

Abstract
Rationale: Mutations in bone morphogenetic protein receptor type II (BMPR-II) underlie most cases of heritable pulmonary arterial hypertension (PAH). However, disease penetrance is only 20–30%, suggesting a requirement for additional triggers. Inflammation is emerging as a key disease-related factor in PAH, but to date there is no clear mechanism linking BMPR-II deficiency and inflammation.
Objectives: To establish a direct link between BMPR-II deficiency, a consequentially heightened inflammatory response, and development of PAH.
Methods: We used pulmonary artery smooth muscle cells from Bmpr2+/− mice and patients with BMPR2 mutations and compared them with wild-type controls. For the in vivo model, we used mice heterozygous for a null allele in Bmpr2 (Bmpr2+/−) and wild-type littermates.
Measurements and Main Results: Acute exposure to LPS increased lung and circulating IL-6 and KC (IL-8 analog) levels in Bmpr2+/− mice to a greater extent than in wild-type controls. Similarly, pulmonary artery smooth muscle cells from Bmpr2+/− mice and patients with BMPR2 mutations produced higher levels of IL-6 and KC/IL-8 after lipopolysaccharide stimulation compared with controls. BMPR-II deficiency in mouse and human pulmonary artery smooth muscle cells was associated with increased phospho-STAT3 and loss of extracellular superoxide dismutase. Chronic lipopolysaccharide administration caused pulmonary hypertension in Bmpr2+/− mice but not in wild-type littermates. Coadministration of tempol, a superoxide dismutase mimetic, ameliorated the exaggerated inflammatory response and prevented development of PAH.
Conclusions: This study demonstrates that BMPR-II deficiency promotes an exaggerated inflammatory response in vitro and in vivo, which can instigate development of pulmonary hypertension.
Keywords: pulmonary hypertension, bone morphogenetic protein receptor type II, cytokine, lipopolysaccharide, inflammation
At a Glance Commentary
Scientific Knowledge on the Subject
Inflammation is strongly linked to the pathogenesis of pulmonary arterial hypertension, but to date there is little knowledge regarding potential underlying mechanisms.
What This Study Adds to the Field
We establish a direct link between bone morphogenetic protein receptor type II deficiency (the genetic defect underpinning heritable pulmonary arterial hypertension) and excessive proinflammatory cytokine production, which in turn leads to a pulmonary hypertensive phenotype in a new mouse model. From this we posit that inflammatory and oxidant pathways may be useful to target in the treatment of this deadly disease.
Pulmonary arterial hypertension (PAH) describes a group of rare conditions characterized by increased pulmonary artery pressures, which, if untreated, lead to progressive right ventricular failure and death (1). Idiopathic PAH does not have an identifiable cause. However, in 6–10% of idiopathic PAH cases there is more than one affected family member (2). More than 70% of cases of familial PAH are caused by mutations in the gene BMPR2, encoding bone morphogenetic protein receptor type II (BMPR-II) (3). Subsequent studies identified that mutations in BMPR2 are also found in approximately 10–26% of patients with sporadic PAH (4). This has led to the reclassification of these individuals as having heritable PAH. BMPR-II deficiency has also been demonstrated in cases of idiopathic PAH in the absence of identifiable mutations in BMPR2 (5) and in animal models of PAH (6, 7). This suggests that a critical reduction in BMPR-II function may promote the development of PAH.
Although BMPR2 mutations are a significant predisposing factor for PAH, the majority of gene carriers do not develop the disease. In large families the penetrance has been estimated to be 10–20% (2). Therefore additional factors must be required for initiation or progression of disease in a manner reminiscent of carcinogenesis (8).
There is considerable circumstantial evidence that inflammation plays a role in the pathobiology of PAH (9). For example, subjects with idiopathic or heritable PAH demonstrate higher levels of inflammatory cytokines than do healthy volunteers (10, 11). Elevated levels of several cytokines are associated with higher mortality in PAH (11). PAH is also strongly associated with certain diseases characterized by an inflammatory and/or immune component, such as limited systemic sclerosis, mixed connective tissue disease, and HIV (12, 13). Transgenic IL-6 overexpression exaggerates hypoxia-induced pulmonary hypertension in mice (14), whereas IL-6–deficient mice are protected from hypoxia-driven pulmonary hypertension (15). Adenoviral overexpression of the antiinflammatory cytokine IL-10 (16), or immunosuppression with corticosteroids (17), protects against monocrotaline-induced pulmonary hypertension.
Therefore inflammation is a plausible trigger for the onset of PAH in subjects harboring BMPR2 mutations. Bmpr2+/− mice have little to no elevation of pulmonary artery pressures at baseline (18), which is similar to the majority of people carrying BMPR2 mutations. However, exposure to serotonin (19) is sufficient to instigate pulmonary hypertension. Transgenic mice overexpressing a kinase-domain mutation in BMPR-II spontaneously develop pulmonary hypertension, and this is associated with increased lung levels of IL-6 (20). However, it is not known whether BMPR-II deficiency directly promotes inflammation, or whether inflammation precedes the development of pulmonary hypertension. To explore this theory, we used two in vitro models of BMPR-II deficiency (mouse Bmpr2+/− and human BMPR2mut pulmonary artery smooth muscle cells) and the in vivo model of the Bmpr2+/− mouse. We demonstrate that haploinsufficiency for BMPR-II is associated with increased cytokine expression and activation of phospho-STAT3 (phosphorylated signal transducer and activator of transcription 3) in vitro and in vivo after exposure to LPS. Furthermore, we link BMPR-II deficiency with increased superoxide levels associated with reduced superoxide dismutase 3 (SOD3) levels. Finally, in a new model of pulmonary hypertension caused by chronic, low-grade inflammation in the presence of BMPR-II deficiency, we show that disease is prevented by a superoxide dismutase mimetic.
Methods
Study Animals
Mice heterozygous for a null allele in Bmpr2 generated on a C57BL/6J strain were kindly provided by H. Beppu (18). The LPS used was derived from Escherichia coli O111:B4 (Sigma-Aldrich, St. Louis, MO). For the acute experiments, mice were injected intraperitoneally with LPS (100 μg/kg) and humanely killed at 3- and 24-hour time points. In the chronic administration experiments, mice were injected with LPS (0.5 mg/kg) three times per week and humanely killed after 6 weeks. The prevention arm used tempol (4-hydroxy-2,2,6,6-tetramethylpiperidine 1-oxyl; Sigma-Aldrich) added to the drinking water at a final concentration of 1 mmol/L and provided ad libitum (21). After finishing the treatment course, mice were anesthetized, and right and left heart catheterization was performed and right ventricular indices measured as previously described (22–24). The local animal care committee reviewed and approved all mouse experiments.
Immunohistochemistry and Pulmonary Vascular Morphometry
Lung sections were stained with hematoxylin and eosin and elastic van Gieson (VWR, Radnor, PA) to assess structure and morphology. Pulmonary vascular morphometry was performed as previously described (23) (see the online supplement).
Cytokine Detection
Serum and lung cytokine levels (IL-1β, -2, -4, -6, -8, and -10; tumor necrosis factor-α; and IFN-γ) were measured with the Fluorokine multianalyte profiling system (R&D Systems, Minneapolis, MN).
Tissue Culture Methods
Pulmonary artery smooth muscle cells (PASMCs) were harvested from peripheral pulmonary arteries (<2 mm in diameter) from patients with a mutation in BMPR2 undergoing lung transplantation as previously described (25). The Papworth Hospital (Cambridge, UK) Ethics Committee approved the study and written informed consent was obtained.
Mouse PASMCs were explant-derived from Bmpr2+/− mice and their wild-type littermates (19). The smooth muscle phenotype was confirmed by positive immunostaining for both smooth muscle–specific actin and myosin and negative staining for fibronectin.
Quantitative RT-PCR
RNA was extracted with an RNeasy mini kit (Qiagen, Venlo, the Netherlands) and reverse-transcribed with an Applied Biosystems cDNA reverse transcription kit (Thermo Fisher Scientific, Waltham, MA). Q-PCRs were prepared with SYBR green JumpStart Taq ReadyMix (Sigma-Aldrich). Reactions were amplified on an Applied Biosystems StepOnePlus cycler (Thermo Fisher Scientific) using primers from Qiagen and Sigma-Aldrich. The expression of target messenger RNA (mRNA) was normalized to β-actin, using the ΔΔCT method. Further details are available in the online supplement.
Western Blotting
Mouse and human PASMCs were snap-frozen in radioimmunoprecipitation assay buffer containing an EDTA-free protease inhibitor (Roche, Indianapolis, IN). Cell lysates were fractionated on sodium dodecyl sulfate–polyacrylamide gel electrophoresis gels and immunoblotted. Blots were blocked with 5% nonfat milk and incubated overnight at 4°C with primary antibodies (see the online supplement). Blots were reprobed with anti-tubulin antibody to provide loading controls.
ELISA Quantification of Cytokines in Conditioned Media
Human IL-6, human IL-8, and mouse KC were analyzed with an in-house ELISA (see the online supplement). Mouse IL-6 levels were assayed with the IL-6 DuoSet ELISA (R&D Systems).
Measurement of Reactive Oxygen Species
For quantitative measurement of reactive oxygen species, control and BMPR2mut PASMCs were stained with dihydroethidium, using a method adapted from Owuso-Ansah and colleagues (26) (see the online supplement).
Comet Assay to Measure DNA Damage
To assess levels of DNA damage, we used a single-gel alkaline electrophoretic assay adapted from Nandhakumar and colleagues (27) (see the online supplement).
Statistical Analysis and Power Calculations
Data were tested for normality by the Kolmogorov-Smirnov method. For two-variable comparisons the Mann-Whitney test was used for nonparametric data and the Student's t test was used for parametric data. For sets of data one-way analyses of variance were performed. As the Kolmogorov-Smirnov method is not always accurate in small sample sizes, we repeated the statistical analyses for all figures, using nonparametric methods (i.e., either Mann-Whitney tests to compare two sets of variables or Wilcoxon signed rank tests to compare a variable against a theoretical median) to confirm the validity of the results. In the overwhelming majority of cases the results of the repeat testing agreed with our original analyses. The Spearman test was performed to assess intergroup correlations. Statistical analysis was performed with Prism 5.0 (GraphPad, San Diego, CA). In bar graphs the mean and standard error of the mean are shown. In dot-plots median values are shown. P values not exceeding 0.05 were considered statistically significant.
The current animal welfare regulations set out in the Animals (Scientific Procedures) Act, United Kingdom, dictate that the number of mice used be minimized whenever possible. Power calculations were performed according to Statistical Solutions, LLC (Cottage Grove, WI) (28). Assuming a mean right ventricular systolic pressure (RVSP) of 20 mm Hg in the control mouse with a standard deviation of 5 mm Hg (29), the number of mice required to detect an increase to a mean of 28 mm Hg in the LPS-treated mouse with a power of 90% to a two-sided P value of 0.05 is five. We have assumed an increase of 8 mm Hg as mice exposed to chronic hypoxia (a proven stimulus for the development of pulmonary hypertension) normally show increases in their right ventricular pressures of 8–10 mm Hg (19, 30).
Results
BMPR-II–Deficient Mice Produce More IL-6 and KC after Exposure to LPS
We initially measured serum levels and examined lung mRNA expression of IL-6 and KC in untreated 3- to 4-month-old Bmpr2+/− and wild-type mice (KC is the mouse analog of IL-8). There was no difference in basal cytokine levels, either in sera or in lung tissue, between Bmpr2+/− mice and their wild-type littermates (see Figure E1 in the online supplement). We injected wild-type and Bmpr2+/− mice with a single intraperitoneal dose of LPS at 100 μg/kg. Twenty-four hours after LPS challenge, expression of IL-6 and KC mRNAs was greater in the lungs of Bmpr2+/− mice compared with wild-type mice (Figures 1A and 1B). Serum IL-6 levels were significantly elevated in both Bmpr2+/− mice and wild-type mice 3 hours after LPS challenge, and there were no differences between genotypes. However, serum IL-6 levels were higher in Bmpr2+/− mice 24 hours after LPS exposure (Figure 1C). Serum KC levels were higher in Bmpr2+/− mice than in wild-type mice at 3 hours post-LPS, but levels in both genotypes subsided to baseline by 24 hours (Figure 1D). Because STAT3 is a major signaling intermediary downstream of IL-6 we focused on phospho-STAT3 and found that phospho-STAT3 levels were higher in the lungs of Bmpr2+/− mice after LPS exposure than in wild-type littermates (Figures 1E and 1F). These results confirmed that BMPR-II deficiency promotes an exaggerated cytokine response to acute LPS exposure in vivo.
Figure 1.

Bone morphogenetic protein receptor type II (Bmpr2)–deficient mice produce more IL-6 and KC after exposure to LPS. (A and B) Expression of (A) IL-6 messenger RNA (mRNA) and (B) KC mRNA in the lungs of wild-type (Bmpr2+/+) and Bmpr2+/− mice at baseline and 24 hours after exposure to LPS (100 μg/kg). KC is the mouse analog of IL-8. These data are normalized to the data of a wild-type mouse at baseline. Five to 10 mice have been used per arm. (C and D) Levels of (C) IL-6 and (D) KC in the sera of wild-type and Bmpr2+/− mice at baseline, 3 hours, and 24 hours after exposure to LPS. Seven to 11 mice have been used per arm. (E) Representative immunoblot showing levels of pSTAT3 (phosphorylated signal transducer and activator of transcription 3) and total STAT3 from the lungs of wild-type and Bmpr2+/− mice exposed to LPS. Lungs from four mice have been used per arm. (F) Densitometry for E normalized to total STAT3. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001. NS = not significant.
BMPR-II Deficiency Is Associated with Increased Expression and Release of IL-6 and KC/IL-8 in Mouse Bmpr2+/− and Human BMPR2mut PASMCs
Having demonstrated that haploinsufficiency for BMPR-II increases the IL-6 and KC response to LPS in vivo, we sought to determine whether an exaggerated inflammatory response was also a feature of a major cell type involved in pulmonary vascular remodeling, the pulmonary artery smooth muscle cell (PASMC). Bmpr2+/− PASMCs expressed greater levels of IL-6 and KC mRNAs at baseline and after LPS exposure compared with the wild-type (Figures 2A and 2B). Bmpr2+/− PASMCs also secreted higher levels of IL-6 and KC after LPS exposure compared with wild-type PASMCs (Figures 2C and 2D). Similar results were obtained in human PASMCs derived from patients with PAH caused by a mutation in BMPR-II (BMPR2mut) compared with control cells (Figure 2E). LPS increased STAT3 phosphorylation in both mouse and human BMPR-II–deficient PASMCs compared with wild-type controls (Figures 2F and 2G). This was ameliorated by preincubation with an anti–IL-6 antibody before LPS exposure (Figures 2G and 2H). Furthermore, BMPR2mut PASMCs proliferated in response to IL-6. This response was not seen in control PASMCs (Figure 2I). Treatment with LPS also resulted in a pro-proliferative effect in BMPR2mut PASMCs, which was blocked by coincubation with a neutralizing antibody to IL-6 (Figure E2A). Interestingly, LPS has an antiproliferative effect on control PASMCs. These findings suggest that BMPR-II deficiency in PASMCs promotes the expression and release of IL-6 and KC/IL-8, and that the resulting IL-6 activates STAT3 and drives proliferation of PASMCs.
Figure 2.

Bone morphogenetic protein receptor type II (Bmpr2) deficiency is associated with increased IL-6 and KC/IL-8 production in mouse Bmpr2+/− and human BMPR2mut pulmonary artery smooth muscle cells (PASMCs). (A and B) Expression of (A) IL-6 messenger RNA (mRNA) and (B) KC mRNA in mouse wild-type (Bmpr2+/+) and Bmpr2+/− PASMCs at baseline and after 4 hours of treatment with LPS at 1, 10, and 20 μg/ml. KC is the mouse analog of IL-8. The data are normalized to the data of wild-type cells at baseline. Each arm represents the result of three to nine independent experiments. (C and D) Secretion of (C) IL-6 and (D) KC by mouse PASMCs at baseline and after 8 hours of treatment with LPS (10 μg/ml). Each arm represents the result of four to six independent experiments. (E) Secretion of IL-6 by human PASMCs at baseline and after 8 hours of treatment with LPS (10 μg/ml). Each arm represents the result of five independent experiments. All ELISA data (C–E) are normalized to the number of cells per well. (F) Representative immunoblot for pSTAT3 (phosphorylated signal transducer and activator of transcription 3) in mouse wild-type and Bmpr2+/− PASMCs after 4 hours of treatment with LPS (10 μg/ml). Data represent the result of six independent experiments. (G) Representative immunoblot for pSTAT3 in human wild-type and BMPR2mut PASMCs with and without LPS in the presence and absence of anti-IL-6 antibody. m = BMPR2mut; wt = wild-type. Data represent the result of seven independent experiments. (H) Densitometry for G. (I) Pro-proliferative effect of IL-6 on human BMPR2mut smooth muscle cells. Proliferation rates in response to cytokines are normalized to proliferation rates in Dulbecco’s modified Eagle’s medium with 10% fetal bovine serum (FBS), as BMPR2mut PASMCs grow faster at baseline. The concentration of IL-6 and IL-8 used was 25 ng/ml. Data represent the results of three independent experiments. *P ≤ 0.05. NS = not significant.
The Proinflammatory Response Associated with BMPR-II Deficiency Is Associated with Increased Levels of Reactive Oxygen Species
We next sought to determine the possible mechanisms linking BMPR-II deficiency to heightened production of IL-6 and IL-8. To achieve this, we studied the expression of Toll-like receptor 4 (TLR4) in BMPR-II–deficient PASMCs and Bmpr2+/− mouse lungs, the pattern of IL-10 production in BMPR-II–deficient mice acutely exposed to LPS, and levels of reactive oxygen species (ROS) in BMPR-II–deficient PASMCs, compared with their wild-type counterparts. We measured levels of ROS because LPS-driven production of IL-6 and IL-8 is dependent on ROS (31); and increased levels of ROS are implicated in the development of PAH associated with BMPR-II mutations (32). Interestingly, BMPR-II–deficient PASMCs expressed greater levels of TLR4 mRNA than did wild-type PASMCs (Figure E2B). A similar result was seen when Bmpr2+/− mouse lungs were compared with wild-type mouse lungs (Figure E2C). Bmpr2+/− mice also exhibited lower levels of IL-10 mRNA in their lungs 24 hours after LPS exposure compared with the wild-type (Figure E2D). However, the most striking finding was that BMPR-II–deficient PASMCs exhibited higher levels of superoxide (as determined with dihydroethidium, which reacts with superoxide species to form ethidium, which emits a red fluorescence) after exposure to LPS (Figure 3A). Quantification of this signal, using a separate plate-based assay, confirmed that BMPR-II–deficient PASMCs demonstrate increased levels of superoxide after exposure to LPS. This was partially inhibited by coincubation with bovine SOD (Figure 3B). We therefore employed the membrane-permeable superoxide scavenger tempol to determine whether superoxide radicals are involved. Tempol significantly reduced the LPS-induced release of IL-6 in human BMPR2mut PASMCs (Figure 3C). These results strongly suggested that altered ROS handling in BMPR-II–deficient cells was a major contributor to increased IL-6 production.
Figure 3.
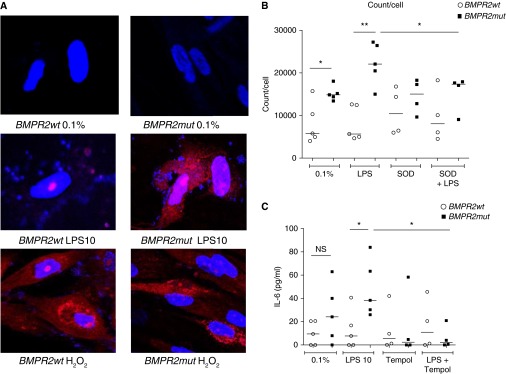
The proinflammatory response associated with bone morphogenetic protein receptor type II (BMPR-II) deficiency is also associated with increased levels of superoxide. (A) Staining for superoxide species in wild-type (BMPR2wt) and BMPR-II mutant (BMPR2mut) pulmonary artery smooth muscle cells (PASMCs) treated with Dulbecco’s modified Eagle’s medium with 0.1% fetal bovine serum (negative control), LPS, or hydrogen peroxide (positive control) with dihydroethidium (pale blue), which reacts with superoxide to form ethidium (red). Slides have been counterstained with 4′,6-diamidino-2-phenylindole (DAPI) to demonstrate the nucleus (bright blue). A is representative of three independent experiments. (B) Quantitation of red light produced by ethidium, using a separate plate-based assay with a luminometer detecting light at 605 nm. Counts have been normalized to the number of cells per well. Each arm represents the result of four or five independent experiments. SOD = superoxide dismutase. (C) Secretion of IL-6 by human BMPR2wt and BMPR2mut PASMCs at baseline and after 8 hours of treatment with LPS (10 μg/ml) in the absence and presence of tempol, a superoxide dismutase mimetic. Each arm represents the result of four or five independent experiments. IL-6 levels have been normalized to the number of cells per well. *P ≤ 0.05, **P ≤ 0.01. NS = not significant.
BMPR-II Deficiency Is Associated with Reduced Superoxide Dismutase 3 Levels
Having identified a potential defect in the ROS-handling mechanisms in BMPR-II mutant cells, we examined the expression of enzymes known to be involved in the generation or removal of ROS; that is, superoxide dismutase isoforms, NADPH oxidases, and catalase (Figure E3). SOD3 mRNA expression was consistently reduced in mouse Bmpr2+/− PASMCs both at baseline and after LPS exposure (Figure 4A), and this was confirmed by reduced SOD3 protein levels in the cell culture supernatant (Figures 4B and 4C). Staining for tubulin was negative, demonstrating that SOD3 had been secreted into the supernatants and was not present simply because of cell death. The loss of SOD3 at both the mRNA and protein levels was also consistently observed in human BMPR2mut PASMCs (Figures 4D–4F). Furthermore, we also observed a loss of SOD3 expression in the lungs of Bmpr2+/− mice at baseline and after exposure to LPS (Figure 4G). If the loss of SOD3 is closely associated with the heightened production of IL-6 one would expect an inverse relationship between levels of SOD3 and levels of IL-6. Therefore we investigated the relationship between SOD3 and IL-6 expression in the lungs of mice exposed to LPS. There was a negative correlation between the expression of SOD3 and IL-6 (r = –0.7, P = 0.05), which was observed only in the Bmpr2+/− mice (Figure 4H). Loss of superoxide dismutase activity is strongly associated with DNA damage and fragmentation (33). Therefore we employed the comet assay (27) to quantify DNA damage in human BMPR2mut PASMCs and wild-type controls. BMPR2mut PASMCs displayed greater levels of DNA damage compared with wild-type controls (Figure E2E).
Figure 4.

Bone morphogenetic protein receptor type II (BMPR-II) deficiency is associated with reduced superoxide dismutase 3 (SOD3). (A) SOD3 expression in mouse wild-type and Bmpr2+/− pulmonary artery smooth muscle cells (PASMCs) after 4 hours of exposure to vehicle or LPS. The data represent the results of five independent experiments. (B) Representative immunoblot showing secreted SOD3 from mouse wild-type and Bmpr2+/− PASMCs after 4 hours of exposure to vehicle or LPS. The data represent the results of three independent experiments. (C) Densitometry for B. (D) SOD3 expression in human wild-type (BMPR2wt) and BMPR-II mutant (BMPR2mut) PASMCs after 4 hours of exposure to vehicle or LPS. The data represent the results of four independent experiments. (E) Representative immunoblot showing secreted SOD3 from human wild-type and BMPR2mut PASMCs after 4 hours of exposure to vehicle or LPS. The data represent the results of three independent experiments. (F) Densitometry for E. (G) SOD3 expression in the lungs of wild-type and Bmpr2+/− mice at baseline and after exposure to LPS. These are normalized to a single randomly chosen heterozygous mouse at baseline. Each arm represents the results from 6 to 10 mice. (H) Correlation between IL-6 and SOD3 expression in the lungs of 10 wild-type and 9 Bmpr2+/− mice exposed to LPS. Spearman’s ρ is shown. (I) Changes in SOD3 expression in mouse PASMCs after exposure for 48 hours to vehicle, LPS, trichostatin A (TSA, 0.4 μM), or LPS + TSA. The data represent the results of five independent experiments. Messenger RNA data are normalized to the wild-type at baseline (A, D, and I). *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001. NS = not significant.
To explore further the relationship between BMPR-II and SOD3, we employed small interfering RNA (siRNA) in human control PASMCs. Despite achieving approximately 80% knockdown of BMPR-II mRNA, SOD3 mRNA levels were unchanged (Figures E4A and E4B). The converse was also true, that is, short-term knockdown of SOD3 did not affect BMPR-II expression (Figures E4C and E4D). Interestingly, short-term knockdown of BMPR-II did not result in an increased IL-6 response to LPS (Figure E5B) when compared with the control arms, which also received LPS (DharmaFECT 1 [GE Healthcare, Pittsburgh, PA] and control pool siRNA). From this we infer that the loss of SOD3, which is associated with the constitutive loss of BMPR-II, is necessary for the proinflammatory phenotype. We reasoned that long-term reduction of BMPR-II expression in vivo may lead to epigenetic repression of SOD3, hence explaining the lack of effect of short-term BMPR2 knockdown. Thus we employed trichostatin A, a histone deacetylase inhibitor, and confirmed that this agent reversed the loss of SOD3 in mouse Bmpr2+/− PASMCs (Figure 4I).
Chronic LPS Administration Promotes Pulmonary Hypertension in BMPR-II–Deficient Mice, Which Is Prevented by Tempol
Having demonstrated that chronic deficiency of BMPR-II in vivo and in vitro promotes IL-6 induction, we questioned whether chronic low-grade inflammation induced by prolonged LPS exposure would cause pulmonary hypertension in BMPR-II–deficient mice. To answer this question, we subjected mice to intraperitoneal injections of LPS (0.5 mg/kg three times weekly for 6 wk) and then performed right and left heart catheterization. Control wild-type and Bmpr2+/− mice had normal right ventricular systolic pressures (RVSPs). Exposure to low-dose LPS for 6 weeks did not alter the RVSPs in wild-type mice whereas Bmpr2+/− mice exhibited a marked increase in RVSP after chronic LPS exposure, which was prevented by coadministration of tempol (Figure 5A). Interestingly, chronic LPS administration increased the right ventricular weight index, Fulton index, and left ventricular weight index in both wild-type and Bmpr2+/− mice (Figures 5B and 5C and Figure E5C). There was no difference in heart rates, left ventricular systolic pressures, or lung BMPR-II levels after exposure to LPS (Figures 5D and 5E, and Figures E5D and E5E). Bmpr2+/− mice exhibited the expected reduction in BMPR-II expression compared with wild-type mice. In addition, STAT3 phosphorylation was increased in the lungs of Bmpr2+/− mice treated with LPS and ameliorated by tempol (Figures 5F and 5G). Furthermore, a loss of SOD3 was observed only in Bmpr2+/− mice exposed to LPS (Figures 5H and 5I). However, we did not find a persistent increase in IL-6 and KC in the lungs or serum of wild-type or Bmpr2+/− mice (Figures E5F–E5I). Histology and pulmonary artery morphometry confirmed an increase in pulmonary vascular remodeling only in Bmpr2+/− mice (Figures 6A and 6B). Figure 6C provides a close-up of smooth muscle actin and elastic van Gieson staining in Bmpr2+/− mouse lungs treated with LPS alone and with LPS and tempol. This indicates that much of the increased wall thickness in the small pulmonary arteries of Bmpr2+/− mice treated with LPS is due to an increased smooth muscle component; and that this is ameliorated by tempol. Ki67 staining also showed an increase in the number of smooth muscle cells undergoing proliferation in Bmpr2+/− mouse lungs treated with LPS.
Figure 5.

Chronic LPS administration promotes pulmonary hypertension in bone morphogenetic protein receptor type II (Bmpr2)–deficient mice, which is prevented by tempol. (A) Right ventricular systolic pressures, (B) right ventricular weight index, (C) Fulton index [right ventricular weight/(left ventricular + septal weight)], and (D) heart rate in wild-type and Bmpr2+/− mice exposed to LPS in the absence and presence of tempol. (E) Left ventricular systolic pressures in wild-type and Bmpr2+/− mice exposed to LPS. For A–E, each arm represents the results from 5 to 15 mice. (F) Representative immunoblot showing p-STAT3 (phosphorylated signal transducer and activator of transcription 3) and total STAT3 levels in lungs from control and LPS-exposed wild-type and Bmpr2+/− mice, with (G) densitometry. Each arm represents the results from four to seven mice. (H) Representative immunoblot showing superoxide dismutase 3 (SOD3) levels in lungs from control and LPS-exposed wild-type and Bmpr2+/− mice, with (I) densitometry. The data represent results from four different mice per arm. *P ≤ 0.05, **P ≤ 0.01. LV = left ventricular weight; LVSP = left ventricular systolic pressure; NS = not significant; RV = right ventricular weight; RVSP = right ventricular systolic pressure; S = septum.
Figure 6.
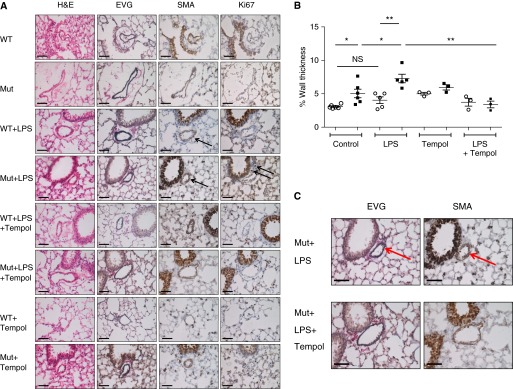
Immunohistochemical and morphological features of LPS-induced pulmonary hypertension. (A) Immunohistochemical studies on lung sections containing at least one distal pulmonary artery from control wild-type mice (WT), control mice heterozygous for a null allele in bone morphogenetic protein receptor (Bmpr2+/− mice) (Mut), wild-type mice receiving LPS for 6 weeks (WT+LPS), Bmpr2+/− mice receiving LPS for 6 weeks (Mut+LPS), wild-type mice receiving LPS and tempol for 6 weeks (WT+LPS+Tempol), Bmpr2+/− mice receiving LPS and tempol for 6 weeks (Mut+LPS+Tempol), wild-type mice receiving tempol for 6 weeks (WT+Tempol), and Bmpr2+/− mice receiving tempol for 6 weeks (Mut+Tempol). Shown are hematoxylin and eosin (H&E), elastic van Gieson (EVG), smooth muscle actin (SMA), and Ki67 staining. Black arrows indicate positively stained cells for SMA and Ki67. Scale bars, 50 μm. The data represent results from four to six different mice per arm. (B) Morphometry showing increased muscularization in pulmonary arteries of Bmpr2+/− mice compared with wild-type controls. This difference is further exaggerated by the administration of chronic LPS and ameliorated by coadministration of tempol. The data represent results from three to six different mice per arm. (C) Close-up of SMA and EVG staining in Bmpr2+/− mice receiving LPS (Mut+LPS) and Bmpr2+/− mice receiving LPS and tempol for 6 weeks (Mut+LPS+Tempol). The majority of the thickened walls of the small arteries of the Bmpr2+/− mice exposed to LPS are due to hypertrophy of the medial smooth muscle cells as indicated by increased smooth muscle (indicated with a red arrow and stained brown by SMA). The EVG stains the elastic fibers comprising the inner and outer elastic lamina (stained blue/black and indicated with a red arrow) and highlights the medial layer of the vessel. *P ≤ 0.05, **P 0.01. NS = not significant.
Discussion
We demonstrate for the first time that BMPR-II deficiency promotes a proinflammatory phenotype, which leads to pulmonary vascular remodeling. Specifically, BMPR-II deficiency increased LPS-stimulated IL-6 and IL-8 production, which appeared to be partly mediated by SOD3 deficiency. The proinflammatory effect of BMPR-II deficiency was confirmed in two in vitro models: in human PASMCs with a disease-causing mutation in BMPR-II (R899X) and in PASMCs from mice with BMPR-II haploinsufficiency. Importantly, in the latter setting, the PASMCs were derived from mice that showed no overt evidence of PAH. This effectively excludes secondary confounding effects of preexisting PAH. The first model is important as it involves a disease-causing mutation that affects patients.
Previous studies have provided evidence linking BMP pathways and inflammation. For example, a negative feedback loop was demonstrated between IL-6 and BMP signaling in vitro (34). PASMCs deficient in BMPR-II are also insensitive to the growth-inhibitory effect of transforming growth factor-β. This resistance is linked to increased production of IL-6 and IL-8, as the normal growth-inhibitory effect of transforming growth factor-β is restored by coincubation with neutralizing antibodies to either IL-6 or IL-8 (35). In addition, siRNA-mediated knockdown of BMPR-II increased monocyte adhesion and induced intercellular adhesion molecule-1 and vascular cell adhesion molecule-1 levels in human umbilical vein endothelial cells (36). Burton and colleagues (37) showed enhanced leukocyte transmigration through BMPR-II–deficient endothelial cells compared with mock-transfected controls. The results of our study are consistent with and extend these findings, as we show that BMPR-II loss is associated with increased inflammatory cytokine production both systemically and in the lung; and that this effect is mediated by increased superoxide species and reduced SOD3 expression. Interestingly, this does not seem to be mediated by canonical BMP signaling, as lung phosphorylated Smad (p-Smad) 1/5/8 levels are unaffected by LPS administration in both wild-type and BMPR2-deficient animals. This fits in with previous published reports which show that LPS treatment per se does not alter the p-Smad 1/5/8 response; but LPS can and does affect p-Smad 1/5/8 when cotreated with BMPs (38, 39). This implies that perhaps the LPS-induced cytokine production pathways (TLR4/MyD88) do not involve p-Smad 1/5/8 directly, but that resulting downstream activation of pathways such as NF-κB interact with BMP signaling. We further show that a persistent low-grade inflammatory insult can induce a pulmonary hypertensive phenotype in the context of BMPR-II deficiency.
Interestingly, the proinflammatory effect of constitutive BMPR-II haploinsufficiency could not be mimicked by short-term BMPR-II knockdown. This implies that long-term BMPR-II deficiency is required to establish the proinflammatory phenotype. A potential mechanism is epigenetic modification of the promoter regions of genes as a consequence of chronic loss of BMPR-II. We demonstrated that the loss of SOD3 expression in Bmpr2+/− cells was reversed by incubation with the histone deacetylase inhibitor trichostatin A. Previous studies have shown that SOD3 is epigenetically regulated in PASMCs via histone deacetylation (40). Studies have shown that histone deacetylase inhibitors can prevent and reverse hypoxia-induced pulmonary hypertension (41). However, the precise mechanism for the loss of SOD3 in association with reduced BMPR-II remains to be established.
SOD3 is the principal extracellular superoxide dismutase and is highly expressed in the lung (42). Rats with a loss-of-function SOD3 mutation demonstrate increased right ventricular pressures compared with wild-type animals after exposure to either hypoxia or monocrotaline (43). Moreover, SOD3 overexpression in the lungs of mice attenuates hypoxia-induced pulmonary hypertension (44). These studies strongly support the possibility that the SOD3 deficiency observed in our study is contributing to pulmonary hypertension in the setting of BMPR-II deficiency.
We have also explored, for the first time, chronic LPS exposure in BMPR-II–deficient mice as a model of PAH. LPS represents a biologically plausible initiating inflammatory stimulus for PAH. TLR4, the main receptor for LPS, is up-regulated in the PASMCs and lungs of BMPR-II–deficient mice, and TLR4 has been shown previously to promote the development of PAH (45). A link between TLR4 and BMPR-II has not previously been demonstrated. The LPS-exposed BMPR-II–deficient mice developed markedly elevated right ventricular systolic pressures, which did not occur in the wild-type. We note that only the BMPR-II–deficient mice develop significant pulmonary artery muscularization at baseline, and that this was further exaggerated after chronic exposure to LPS. Nevertheless, both wild-type and BMPR-II heterozygotes exposed to LPS developed right and left ventricular hypertrophy. Thus the cardiac response in this model appears to be related to LPS exposure. We suggest that increased STAT3 activation in the lungs of BMPR-II–deficient mice drives the process of vascular remodeling. The resulting combination of right ventricular hypertrophy and increased pulmonary vascular resistance is the reason for the pulmonary hypertensive phenotype. Pulmonary STAT3 activation has been shown to be associated with increased PASMC and pulmonary artery endothelial cell proliferation, vascular epidermal growth factor production, and disordered angiogenesis (46). We note that the doses of LPS used in our experiments were much lower than those used in models of left ventricular dysfunction. For example, we used LPS at 0.5 mg/kg compared with 6–25 mg/kg in studies examining the effect of LPS on the left ventricle (47, 48). Also, left ventricular systolic pressures were unchanged and none of the mice died unexpectedly before the planned time of sacrifice. Furthermore, there were no noticeable emphysematous changes or abnormalities in the alveolar spaces or alveolar wall thickness in any of the groups. LPS challenge has been previously used to induce a chronic obstructive pulmonary disease–like phenotype in mice, but these studies have been conducted primarily with endotracheal LPS at a dose greater than used in these experiments (49).
Although we demonstrated increased IL-6 and IL-8 expression and release after acute LPS exposure in Bmpr2+/− mice, we were unable to detect elevated cytokine expression after 6 weeks of LPS, despite evidence of continued STAT3 activation. Repeated exposure to LPS induces endotoxin tolerance, which is associated with reduced inflammatory cytokine production (50). For example, Sun and colleagues demonstrated that pretreatment of macrophages with E. coli LPS decreased tumor necrosis factor-α production from 1,700 to 100 pg/ml (51). Similar effects were observed in vivo (52). Therefore, it is likely that a similar hyporesponsiveness to LPS developed in our studies.
The comet assay that we used to examine DNA damage has a number of drawbacks—the most important being that it represents a snapshot of the amount of DNA breaks present at a specific point in time but does not specifically indicate the fate of the cell—that is, whether it will undergo apoptosis or whether it will continue to divide in an uncontrolled fashion and endow its progeny with an increasing burden of damaged DNA. We suspect the latter as Meloche and colleagues (53) have independently shown that distal pulmonary arteries and PASMCs from patients with PAH exhibit not only increased DNA damage but also increased poly(ADP-ribose) polymerase-1, a trigger of DNA repair; which leads to a pro-proliferative and antiapoptotic phenotype. Li and colleagues (54) have demonstrated a similar phenotype of altered DNA repair and loss of genomic stability in pulmonary artery endothelial cells. Furthermore, accumulation of DNA damage led to a significant down-regulation of BMPR2 which may contribute to the PAH phenotype.
It is true that not all patients with connective tissue disease or HIV develop PAH (12, 13), and not all patients with idiopathic or heritable PAH have a higher burden of inflammatory cytokines (11). It may be that inflammation is a trigger (a “first hit”) and is then not required for progression of disease; or an inflammatory environment may permit progression of PAH (a “second hit” [9]). There is some circumstantial evidence supporting this; as increased IL-6 levels are specifically associated with pulmonary hypertension in patients with systemic sclerosis (55) and in those with COPD (56). The most attractive feature of the “multiple-hit” hypothesis is that inflammation represents a modifiable risk factor, and that if PAH does require “multiple hits” to progress, perhaps modifying one of them would be sufficient to delay or even stop its progress. It was shown that patients with scleroderma and PAH have reduced expression of BMPR2 (57). Thus it is plausible, even in the absence of BMPR2 mutations, that suppression of BMPR-II levels, by whatever mechanism, may trigger disease by allowing an abnormal response of PASMCs to inflammatory stimuli.
Our working hypothesis is summarized in Figure 7. It is proposed that BMPR-II deficiency and the associated increase in TLR4 and loss of SOD3 result in a greater susceptibility to inflammatory stimuli. In the presence of such stimuli, levels of IL-6 and IL-8 are increased and levels of IL-10 are reduced. This contributes to an abnormal proliferative response in PASMCs and increased STAT3 signaling. Uncontrolled inflammation and increased oxidative stress are also linked to a loss of endothelial barrier integrity (37), dysregulation of the nitric oxide synthase system leading to abnormal vasoconstriction (58), and excessive DNA damage (53). It is likely that a combination of these mechanisms leads to the pulmonary hypertensive phenotype. We have not identified the specific details by which BMPR-II deficiency leads to the proinflammatory effect, and we view this study as a “gate opener” that invites further examination of these mechanisms. Our findings also suggest that epigenetic modification of gene promoters involved in these pathways would be worthy of further investigation.
Figure 7.

Proposed sequence of events arising from loss of bone morphogenetic protein receptor type II (BMPR-II). When pulmonary artery smooth muscle cells (PASMCs) deficient in BMPR-II face a proinflammatory insult, they react by producing abnormally high levels of IL-6 and IL-8, and abnormally low levels of IL-10. The concomitant reduction in superoxide dismutase 3 (SOD3) associated with BMPR-II deficiency also predisposes these PASMCs to increased oxidative stress. The net result of this abnormal proinflammatory response is a sustained increase in STAT3 (signal transducer and activator of transcription 3) signaling, increased proliferation of PASMCs, and accumulation of DNA damage within the PASMCs. We hypothesize that this sequence of events leads to pulmonary artery muscularization and a pulmonary hypertensive phenotype. PAH = pulmonary arterial hypertension; TLR4 = Toll-like receptor 4.
Finally, our study also supports the possibility that specific antiinflammatory or antioxidant strategies might be worth pursuing in the clinical setting. The current therapies licensed for PAH (59) focus on vasodilation. Reports have suggested that anti–IL-6 therapy might be of value in PAH, especially in connective tissue disease (60). Dietary antioxidants such as tempol may have therapeutic potential in patients with BMPR2 mutations because these patients have evidence of greater than normal oxidative injury (32). Histone deacetylase inhibition, as a means of up-regulating SOD3 expression, is also a potential therapeutic avenue (41).
In conclusion, this work demonstrates a direct link between BMPR-II deficiency and a proinflammatory phenotype, which predisposes to pulmonary hypertension. We also describe for the first time an LPS-induced model for PAH in the setting of BMPR-II deficiency. It is likely that multiple mechanisms are involved, including reduced superoxide metabolism due to loss of SOD3, which may be due to epigenetic silencing. These findings support the pursuit of antiinflammatory and antioxidant strategies in the prevention and treatment of disease.
Acknowledgments
Acknowledgment
John Deighton developed the in-house ELISA protocols. Special thanks to the patients and staff at Papworth Hospital for donating their time, effort, and tissue.
Footnotes
Supported by a Medical Research Council training fellowship and Sackler scholarship to E.S., a Wellcome Trust fellowship to P.Y., a National Institute for Health Research (NIHR) Healthcare Scientist fellowship to M.S., a British Heart Foundation program grant to N.W.M., and a Fondation Leducq network award to K.D.B. and N.W.M. Infrastructure support was provided by the Cambridge NIHR Biomedical Research Centre.
Author Contributions: E.S. designed and performed experiments, analyzed and interpreted data, and wrote the manuscript. A.C. performed experiments, analyzed and interpreted data, and helped write the manuscript. M.S., P.Y., T.T., M.T., and S.A. performed experiments and analyzed and interpreted data. C.M.S., K.D.B., J.P.-Z., P.U., and N.W.M. planned experiments, interpreted data, and helped write the manuscript. All authors were involved in reading and critically revising the manuscript.
This article has an online supplement, which is accessible from this issue’s table of contents at www.atsjournals.org
Originally Published in Press as DOI: 10.1164/rccm.201408-1509OC on June 13, 2015
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.Simonneau G, Gatzoulis MA, Adatia I, Celermajer D, Denton C, Ghofrani A, Gomez Sanchez MA, Krishna Kumar R, Landzberg M, Machado RF, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2013;62(25) Suppl:D34–D41. doi: 10.1016/j.jacc.2013.10.029. [DOI] [PubMed] [Google Scholar]
- 2.Newman JH, Wheeler L, Lane KB, Loyd E, Gaddipati R, Phillips JA, III, Loyd JE. Mutation in the gene for bone morphogenetic protein receptor II as a cause of primary pulmonary hypertension in a large kindred. N Engl J Med. 2001;345:319–324. doi: 10.1056/NEJM200108023450502. [DOI] [PubMed] [Google Scholar]
- 3.Lane KB, Machado RD, Pauciulo MW, Thomson JR, Phillips JA, III, Loyd JE, Nichols WC, Trembath RC International PPH Consortium. Heterozygous germline mutations in BMPR2, encoding a TGF-β receptor, cause familial primary pulmonary hypertension. Nat Genet. 2000;26:81–84. doi: 10.1038/79226. [DOI] [PubMed] [Google Scholar]
- 4.Machado RD, Aldred MA, James V, Harrison RE, Patel B, Schwalbe EC, Gruenig E, Janssen B, Koehler R, Seeger W, et al. Mutations of the TGF-β type II receptor BMPR2 in pulmonary arterial hypertension. Hum Mutat. 2006;27:121–132. doi: 10.1002/humu.20285. [DOI] [PubMed] [Google Scholar]
- 5.Atkinson C, Stewart S, Upton PD, Machado R, Thomson JR, Trembath RC, Morrell NW. Primary pulmonary hypertension is associated with reduced pulmonary vascular expression of type II bone morphogenetic protein receptor. Circulation. 2002;105:1672–1678. doi: 10.1161/01.cir.0000012754.72951.3d. [DOI] [PubMed] [Google Scholar]
- 6.Takahashi H, Goto N, Kojima Y, Tsuda Y, Morio Y, Muramatsu M, Fukuchi Y. Downregulation of type II bone morphogenetic protein receptor in hypoxic pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2006;290:L450–L458. doi: 10.1152/ajplung.00206.2005. [DOI] [PubMed] [Google Scholar]
- 7.Morty RE, Nejman B, Kwapiszewska G, Hecker M, Zakrzewicz A, Kouri FM, Peters DM, Dumitrascu R, Seeger W, Knaus P, et al. Dysregulated bone morphogenetic protein signaling in monocrotaline-induced pulmonary arterial hypertension. Arterioscler Thromb Vasc Biol. 2007;27:1072–1078. doi: 10.1161/ATVBAHA.107.141200. [DOI] [PubMed] [Google Scholar]
- 8.Bignold LP, Coghlan BL, Jersmann HP. Cancer morphology, carcinogenesis and genetic instability: a background. EXS. 2006;96:1–24. doi: 10.1007/3-7643-7378-4_1. [DOI] [PubMed] [Google Scholar]
- 9.Price LC, Wort SJ, Perros F, Dorfmüller P, Huertas A, Montani D, Cohen-Kaminsky S, Humbert M. Inflammation in pulmonary arterial hypertension. Chest. 2012;141:210–221. doi: 10.1378/chest.11-0793. [DOI] [PubMed] [Google Scholar]
- 10.Humbert M, Monti G, Brenot F, Sitbon O, Portier A, Grangeot-Keros L, Duroux P, Galanaud P, Simonneau G, Emilie D. Increased interleukin-1 and interleukin-6 serum concentrations in severe primary pulmonary hypertension. Am J Respir Crit Care Med. 1995;151:1628–1631. doi: 10.1164/ajrccm.151.5.7735624. [DOI] [PubMed] [Google Scholar]
- 11.Soon E, Holmes AM, Treacy CM, Doughty NJ, Southgate L, Machado RD, Trembath RC, Jennings S, Barker L, Nicklin P, et al. Elevated levels of inflammatory cytokines predict survival in idiopathic and familial pulmonary arterial hypertension. Circulation. 2010;122:920–927. doi: 10.1161/CIRCULATIONAHA.109.933762. [DOI] [PubMed] [Google Scholar]
- 12.Fagan KA, Badesch DB. Pulmonary hypertension associated with connective tissue disease. Prog Cardiovasc Dis. 2002;45:225–234. doi: 10.1053/pcad.2002.129975. [DOI] [PubMed] [Google Scholar]
- 13.Quezada M, Martin-Carbonero L, Soriano V, Vispo E, Valencia E, Moreno V, de Isla LP, Lennie V, Almería C, Zamorano JL. Prevalence and risk factors associated with pulmonary hypertension in HIV-infected patients on regular follow-up. AIDS. 2012;26:1387–1392. doi: 10.1097/QAD.0b013e328354f5a1. [DOI] [PubMed] [Google Scholar]
- 14.Steiner MK, Syrkina OL, Kolliputi N, Mark EJ, Hales CA, Waxman AB. Interleukin-6 overexpression induces pulmonary hypertension. Circ Res. 2009;104:236–244, 28p, 244. doi: 10.1161/CIRCRESAHA.108.182014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Savale L, Tu L, Rideau D, Izziki M, Maitre B, Adnot S, Eddahibi S. Impact of interleukin-6 on hypoxia-induced pulmonary hypertension and lung inflammation in mice. Respir Res. 2009;10:6. doi: 10.1186/1465-9921-10-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ito T, Okada T, Miyashita H, Nomoto T, Nonaka-Sarukawa M, Uchibori R, Maeda Y, Urabe M, Mizukami H, Kume A, et al. Interleukin-10 expression mediated by an adeno-associated virus vector prevents monocrotaline-induced pulmonary arterial hypertension in rats. Circ Res. 2007;101:734–741. doi: 10.1161/CIRCRESAHA.107.153023. [DOI] [PubMed] [Google Scholar]
- 17.Price LC, Montani D, Tcherakian C, Dorfmüller P, Souza R, Gambaryan N, Chaumais MC, Shao DM, Simonneau G, Howard LS, et al. Dexamethasone reverses monocrotaline-induced pulmonary arterial hypertension in rats. Eur Respir J. 2011;37:813–822. doi: 10.1183/09031936.00028310. [DOI] [PubMed] [Google Scholar]
- 18.Beppu H, Ichinose F, Kawai N, Jones RC, Yu PB, Zapol WM, Miyazono K, Li E, Bloch KD. BMPR-II heterozygous mice have mild pulmonary hypertension and an impaired pulmonary vascular remodeling response to prolonged hypoxia. Am J Physiol Lung Cell Mol Physiol. 2004;287:L1241–L1247. doi: 10.1152/ajplung.00239.2004. [DOI] [PubMed] [Google Scholar]
- 19.Long L, MacLean MR, Jeffery TK, Morecroft I, Yang X, Rudarakanchana N, Southwood M, James V, Trembath RC, Morrell NW. Serotonin increases susceptibility to pulmonary hypertension in BMPR2-deficient mice. Circ Res. 2006;98:818–827. doi: 10.1161/01.RES.0000215809.47923.fd. [DOI] [PubMed] [Google Scholar]
- 20.Tada Y, Majka S, Carr M, Harral J, Crona D, Kuriyama T, West J. Molecular effects of loss of BMPR2 signaling in smooth muscle in a transgenic mouse model of PAH. Am J Physiol Lung Cell Mol Physiol. 2007;292:L1556–L1563. doi: 10.1152/ajplung.00305.2006. [DOI] [PubMed] [Google Scholar]
- 21.Jerkic M, Kabir MG, Davies A, Yu LX, McIntyre BA, Husain NW, Enomoto M, Sotov V, Husain M, Henkelman M, et al. Pulmonary hypertension in adult Alk1 heterozygous mice due to oxidative stress. Cardiovasc Res. 2011;92:375–384. doi: 10.1093/cvr/cvr232. [DOI] [PubMed] [Google Scholar]
- 22.Lorenz JN, Robbins J. Measurement of intraventricular pressure and cardiac performance in the intact closed-chest anesthetized mouse. Am J Physiol. 1997;272:H1137–H1146. doi: 10.1152/ajpheart.1997.272.3.H1137. [DOI] [PubMed] [Google Scholar]
- 23.Morrell NW, Atochina EN, Morris KG, Danilov SM, Stenmark KR. Angiotensin converting enzyme expression is increased in small pulmonary arteries of rats with hypoxia-induced pulmonary hypertension. J Clin Invest. 1995;96:1823–1833. doi: 10.1172/JCI118228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schermuly RT, Dony E, Ghofrani HA, Pullamsetti S, Savai R, Roth M, Sydykov A, Lai YJ, Weissmann N, Seeger W, et al. Reversal of experimental pulmonary hypertension by PDGF inhibition. J Clin Invest. 2005;115:2811–2821. doi: 10.1172/JCI24838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang X, Long L, Southwood M, Rudarakanchana N, Upton PD, Jeffery TK, Atkinson C, Chen H, Trembath RC, Morrell NW. Dysfunctional Smad signaling contributes to abnormal smooth muscle cell proliferation in familial pulmonary arterial hypertension. Circ Res. 2005;96:1053–1063. doi: 10.1161/01.RES.0000166926.54293.68. [DOI] [PubMed] [Google Scholar]
- 26.Owuso-Ansah E, Yavari A, Banerjee U.A protocol for in vivo detection of reactive oxygen species [Protocol Exchange/Community Contributed]. 2008 Feb 27 [accessed 2014 Aug 18]. Available from: http://www.nature.com/protocolexchange/protocols/414#/anticipated_results
- 27.Nandhakumar S, Parasuraman S, Shanmugam MM, Rao KR, Chand P, Bhat BV. Evaluation of DNA damage using single-cell gel electrophoresis (Comet Assay) J Pharmacol Pharmacother. 2011;2:107–111. doi: 10.4103/0976-500X.81903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Statistical Solutions, LLC. Power & sample size calculator [accessed 2015 May 31]. Available from: http://www.statisticalsolutions.net/pss_calc.php
- 29.Crosby A, Jones FM, Kolosionek E, Southwood M, Purvis I, Soon E, Butrous G, Dunne DE, Morrell NW. Praziquantel reverses pulmonary hypertension and vascular remodeling in murine schistosomiasis. Am J Respir Crit Care Med. 2011;184:467–473. doi: 10.1164/rccm.201101-0146OC. [DOI] [PubMed] [Google Scholar]
- 30.Zhao L, Long L, Morrell NW, Wilkins MR. NPR-A-Deficient mice show increased susceptibility to hypoxia-induced pulmonary hypertension. Circulation. 1999;99:605–607. doi: 10.1161/01.cir.99.5.605. [DOI] [PubMed] [Google Scholar]
- 31.Bulua AC, Simon A, Maddipati R, Pelletier M, Park H, Kim KY, Sack MN, Kastner DL, Siegel RM. Mitochondrial reactive oxygen species promote production of proinflammatory cytokines and are elevated in TNFR1-associated periodic syndrome (TRAPS) J Exp Med. 2011;208:519–533. doi: 10.1084/jem.20102049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lane KL, Talati M, Austin E, Hemnes AR, Johnson JA, Fessel JP, Blackwell T, Mernaugh RL, Robinson L, Fike C, et al. Oxidative injury is a common consequence of BMPR2 mutations. Pulm Circ. 2011;1:72–83. doi: 10.4103/2045-8932.78107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Muid KA, Karakaya HC, Koc A. Absence of superoxide dismutase activity causes nuclear DNA fragmentation during the aging process. Biochem Biophys Res Commun. 2014;444:260–263. doi: 10.1016/j.bbrc.2014.01.056. [DOI] [PubMed] [Google Scholar]
- 34.Hagen M, Fagan K, Steudel W, Carr M, Lane K, Rodman DM, West J. Interaction of interleukin-6 and the BMP pathway in pulmonary smooth muscle. Am J Physiol Lung Cell Mol Physiol. 2007;292:L1473–L1479. doi: 10.1152/ajplung.00197.2006. [DOI] [PubMed] [Google Scholar]
- 35.Davies RJ, Holmes AM, Deighton J, Long L, Yang X, Barker L, Walker C, Budd DC, Upton PD, Morrell NW. BMP type II receptor deficiency confers resistance to growth inhibition by TGF-β in pulmonary artery smooth muscle cells: role of proinflammatory cytokines. Am J Physiol Lung Cell Mol Physiol. 2012;302:L604–L615. doi: 10.1152/ajplung.00309.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim CW, Song H, Kumar S, Nam D, Kwon HS, Chang KH, Son DJ, Kang DW, Brodie SA, Weiss D, et al. Anti-inflammatory and antiatherogenic role of BMP receptor II in endothelial cells. Arterioscler Thromb Vasc Biol. 2013;33:1350–1359. doi: 10.1161/ATVBAHA.112.300287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Burton VJ, Ciuclan LI, Holmes AM, Rodman DM, Walker C, Budd DC. Bone morphogenetic protein receptor II regulates pulmonary artery endothelial cell barrier function. Blood. 2011;117:333–341. doi: 10.1182/blood-2010-05-285973. [DOI] [PubMed] [Google Scholar]
- 38.Huang RL, Yuan Y, Zou GM, Liu G, Tu J, Li Q. LPS-stimulated inflammatory environment inhibits BMP-2-induced osteoblastic differentiation through crosstalk between TLR4/MyD88/NF-κB and BMP/Smad signaling. Stem Cells Dev. 2014;23:277–289. doi: 10.1089/scd.2013.0345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu X, Yung LM, Cheng WH, Yu PB, Babitt JL, Lin HY, Xia Y. Hepcidin regulation by BMP signaling in macrophages is lipopolysaccharide dependent. PLoS One. 2012;7:e44622. doi: 10.1371/journal.pone.0044622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zelko IN, Stepp MW, Vorst AL, Folz RJ. Histone acetylation regulates the cell-specific and interferon-γ-inducible expression of extracellular superoxide dismutase in human pulmonary arteries. Am J Respir Cell Mol Biol. 2011;45:953–961. doi: 10.1165/rcmb.2011-0012OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhao L, Chen CN, Hajji N, Oliver E, Cotroneo E, Wharton J, Wang D, Li M, McKinsey TA, Stenmark KR, et al. Histone deacetylation inhibition in pulmonary hypertension: therapeutic potential of valproic acid and suberoylanilide hydroxamic acid. Circulation. 2012;126:455–467. doi: 10.1161/CIRCULATIONAHA.112.103176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Folz RJ, Crapo JD. Extracellular superoxide dismutase (SOD3): tissue-specific expression, genomic characterization, and computer-assisted sequence analysis of the human EC SOD gene. Genomics. 1994;22:162–171. doi: 10.1006/geno.1994.1357. [DOI] [PubMed] [Google Scholar]
- 43.Xu D, Guo H, Xu X, Lu Z, Fassett J, Hu X, Xu Y, Tang Q, Hu D, Somani A, et al. Exacerbated pulmonary arterial hypertension and right ventricular hypertrophy in animals with loss of function of extracellular superoxide dismutase. Hypertension. 2011;58:303–309. doi: 10.1161/HYPERTENSIONAHA.110.166819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nozik-Grayck E, Suliman HB, Majka S, Albietz J, Van Rheen Z, Roush K, Stenmark KR. Lung EC-SOD overexpression attenuates hypoxic induction of Egr-1 and chronic hypoxic pulmonary vascular remodeling. Am J Physiol Lung Cell Mol Physiol. 2008;295:L422–L430. doi: 10.1152/ajplung.90293.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bauer EM, Shapiro R, Zheng H, Ahmad F, Ishizawar D, Comhair SA, Erzurum SC, Billiar TR, Bauer PM. High mobility group box 1 contributes to the pathogenesis of experimental pulmonary hypertension via activation of Toll-like receptor 4. Mol Med. 2012;18:1509–1518. doi: 10.2119/molmed.2012.00283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Paulin R, Meloche J, Bonnet S. STAT3 signaling in pulmonary arterial hypertension. JAK-STAT. 2012;1:223–233. doi: 10.4161/jkst.22366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Buys ES, Cauwels A, Raher MJ, Passeri JJ, Hobai I, Cawley SM, Rauwerdink KM, Thibault H, Sips PY, Thoonen R, et al. sGC(α)1(β)1 attenuates cardiac dysfunction and mortality in murine inflammatory shock models. Am J Physiol Heart Circ Physiol. 2009;297:H654–H663. doi: 10.1152/ajpheart.00367.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Turdi S, Han X, Huff AF, Roe ND, Hu N, Gao F, Ren J. Cardiac-specific overexpression of catalase attenuates lipopolysaccharide-induced myocardial contractile dysfunction: role of autophagy. Free Radic Biol Med. 2012;53:1327–1338. doi: 10.1016/j.freeradbiomed.2012.07.084. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 49.Khedoe PP, Wong MC, Wagenaar GT, Plomp JJ, van Eck M, Havekes LM, Rensen PC, Hiemstra PS, Berbée JF. The effect of PPE-induced emphysema and chronic LPS-induced pulmonary inflammation on atherosclerosis development in APOE*3-LEIDEN mice. PLoS One. 2013;8:e80196. doi: 10.1371/journal.pone.0080196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Biswas SK, Lopez-Collazo E. Endotoxin tolerance: new mechanisms, molecules and clinical significance. Trends Immunol. 2009;30:475–487. doi: 10.1016/j.it.2009.07.009. [DOI] [PubMed] [Google Scholar]
- 51.Sun Y, Li H, Yang MF, Shu W, Sun MJ, Xu Y. Effects of aging on endotoxin tolerance induced by lipopolysaccharides derived from Porphyromonas gingivalis and Escherichia coli. PLoS One. 2012;7:e39224. doi: 10.1371/journal.pone.0039224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xiong Y, Medvedev AE. Induction of endotoxin tolerance in vivo inhibits activation of IRAK4 and increases negative regulators IRAK-M, SHIP-1, and A20. J Leukoc Biol. 2011;90:1141–1148. doi: 10.1189/jlb.0611273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Meloche J, Pflieger A, Vaillancourt M, Paulin R, Potus F, Zervopoulos S, Graydon C, Courboulin A, Breuils-Bonnet S, Tremblay E, et al. Role for DNA damage signaling in pulmonary arterial hypertension. Circulation. 2014;129:786–797. doi: 10.1161/CIRCULATIONAHA.113.006167. [DOI] [PubMed] [Google Scholar]
- 54.Li M, Vattulainen S, Aho J, Orcholski M, Rojas V, Yuan K, Helenius M, Taimen P, Myllykangas S, De Jesus Perez V, et al. Loss of bone morphogenetic protein receptor 2 is associated with abnormal DNA repair in pulmonary arterial hypertension. Am J Respir Cell Mol Biol. 2014;50:1118–1128. doi: 10.1165/rcmb.2013-0349OC. [DOI] [PubMed] [Google Scholar]
- 55.Gourh P, Arnett FC, Assassi S, Tan FK, Huang M, Diekman L, Mayes MD, Reveille JD, Agarwal SK. Plasma cytokine profiles in systemic sclerosis: associations with autoantibody subsets and clinical manifestations. Arthritis Res Ther. 2009;11:R147. doi: 10.1186/ar2821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chaouat A, Savale L, Chouaid C, Tu L, Sztrymf B, Canuet M, Maitre B, Housset B, Brandt C, Le Corvoisier P, et al. Role for interleukin-6 in COPD-related pulmonary hypertension. Chest. 2009;136:678–687. doi: 10.1378/chest.08-2420. [DOI] [PubMed] [Google Scholar]
- 57.Gilbane AJ, Derrett-Smith E, Trinder SL, Good RB, Pearce A, Denton CP, Holmes AM. Impaired bone morphogenetic protein receptor II signaling in a transforming growth factor-β-dependent mouse model of pulmonary hypertension and in systemic sclerosis. Am J Respir Crit Care Med. 2015;191:665–677. doi: 10.1164/rccm.201408-1464OC. [DOI] [PubMed] [Google Scholar]
- 58.Dubois M, Delannoy E, Duluc L, Closs E, Li H, Toussaint C, Gadeau AP, Gödecke A, Freund-Michel V, Courtois A, et al. Biopterin metabolism and eNOS expression during hypoxic pulmonary hypertension in mice. PLoS One. 2013;8:e82594. doi: 10.1371/journal.pone.0082594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Humbert M, Sitbon O, Simonneau G. Treatment of pulmonary arterial hypertension. N Engl J Med. 2004;351:1425–1436. doi: 10.1056/NEJMra040291. [DOI] [PubMed] [Google Scholar]
- 60.Furuya Y, Satoh T, Kuwana M. Interleukin-6 as a potential therapeutic target for pulmonary arterial hypertension. Int J Rheumatol. 2010;2010:720305. doi: 10.1155/2010/720305. [DOI] [PMC free article] [PubMed] [Google Scholar]