann D.; Kershaw N. J.; Granatino N.; Schofield C. J. (2006) Structural studies on 2-oxoglutarate oxygenases and related double-stranded beta-helix fold proteins. J. Inorg. Biochem. 100, 644–669. 10.1016/j.jinorgbio.2006.01.024. [DOI] [PubMed] [Google Scholar]
- Loenarz C.; Sekirnik R.; Thalhammer A.; Ge W.; Spivakovsky E.; Mackeen M. M.; McDonough M. A.; Cockman M. E.; Kessler B. M.; Ratcliffe P. J.; Wolf A.; Schofield C. J. (2014) Hydroxylation of the eukaryotic ribosomal decoding center affects translational accuracy. Proc. Natl. Acad. Sci. U. S. A. 111, 4019–4024. 10.1073/pnas.1311750111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schofield C. J.; Ratcliffe P. J. (2004) Oxygen sensing by HIF hydroxylases. Nat. Rev. Mol. Cell Biol. 5, 343–354. 10.1038/nrm1366. [DOI] [PubMed] [Google Scholar]
- Myllyharju J. (2013) Prolyl 4-hydroxylases, master regulators of the hypoxia response. Acta Physiol. 208, 148–165. 10.1111/apha.12096. [DOI] [PubMed] [Google Scholar]
- Berra E.; Benizri E.; Ginouvès A.; Volmat V.; Roux D.; Pouysségur J. (2003) HIF prolyl-hydroxylase 2 is the key oxygen sensor setting low steady-state levels of HIF-1alpha in normoxia. EMBO J. 22, 4082–4090. 10.1093/emboj/cdg392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda K.; Cowan A.; Fong G. H. (2007) Essential role for prolyl hydroxylase domain protein 2 in oxygen homeostasis of the adult vascular system. Circulation 116, 774–781. 10.1161/CIRCULATIONAHA.107.701516. [DOI] [PubMed] [Google Scholar]
- Flashman E.; Hoffart L. M.; Hamed R. B.; Bollinger J. M. Jr.; Krebs C.; Schofield C. J. (2010) Evidence for the slow reaction of hypoxia-inducible factor prolyl hydroxylase 2 with oxygen. FEBS J. 277, 4089–4099. 10.1111/j.1742-4658.2010.07804.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorres K. L.; Raines R. T. (2010) Prolyl 4-hydroxylase. Crit. Rev. Biochem. Mol. Biol. 45, 106–124. 10.3109/10409231003627991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksson M.; Myllyharju J.; Tu H.; Hellman M.; Kivirikko K. I. (1999) Evidence for 4-hydroxyproline in viral proteins. Characterization of a viral prolyl 4-hydroxylase and its peptide substrates. J. Biol. Chem. 274, 22131–22134. 10.1074/jbc.274.32.22131. [DOI] [PubMed] [Google Scholar]
- Loenarz C.; Mecinović J.; Chowdhury R.; McNeill L. A.; Flashman E.; Schofield C. J. (2009) Evidence for a stereoelectronic effect in human oxygen sensing. Angew. Chem., Int. Ed. 48, 1784–1787. 10.1002/anie.200805427. [DOI] [PubMed] [Google Scholar]
- Flashman E.; Bagg E. A. L.; Chowdhury R.; Mecinović J.; Loenarz C.; McDonough M. A.; Hewitson K. S.; Schofield C. J. (2008) Kinetic rationale for selectivity toward N- and C-terminal oxygen-dependent degradation domain substrates mediated by a loop region of hypoxia-inducible factor prolyl hydroxylases. J. Biol. Chem. 283, 3808–3815. 10.1074/jbc.M707411200. [DOI] [PubMed] [Google Scholar]
- Kabsch W. (2010) XDS. Acta Crystallogr., Sect. D: Biol. Crystallogr. 66, 125–132. 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy A. J.; Grosse-Kunstleve R. W.; Adams P. D.; Winn M. D.; Storoni L. C.; Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674. 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams P. D.; Afonine P. V.; Bunkóczi G.; Chen V. B.; Davis I. W.; Echols N.; Headd J. J.; Hung L. W.; Kapral G. J.; Grosse-Kunstleve R. W.; McCoy A. J.; Moriarty N. W.; Oeffner R.; Read R. J.; Richardson D. C.; Richardson J. S.; Terwilliger T. C.; Zwart P. H. (2010) PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr., Sect. D: Biol. Crystallogr. 66, 213–221. 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Lohkamp B.; Scott W. G.; Cowtan K. (2010) Features and development of Coot. Acta Crystallogr., Sect. D: Biol. Crystallogr. 66, 486–501. 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen V. B.; Arendall W. B.; Headd J. J.; Keedy D. A.; Immormino R. M.; Kapral G. J.; Murray L. W.; Richardson J. S.; Richardson D. C. (2010) MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr., Sect. D: Biol. Crystallogr. 66, 12–21. 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joosten R. P.; Long F.; Murshudov G. N.; Perrakis A. (2014) The {\it PDB_REDO} server for macromolecular structure model optimization. IUCrJ 1, 213–220. 10.1107/S2052252514009324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krissinel E.; Henrick K. (2004) Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr., Sect. D: Biol. Crystallogr. 60, 2256–2268. 10.1107/S0907444904026460. [DOI] [PubMed] [Google Scholar]
- London N.; Raveh B.; Cohen E.; Fathi G.; Schueler-Furman O. (2011) Rosetta FlexPepDock web server--high resolution modeling of peptide-protein interactions. Nucleic Acids Res. 39, W249–253. 10.1093/nar/gkr431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koski M. K.; Hieta R.; Böllner C.; Kivirikko K. I.; Myllyharju J.; Wierenga R. K. (2007) The active site of an algal prolyl 4-hydroxylase has a large structural plasticity. J. Biol. Chem. 282, 37112–37123. 10.1074/jbc.M706554200. [DOI] [PubMed] [Google Scholar]
- Biasini M.; Bienert S.; Waterhouse A.; Arnold K.; Studer G.; Schmidt T.; Kiefer F.; Cassarino T. G.; Bertoni M.; Bordoli L.; Schwede T. (2014) SWISS-MODEL: Modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 42, W252–258. 10.1093/nar/gku340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold K.; Bordoli L.; Kopp J.; Schwede T. (2006) The SWISS-MODEL workspace: A web-based environment for protein structure homology modelling. Bioinformatics 22, 195–201. 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
- Hess B.; Kutzner C.; Van Der Spoel D.; Lindahl E. (2008) GRGMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 4, 435–447. 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- Van Der Spoel D.; Lindahl E.; Hess B.; Groenhof G.; Mark A. E.; Berendsen H. J. C. (2005) GROMACS: Fast, flexible, and free. J. Comput. Chem. 26, 1701–1718. 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- Oostenbrink C.; Villa A.; Mark A. E.; Van Gunsteren W. F. (2004) A biomolecular force field based on the free enthalpy of hydration and solvation: The GROMOS force-field parameter sets 53A5 and 53A6. J. Comput. Chem. 25, 1656–1676. 10.1002/jcc.20090. [DOI] [PubMed] [Google Scholar]
- Zheng H.; Chordia M. D.; Cooper D. R.; Chruszcz M.; Müller P.; Sheldrick G. M.; Minor W. (2013) Validation of metal-binding sites in macromolecular structures with the CheckMyMetal web server. Nat. Protoc. 9, 156–170. 10.1038/nprot.2013.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdhury R.; McDonough M. A.; Mecinović J.; Loenarz C.; Flashman E.; Hewitson K. S.; Domene C.; Schofield C. J. (2009) Structural basis for binding of hypoxia-inducible factor to the oxygen-sensing prolyl hydroxylases. Structure 17, 981–989. 10.1016/j.str.2009.06.002. [DOI] [PubMed] [Google Scholar]
- Koski M. K.; Hieta R.; Hirsilä M.; Rönkä A.; Myllyharju J.; Wierenga R. K. (2009) The crystal structure of an algal prolyl 4-hydroxylase complexed with a proline-rich peptide reveals a novel buried tripeptide binding motif. J. Biol. Chem. 284, 25290–25301. 10.1074/jbc.M109.014050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y.; Lu Z.; Burbank D. E.; Kutish G. F.; Rock D. L.; Van Etten J. L. (1995) Analysis of 43 kb of the Chlorella Virus PBCV-1 330-kb Genome: Map Positions 45 to 88. Virology 212, 134–150. 10.1006/viro.1995.1462. [DOI] [PubMed] [Google Scholar]
- McDonough M. A.; Li V.; Flashman E.; Chowdhury R.; Mohr C.; Liénard B. M. R.; Zondlo J.; Oldham N. J.; Clifton I. J.; Lewis J.; McNeill L. A.; Kurzeja R. J. M.; Hewitson K. S.; Yang E.; Jordan S.; Syed R. S.; Schofield C. J. (2006) Cellular oxygen sensing: Crystal structure of hypoxia-inducible factor prolyl hydroxylase (PHD2). Proc. Natl. Acad. Sci. U. S. A. 103, 9814–9819. 10.1073/pnas.0601283103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culpepper M. A.; Scott E. E.; Limburg J. (2010) Crystal structure of prolyl 4-hydroxylase from Bacillus anthracis. Biochemistry 49, 124–133. 10.1021/bi901771z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scotti J. S.; Leung I. K. H.; Ge W.; Bentley M. A.; Paps J.; Kramer H. B.; Lee J.; Aik W.; Choi H.; Paulsen S. M.; Bowman L. A. H.; Loik N. D.; Horita S.; Ho C.; Kershaw N. J.; Tang C. M.; Claridge T. D. W.; Preston G. M.; McDonough M. A.; Schofield C. J. (2014) Human oxygen sensing may have origins in prokaryotic elongation factor Tu prolyl-hydroxylation. Proc. Natl. Acad. Sci. U. S. A. 111, 13331–13336. 10.1073/pnas.1409916111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horita S.; Scotti J. S.; Thinnes C.; Mottaghi-Taromsari Y. S.; Thalhammer A.; Ge W.; Aik W.; Loenarz C.; Schofield C. J.; McDonough M. A. (2015) Structure of the Ribosomal Oxygenase OGFOD1 Provides Insights into the Regio- and Stereoselectivity of Prolyl Hydroxylases. Structure 23, 639–652. 10.1016/j.str.2015.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henri J.; Rispal D.; Bayart E.; Van Tilbeurgh H.; Séraphin B.; Graille M. (2010) Structural and functional insights into Saccharomyces cerevisiae Tpa1, a putative prolylhydroxylase influencing translation termination and transcription. J. Biol. Chem. 285, 30767–30778. 10.1074/jbc.M110.106864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNeill L. A.; Flashman E.; Buck M. R. G.; Hewitson K. S.; Clifton I. J.; Jeschke G.; Claridge T. D. W.; Ehrismann D.; Oldham N. J.; Schofield C. J. (2005) Hypoxia-inducible factor prolyl hydroxylase 2 has a high affinity for ferrous iron and 2-oxoglutarate. Mol. BioSyst. 1, 321–324. 10.1039/b511249b. [DOI] [PubMed] [Google Scholar]
- Tarhonskaya H.; Chowdhury R.; Leung I. K. H.; Loik N. D.; McCullagh J. S. O.; Claridge T. D. W.; Schofield C. J.; Flashman E. (2014) Investigating the contribution of the active site environment to the slow reaction of hypoxia-inducible factor prolyl hydroxylase domain 2 with oxygen. Biochem. J. 463, 363–372. 10.1042/BJ20140779. [DOI] [PubMed] [Google Scholar]
- Clifton I. J.; Doan L. X.; Sleeman M. C.; Topf M.; Suzuki H.; Wilmouth R. C.; Schofield C. J. (2003) Crystal structure of carbapenem synthase (CarC). J. Biol. Chem. 278, 20843–20850. 10.1074/jbc.M213054200. [DOI] [PubMed] [Google Scholar]
- Wilmouth R. C.; Turnbull J. J.; Welford R. W. D.; Clifton I. J.; Prescott A. G.; Schofield C. J. (2002) Structure and mechanism of anthocyanidin synthase from Arabidopsis thaliana. Structure 10, 93–103. 10.1016/S0969-2126(01)00695-5. [DOI] [PubMed] [Google Scholar]
- Zhang Z.; Ren J.-s.; Harlos K.; McKinnon C. H.; Clifton I. J.; Schofield C. J. (2002) Crystal structure of a clavaminate synthase-Fe(II)-2-oxoglutarate-substrate-NO complex: evidence for metal centered rearrangements. FEBS Lett. 517, 7–12. 10.1016/S0014-5793(02)02520-6. [DOI] [PubMed] [Google Scholar]
- Price J. C.; Barr E. W.; Hoffart L. M.; Krebs C.; Bollinger J. M. (2005) Kinetic dissection of the catalytic mechanism of taurine:alpha-ketoglutarate dioxygenase (TauD) from Escherichia coli. Biochemistry 44, 8138–8147. 10.1021/bi050227c. [DOI] [PubMed] [Google Scholar]
- Bleijlevens B.; Shivarattan T.; van den Boom K. S.; de Haan A.; van der Zwan G.; Simpson P. J.; Matthews S. J. (2012) Changes in protein dynamics of the DNA repair dioxygenase AlkB upon binding of Fe(2+) and 2-oxoglutarate. Biochemistry 51, 3334–3341. 10.1021/bi201699e. [DOI] [PubMed] [Google Scholar]
- Bleijlevens B.; Shivarattan T.; Flashman E.; Yang Y.; Simpson P. J.; Koivisto P.; Sedgwick B.; Schofield C. J.; Matthews S. J. (2008) Dynamic states of the DNA repair enzyme AlkB regulate product release. EMBO Rep. 9, 872–877. 10.1038/embor.2008.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stubbs C. J.; Loenarz C.; Mecinović J.; Yeoh K. K.; Hindley N.; Liénard B. M.; Sobott F.; Schofield C. J.; Flashman E. (2009) Application of a proteolysis/mass spectrometry method for investigating the effects of inhibitors on hydroxylase structure. J. Med. Chem. 52, 2799–2805. 10.1021/jm900285r. [DOI] [PubMed] [Google Scholar]
- Flagg S. C.; Giri N.; Pektas S.; Maroney M. J.; Knapp M. J. (2012) Inverse solvent isotope effects demonstrate slow aquo release from hypoxia inducible factor-prolyl hydroxylase (PHD2). Biochemistry 51, 6654–6666. 10.1021/bi300229y. [DOI] [PMC free article] [PubMed] [Google Scholar]