

Abstract
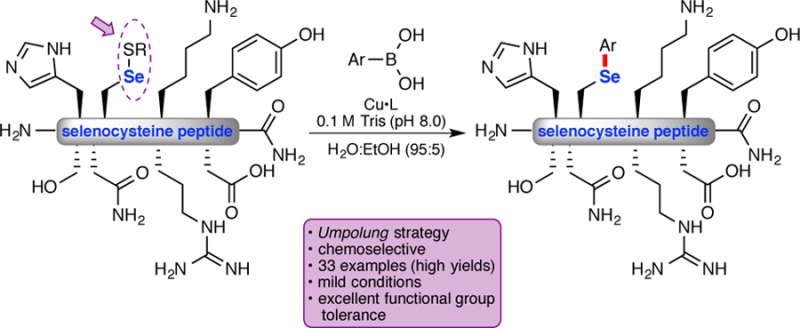
Herein we report an umpolung strategy for the bioconjugation of selenocysteine in unprotected peptides. This mild and operationally simple approach takes advantage of the electrophilic character of an oxidized selenocysteine (Se–S bond) to react with a nucleophilic arylboronic acid to provide the arylated selenocysteine within hours. This reaction is amenable to a wide range of boronic acids with different biorelevant functional groups and is unique to selenocysteine. Experimental evidence indicates that under oxidative conditions the arylated derivatives are more stable than the corresponding alkylated selenocysteine.
In 1973, Ganther reported the presence of selenium within the natural enzyme glutathione peroxidase.1 While it was strongly suspected that the detection of selenium in this natural enzyme was the result of a parent amino acid, the existence of selenocysteine was not confirmed until the subsequent reports by Stadtman2 and Tappel.3 Selenocysteine (Sec or U), the 21st proteinogenic amino acid,4 is a structural analogue to cysteine (Cys) with a selenol in place of the thiol.5 There are several notable differences between Cys and Sec. The greater acidity of Sec (pKa = 5.47)6 versus Cys (pKa = 8.14)7 causes it to be deprotonated at physiological pH, and its lower reduction potential makes it an integral part of antioxidant proteins.8 Sec is essential for activity in several enzymes, including glutathione peroxidases, iodothyronine deiodinases, formate dehydrogenases, and methionine-R-sulfoxide reductase. Mutation of the catalytic Sec to Cys in the aforementioned enzymes results in >100-fold decreases in activity.9
The inherent nucleophilicity of selenols makes selenocysteine an appealing handle for chemoselective bioconjugation in peptides and proteins.10 Nevertheless, reports on bioconjugation with this amino acid have been sparse because of several challenges associated with its functionalization.11 Specifically, the low selenol redox potential promotes facile oxidation to the diselenide or seleninic acid, and the high polarizability of selenium leads to elimination to generate dehydroalanine.11e,12 Reports of Sec functionalization have paralleled methods to modify Cys13 and relied upon alkylation and maleimide conjugate addition with the selenol group (Figure 1, eq 1). In contrast to common Cys conjugation methods, a reducing agent [i.e., tris(2-carboxyethyl)phosphine (TCEP)] and exclusion of oxygen are needed to generate the selenol in situ prior to reaction with the electrophile. The development of a direct, robust, and selective method for the functionalization of Sec that overcomes these limitations would be an important breakthrough for site-selective modification of unprotected peptides.
Figure 1.
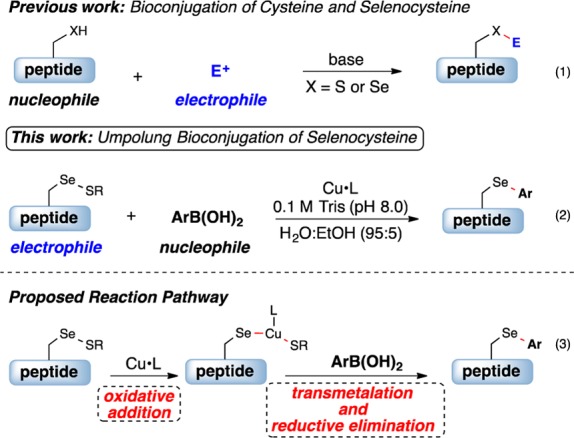
Bioconjugation of cysteine and selenocysteine.
Herein we disclose an umpolung approach14 that utilizes the electrophilic character of the oxidized selenocysteine15 together with a copper, ligand, and nucleophilic boronic acid combination to provide the arylated selenocysteine in unprotected peptides (eq 2). The proposed reaction pathway involves initial oxidative addition of copper to the Se–S bond (eq 3). Subsequent transmetalation with an arylboronic acid and reductive elimination furnishes the arylated selenocysteine.16 This method does not require oxygen-free conditions or a reducing agent and takes place in water-rich media [0.1 M Tris buffer (pH 8.0), 95:5 H2O/EtOH]. Reaction yields are excellent for a wide range of substrates, and this approach is unique to selenocysteine.
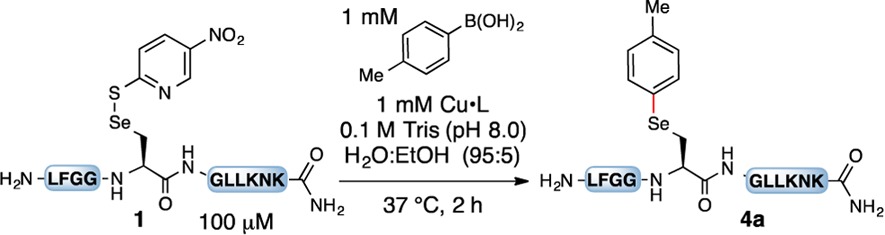
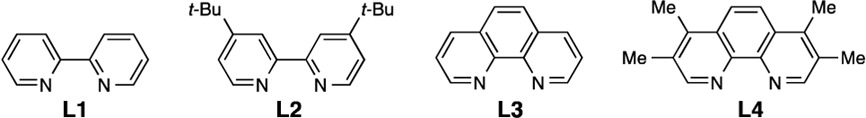
Our optimization of the synthesis for arylated selenocysteine began by combining unprotected peptide 1, p-tolylboronic acid, CuSO4, and a ligand in an aqueous buffer [0.1 M Tris (pH 8.0), 95:5 H2O/EtOH] at 37 °C. We chose to begin our initial studies with 2-thiol-5-nitropyridine (TNP) protected selenocysteine-containing peptide 1, as it is the reaction product from the deprotection of the p-methoxybenzyl protecting group in solid-phase peptide synthesis.17 First we tested several bipyridine and phenanthroline ligands (Table 1, entries 1–4) and found 4,4′-di-tert-butyl-2,2′-bipyridine (L2) to be the best ligand for conversion and yield of arylated peptide 4a, as observed by LC–MS. The diselenide, seleninic acid, and the elimination product dehydroalanine were the observed side products.18 In the absence of ligand, only 30% conversion of 1 and 5% arylation to give 4a were observed (entry 5). Other sources of copper provided similar yields (entries 6–8). In the absence of copper, no arylation was observed (entry 9). Lastly, decreasing the amount of copper, ligand, or boronic acid resulted in lower levels of conversion and increased side reactions (entries 10 and 11).18 No product was observed in a control reaction with the Sec-to-Ser variant (entry 12), confirming that arylation occurs exclusively at selenium. To probe the reactivity of selenocysteine versus cysteine, we investigated two peptides in which the Sec was replaced with Cys (free thiol) or Cys linked with TNP as the masking agent (entries 13 and 14). For both peptides we observed low conversion to the arylated cysteine; rather, the starting material was converted to the respective disulfide or sulfinic acid.19 This result demonstrates the enhanced reactivity of Sec over Cys toward Cu-mediated arylation. It also indicates that Cys is not compatible with these reaction conditions.20 Lastly, no oxidation was detected for the Sec-to-Met variant (entry 15), proving that this method could be applied to methionine-containing peptides.
Table 1. Optimization of the Reaction Parametersa.
| entry | Cu | ligand | % yield (% conversion)b |
|---|---|---|---|
| 1 | CuSO4 | L1 | 79 (99) |
| 2 | CuSO4 | L2 | 81 (99) |
| 3 | CuSO4 | L3 | 66 (99) |
| 4 | CuSO4 | L4 | 17 (91) |
| 5 | CuSO4 | – | 5 (30) |
| 6 | CuCl2·2H2O | L2 | 78 (99) |
| 7 | CuBr2 | L2 | 79 (99) |
| 8 | Cu(OAc)2 | L2 | 78 (99) |
| 9 | – | L2 | 0 (22)c |
| 10d | CuSO4 | L2 | 44 (72) |
| 11e | CuSO4 | L2 | 7 (30) |
| 12f | CuSO4 | L2 | NR |
| 13g | CuSO4 | L2 | 5 (99) |
| 14h | CuSO4 | L2 | 4 (99) |
| 15i | CuSO4 | L2 | NR |
See the Supporting Information for details. Amino acids are shown in a one-letter code. NR = no reaction.
Yields were determined by integration of the total ion currents (TICs) from LC–MS analyses of the unpurified reaction mixtures.
Elimination and diselenide were the only observable products.
0.5 mM CuSO4, 0.5 mM L2, and 0.5 mM boronic acid were used.
0.25 mM CuSO4, 0.25 mM L2, and 0.25 mM boronic acid were used.
The Sec-TNP residue replaced with Ser.
The Sec-TNP residue was replaced with Cys.
The Sec-TNP residue was replaced with Cys-TNP.
The Sec-TNP residue was replaced with Met.
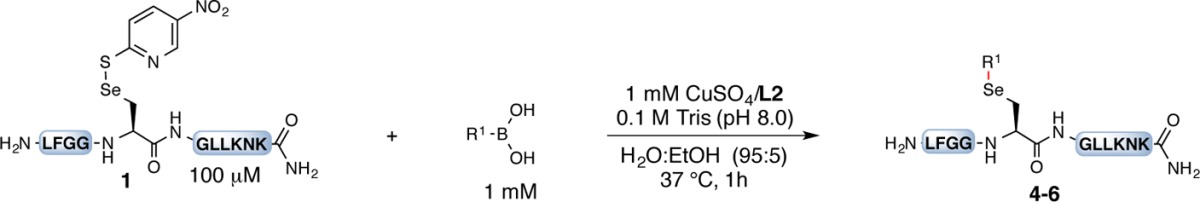
Having found the optimized conditions, we explored the substrate scope of this copper-mediated synthesis of arylated selenocysteine in unprotected peptides (Table 2). Arylboronic acids with electron-donating functional groups were first examined (Table 2, section a). Methoxy (4b), o-phenol (4c), dioxolane (4d), −CH2N(H)Boc (4e), and p-dimethylamine (4f) groups were readily tolerated. Under these reaction conditions, Boc deprotection was not observed (4e), thus demonstrating the mild conditions needed to achieve the arylation.
Table 2. Substrate Scope of Arylated Selenocysteinea.
See the Supporting Information for details. Amino acids are shown in a one-letter code. Yields were determined by integration of TICs from LC–MS analyses of the unpurified reaction mixtures (averages of two runs).
The reaction time was 2 h.
The reaction time was 1.5 h.
2 mM CuSO4, 2 mM L2, and 2 mM boronic acid were used.
2 mM CuSO4, 2 mM L2, 2 mM boronic acid, and 90:10 H2O/DMF for 3 h.
Next we surveyed arylboronic acids with electron-withdrawing functional groups (Table 2, section a). Despite the decreased nucleophilicity of these electron-poor boronic acids, the corresponding selenium conjugates were formed in good to excellent yields. In some cases, the amounts of Cu/L and boronic acid had to be increased to provide full conversion to product while suppressing side reactions. Ester (4g), amide (4h), nitro (4i), nitrile (4j), and halogen (4k, 4l) groups were compatible with these conditions. Peptide 4l was formed quantitatively even though 2,6-difluorophenylboronic acid tends to undergo protodeboronation under basic conditions.21
We next examined the conjugation of heteroaryl substrates (Table 2, section b). The known bioactivities of heterocycles make them an important class of molecules for conjugation to selenocysteine.22 The arylation of five-membered heterocycles such as indole (5a, 5b), benzofuran (5c), pyrazole (5d), 2,5-disubstituted isoxazole (5e), and thiophene (5f, 5g) proceeded with excellent yields. Six-membered heterocycles such as dibenzofuran (5h), phenoxathiine (5i), and pyrimidine (5j) furnished the corresponding conjugates in >90% yield.
To demonstrate the utility of this reaction, biorelevant molecules with pendent boronic acids were investigated (Table 2, section c). The linking of protected phenylalanine and coumarin proceeded in good yield (6a and 6b, respectively). The boronic acids of the anti-inflammatory drugs tolfenamic ester and paracetamol furnished the corresponding conjugates in moderate to good yields (6c and 6d, respectively). Lastly, estrone was coupled to selenium in moderate yield (6e).
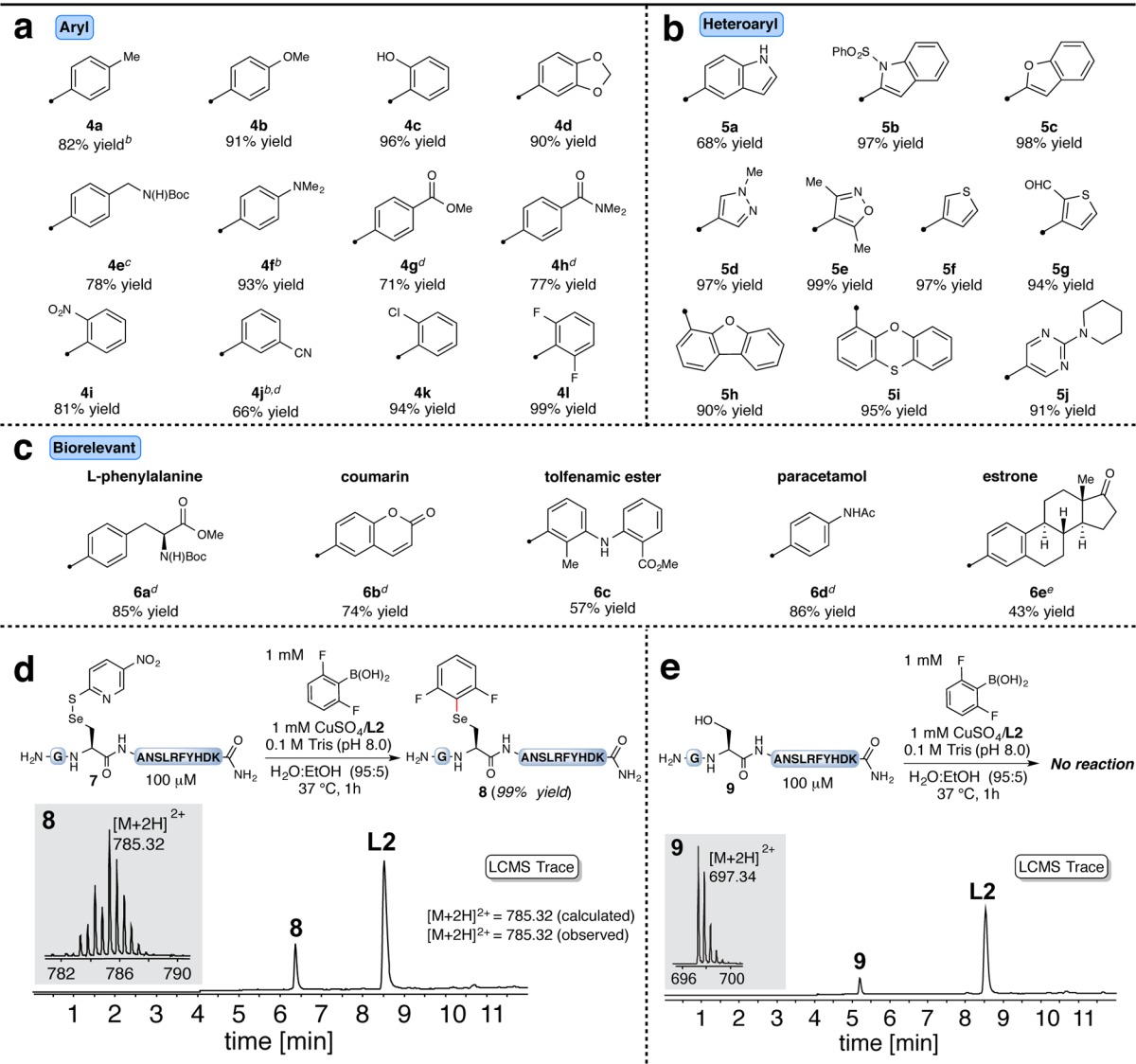
To investigate the robustness and selectivity of this strategy, we prepared peptide 7, which contained most of the key functional groups found in polyamides (e.g., histidine, arginine, aspartic acid, tyrosine, and asparagine; Table 2, section d). Exposure of 7 to our optimized reaction conditions with 2,6-difluorophenylboronic acid as the nucleophile provided the corresponding arylated product 8 in quantitative yield based on LC–MS analysis. No other peptidic species were observed (see the LC–MS trace in section d).23 The arylation occurred exclusively on the selenocysteine, as a Sec-to-Ser control variant (9) provided no product under identical reaction conditions (Table 2, section e).
The stability of these aryl conjugates relative to the corresponding benzylated variants under basic conditions was investigated. In a solution buffered at pH 8.0 at room temperature or 37 °C, neither the arylated nor alkylated selenocysteine peptide underwent elimination to give dehydroalanine (Dha)-containing peptide 1a even after 11 h.18 However, in a solution buffered at pH 10.0, appreciable amounts of elimination were observed for both the alkylated and arylated derivatives depending on the substituent on the selenium.18
We next focused on the stability of these functionalized selenocysteine peptides under oxidative conditions. Exposure of these peptide derivatives to a large excess of H2O2 (10 mM) at room temperature led to the formation of varying amounts of 1a depending on the R group on the selenocysteine (Figure 2). In general, the arylated selenocysteine derivatives were less prone to oxidation/elimination than the benzylated selenocysteine (4n). This result is in agreement with previous reports that alkylated or arylated selenocysteine can be readily converted to Dha even under mildly oxidative conditions.5,11e,12a,12c−12f With regard to the different arenes, the more electron-deficient the aryl ring on the selenium, the slower was the formation of 1a. This observation points to our ability to stabilize the arylated selenocysteine by modulating the electronic nature of the arene substituent.
Figure 2.
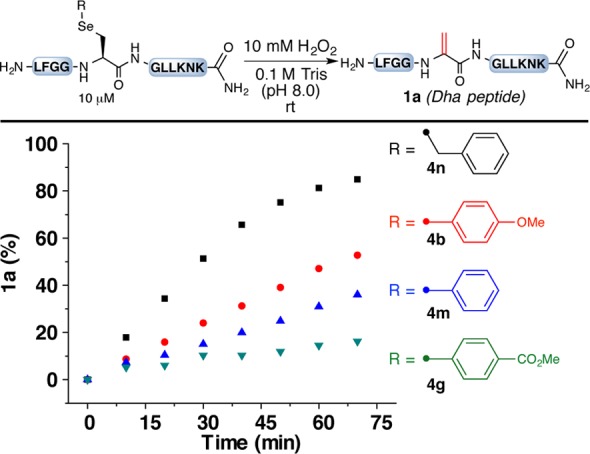
Stability study of functionalized selenocysteine in peptides. Yields were determined by integration of total ion currents from LC–MS analyses of the unpurified reaction mixtures.
In summary, we have developed a chemoselective and general strategy for the synthesis of arylated selenocysteine in unprotected peptides. Exploitation of this umpolung approach avoids the need for oxygen-free conditions and the use of a reducing agent to generate the selenol in situ. Instead, it relies on the electrophilic character of the Se–S bond to provide the respective conjugate. This versatile protocol tolerates a wide range of boronic acids, is compatible with most of the amino acids, is run under buffered aqueous conditions, and is unique to selenocysteine. Lastly, we have demonstrated that varying the substitution on the aryl ring can control the stability of the conjugate toward oxidation/elimination. Although this strategy is effective in peptide-based conjugation, the technology needed to apply this to proteins requires further exploration. Further work to investigate and utilize this umpolung strategy for the selective functionalization of unprotected peptides is ongoing and will be reported in due course.
Acknowledgments
Financial support was provided by NIH Awards GM46059 (S.L.B.), GM108294 (D.T.C.), and GM110535 (B.L.P.) and also by MIT startup funds, a Damon Runyon Cancer Research Foundation Award, and a Sontag Distinguished Scientist Award for B.L.P. C.Z. is a recipient of a George Buchi Summer Research Fellowship and a Koch Graduate Fellowship in Cancer Research from MIT. We thank Dr. Michael Pirnot (MIT) for help in preparing this article and Aldo Rancier (Merck) for Cu ICP-MS analysis of purified peptides 4b, 4m, and 4g. Dr. Alexander M. Spokony (UCLA) is thanked for insightful discussions.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.5b05447.
Procedures and characterization data (PDF)
Author Contributions
† D.T.C. and C.Z. contributed equally.
The authors declare the following competing financial interest(s): MIT has filed a provisional patent based on this technology of which S.L.B, B.L.P, D.T.C., and C.Z. will receive possible royalties.
Supplementary Material
References
- Rotruck J. T.; Pope A. L.; Ganther H. E.; Swanson A. B.; Hafeman D. G.; Hoekstra W. G. Science 1973, 179, 588. 10.1126/science.179.4073.588. [DOI] [PubMed] [Google Scholar]
- Cone J. E.; Del Río R. M.; Davis J. N.; Stadtman T. C. Proc. Natl. Acad. Sci. U. S. A. 1976, 73, 2659. 10.1073/pnas.73.8.2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forstrom J. W.; Zakowski J. J.; Tappel A. L. Biochemistry 1978, 17, 2639. 10.1021/bi00606a028. [DOI] [PubMed] [Google Scholar]
- For reports showing that selenocysteine is encoded by the DNA using a UGA codon, see:; a Chambers I.; Frampton J.; Goldfarb P.; Affara N.; McBain W.; Harrison P. R. EMBO J. 1986, 5, 1221. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Zinoni F.; Birkmann A.; Stadtman T. C.; Böck A. Proc. Natl. Acad. Sci. U. S. A. 1986, 83, 4650. 10.1073/pnas.83.13.4650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metanis N.; Beld J.; Hilvert D. In PATAI’S Chemistry of Functional Groups; Wiley: Chichester, U.K., 2009. [Google Scholar]
- Byun B. J.; Kang Y. K. Biopolymers 2011, 95, 345. 10.1002/bip.21581. [DOI] [PubMed] [Google Scholar]
- Nygard B. Ark. Kemi 1967, 27, 341. [Google Scholar]
- a Moroder L. J. Pept. Sci. 2005, 11, 187. 10.1002/psc.654. [DOI] [PubMed] [Google Scholar]; b Muttenthaler M.; Alewood P. F. J. Pept. Sci. 2008, 14, 1223. 10.1002/psc.1075. [DOI] [PubMed] [Google Scholar]
- Johansson L.; Gafvelin G.; Arnér E. S. J. Biochim. Biophys. Acta, Gen. Subj. 2005, 1726, 1. 10.1016/j.bbagen.2005.05.010. [DOI] [PubMed] [Google Scholar]
- For examples of selenocysteine in native chemical ligation, see:; a Gieselman M. D.; Xie L.; van der Donk W. A. Org. Lett. 2001, 3, 1331. 10.1021/ol015712o. [DOI] [PubMed] [Google Scholar]; b Hondal R. J.; Nilsson B. L.; Raines R. T. J. Am. Chem. Soc. 2001, 123, 5140. 10.1021/ja005885t. [DOI] [PubMed] [Google Scholar]; c Ollivier N.; Blanpain A.; Boll E.; Raibaut L.; Drobecq H.; Melnyk O. Org. Lett. 2014, 16, 4032. 10.1021/ol501866j. [DOI] [PMC free article] [PubMed] [Google Scholar]; For a review of this topic, see:; d Dawson P. E. Isr. J. Chem. 2011, 51, 862. 10.1002/ijch.201100128. [DOI] [Google Scholar]
- a Gieselman M. D.; Zhu Y.; Zhou H.; Galonic D.; van der Donk W. A. ChemBioChem 2002, 3, 709.. [DOI] [PubMed] [Google Scholar]; b Hofer T.; Thomas J. D.; Burke T. R.; Rader C. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 12451. 10.1073/pnas.0800800105. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Hofer T.; Skeffington L. R.; Chapman C. M.; Rader C. Biochemistry 2009, 48, 12047. 10.1021/bi901744t. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Thomas J. D.; Cui H.; North P. J.; Hofer T.; Rader C.; Burke T. R. Bioconjugate Chem. 2012, 23, 2007. 10.1021/bc300052u. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Lin Y. A.; Boutureira O.; Lercher L.; Bhushan B.; Paton R. S.; Davis B. G. J. Am. Chem. Soc. 2013, 135, 12156. 10.1021/ja403191g. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Li X.; Yang J.; Rader C. Methods 2014, 65, 133. 10.1016/j.ymeth.2013.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Hashimoto K.; Sakai M.; Okuno T.; Shirahama H. Chem. Commun. 1996, 1139. 10.1039/cc9960001139. [DOI] [Google Scholar]; b Ma S.; Caprioli R. M.; Hill K. E.; Burk R. F. J. Am. Soc. Mass Spectrom. 2003, 14, 593. 10.1016/S1044-0305(03)00141-7. [DOI] [PubMed] [Google Scholar]; c Seebeck F. P.; Szostak J. W. J. Am. Chem. Soc. 2006, 128, 7150. 10.1021/ja060966w. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Levengood M. R.; van der Donk W. A. Nat. Protocols 2007, 1, 3001. 10.1038/nprot.2006.470. [DOI] [PubMed] [Google Scholar]; e Wang J.; Schiller S. M.; Schultz P. G. Angew. Chem., Int. Ed. 2007, 46, 6849. 10.1002/anie.200702305. [DOI] [PubMed] [Google Scholar]; f Lin S.; He D.; Long T.; Zhang S.; Meng R.; Chen P. R. J. Am. Chem. Soc. 2014, 136, 11860. 10.1021/ja504371w. [DOI] [PubMed] [Google Scholar]
- a Hermanson G. T.Bioconjugate Teachniques; Academic Press: London, 2008. [Google Scholar]; b Chalker J. M.; Bernardes G. J. L.; Lin Y. A.; Davis B. G. Chem. - Asian J. 2009, 4, 630. 10.1002/asia.200800427. [DOI] [PubMed] [Google Scholar]; c Heinis C.; Rutherford T.; Freund S.; Winter G. Nat. Chem. Biol. 2009, 5, 502. 10.1038/nchembio.184. [DOI] [PubMed] [Google Scholar]; d Smeenk L. E. J.; Dailly N.; Hiemstra H.; van Maarseveen J. H.; Timmerman P. Org. Lett. 2012, 14, 1194. 10.1021/ol203259a. [DOI] [PubMed] [Google Scholar]; e Spokoyny A. M.; Zou Y.; Ling J. J.; Yu H.; Lin Y.-S.; Pentelute B. L. J. Am. Chem. Soc. 2013, 135, 5946. 10.1021/ja400119t. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Toda N.; Asano S.; Barbas C. F. Angew. Chem., Int. Ed. 2013, 52, 12592. 10.1002/anie.201306241. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Zhang C.; Spokoyny A. M.; Zou Y.; Simon M. D.; Pentelute B. L. Angew. Chem., Int. Ed. 2013, 52, 14001. 10.1002/anie.201306430. [DOI] [PMC free article] [PubMed] [Google Scholar]; h Cal P. M. S. D.; Bernardes G. J. L.; Gois P. M. P. Angew. Chem., Int. Ed. 2014, 53, 10585. 10.1002/anie.201405702. [DOI] [PubMed] [Google Scholar]
- a Wittig G.; Davis P.; Koenig G. Chem. Ber. 1951, 84, 627. 10.1002/cber.19510840713. [DOI] [Google Scholar]; b Gröbel B.-T.; Seebach D. Synthesis 1977, 357. 10.1055/s-1977-24412. [DOI] [Google Scholar]; c Seebach D. Angew. Chem., Int. Ed. Engl. 1979, 18, 239. 10.1002/anie.197902393. [DOI] [Google Scholar]
- For examples of selenium as an electrophile, see:; a Tiecco M. In Organoselenium Chemistry; Wirth T., Ed.; Springer: Berlin, 2000. [Google Scholar]; b Santi C.; Tidei C. In PATAI’S Chemistry of Functional Groups; Wiley: Chichester, U.K., 2009. [Google Scholar]; c Freudendahl D.; Wirth T. In Selenium and Tellurium Chemistry; Woollins J. D., Laitinen R., Eds.; Springer: Berlin, 2011. [Google Scholar]
- For a review of aerobic Cu-catalyzed reactions and plausible mechanisms, see:; a Allen S. E.; Walvoord R. R.; Padilla-Salinas R.; Kozlowski M. C. Chem. Rev. 2013, 113, 6234. 10.1021/cr300527g. [DOI] [PMC free article] [PubMed] [Google Scholar]; For the first report of a Cu-catalyzed C–Se coupling of diselenides and boronic acids, see:; b Taniguchi N. J. Org. Chem. 2007, 72, 1241. 10.1021/jo062131+. [DOI] [PubMed] [Google Scholar]; For a room-temperature Cu-catalyzed C–Se coupling of aryl diselenides and arylboronic acids, see:; c Zheng B.; Gong Y.; Xu H.-J. Tetrahedron 2013, 69, 5342. 10.1016/j.tet.2013.04.124. [DOI] [Google Scholar]
- Harris K. M.; Flemer S.; Hondal R. J. J. Pept. Sci. 2007, 13, 81. 10.1002/psc.795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- See the Supporting Information (SI) for more details.
- The free cysteine gave disulfide exclusively with <5% arylation. The cysteine linked with TNP gave an inseparable mixture of disulfide and sulfinic acid and <5% arylation. See the SI for more details.
- Attempts to carry out this Cu-mediated arylation on a peptide containing both cysteine and selenocysteine resulted in mostly Se–S intramolecular cross-linking. Further investigation is ongoing.
- Lozada J.; Liu Z.; Perrin D. M. J. Org. Chem. 2014, 79, 5365. 10.1021/jo500734z. [DOI] [PubMed] [Google Scholar]
- Bioactive Heterocyclic Compound Classes: Phamaceuticals; Wiley-VCH: Weinheim, Germany, 2012. [Google Scholar]
- Running this reaction with a 0.1 M phosphate buffer (pH 8.0) furnished the product in 99% yield. Examination of the reaction with a decreased concentration of peptide (10 μM) gave the arylated product in 36% yield. See the SI for details.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.