

Abstract
Phosphodiesterase-9 (PDE9) inhibitors have been studied as potential therapeutics for treatment of central nervous system diseases and diabetes. Here, we report the discovery of a new category of PDE9 inhibitors by rational design on the basis of the crystal structures. The best compound, (S)-6-((1-(4-chlorophenyl)ethyl)amino)-1-cyclopentyl-1,5,6,7-tetrahydro-4H-pyrazolo[3,4-day]pyrimidin-4-one [(S)-C33], has an IC50 value of 11 nM against PDE9 and the racemic C33 has bioavailability of 56.5% in the rat pharmacokinetic model. The crystal structures of PDE9 in the complex with racemic C33, (R)-C33, and (S)-C33 reveal subtle conformational asymmetry of two M-loops in the PDE9 dimer and different conformations of two C33 enantiomers. The structures also identified a small hydrophobic pocket that interacts with the tyrosyl tail of (S)-C33 but not with (R)-C33, and is thus possibly useful for improvement of selectivity of PDE9 inhibitors. The asymmetry of the M-loop and the different interactions of the C33 enantiomers imply the necessity to consider the whole PDE9 dimer in the design of inhibitors.
Introduction
Phosphodiesterase (PDE) is a superfamily of enzymes hydrolyzing the second messengers, cGMP and cAMP. For the critical roles of cAMP and cGMP in physiologic processes, PDEs have been studied as drug targets for treatment of various diseases (Conti and Beavo, 2007; Maurice et al., 2014). The human genome contains 21 genes that are categorized into 11 PDE families and express >100 isoforms (Conti and Beavo, 2007; Maurice et al., 2014). PDE5, PDE6, and PDE9 specifically recognize cGMP as their substrate, while PDE4, PDE7, and PDE8 are cAMP-specific. The remaining PDE families are capable of degrading both substrates. PDE9 inhibitors have been studied for their potential applications to treat diabetes (Deninno et al., 2009; Shao et al., 2014) and central nervous system diseases such as Alzheimer’s disease (Wunder et al., 2005; van der Staay et al., 2008; Verhoest et al., 2009, 2012; Hutson et al., 2011; Vardigan et al., 2011; Claffey et al., 2012; Kleiman et al., 2012; Kroker et al., 2012, 2014; Liddie et al., 2012; Schwam et al., 2014; Singh and Patra, 2014; Heckman et al., 2015; Nagy et al., 2015).
On the other hand, since almost all important biomacromolecules such as proteins and nucleic acids exist in chiral forms, enantiomeric molecular recognition is the most important biologic process in nature. Similarly, when exogenous compounds such as drugs are introduced into the human body, chiral discrimination plays a fundamental role in determining the pharmacokinetic properties and biorecognition of drugs in physiologic processes (Agranat et al., 2002). The market share for single-enantiomer drugs increased from 27% (US$74.4 billion) in 1996 to 40%–50% in the pharmaceutical market today (Shaner et al., 2005; Sekhon, 2013). Recently, legal regulations have intended to allow only single-enantiomer drugs to be marketed (Mentel et al., 2009). Despite the importance of the drug chirality, the impacts of potentially different bindings of enantiomers on biologic effects have not been completely illustrated (Londesborough, 1985).
In this paper, we report a novel category of PDE9 inhibitors that have been discovered by using the rational structure-based design and docking on the basis of our early analogs of 28s and 3r (Meng et al., 2012; Shao et al., 2014). The crystal structures of PDE9 in the complex with the best compound, (S)-6-((1-(4-chlorophenyl)ethyl)amino)-1-cyclopentyl-1,5,6,7-tetrahydro-4H-pyrazolo[3,4-day]pyrimidin-4-one [(S)-C33], and a systematic comparison among PDE9 structures in the RCSB Protein Data Bank (www.rcsb.org/pdb) revealed subtle but significant conformation differences between two M-loops in the PDE9 dimers. In addition, the crystal structure of PDE9A-C33 identified a small hydrophobic pocket that interacted with (S)-C33 only, and may thus play a critical role in determination of inhibitor selectivity.
Materials and Methods
Molecular Docking
PDE9A (PDB ID: 4GH6) was used for docking by CDOCKER (Wu et al., 2003) and LigandFit (Venkatachalam et al., 2003) embedded in Accelrys Discovery Studio 2.5.5. Hydrogen atoms and charges were generated by the CHARMM force field and the Momany-Rone partial charge method. All ionizable residues were set to their protonation states. Charges of zinc and magnesium ions were assigned to +2. The radius of the docking sphere was set to 10 Å. The default values were used for the rest of the docking parameters. The PDE9A inhibitor 28s (Meng et al., 2012) was used as a reference for the docking test. The reliability of the docking results is also confirmed by a comparison between the docking poses and the crystal structure of PDE9-(S)-C33 (Supplemental Fig. 1). Fifty conformations of each ligand were randomly generated, docked, and output for evaluation. The candidates with high scores and reasonable binding patterns were chemically synthesized.
Synthesis
1H NMR and 13C NMR spectra were recorded at room temperature on a Bruker AVANCE III 400 (Karlsruhe, Germany) instrument with tetramethylsilane as an internal reference. The abbreviations of s, d, t, q, p, hept, m, and brs represent singlet, doublet, triplet, quartet, pentet, heptet, multiplet, and broad singlet, respectively. Optical rotations were measured using a Bellingham + Stanley ADP 440+ polarimeter (Kent, UK). Liquid chromatography mass spectrometry (MS) equipment LCMS-2010A (Shimadzu, Japan) was used for mass analysis. Thin-layer chromatography was performed on precoated silica gel F-254 plates (0.25 mm) (Merck kGaA, Darmstadt, Germany). Elemental analysis was carried out with a Vario EL cube series analyzer (Elementar, Germany). The m.p. values were determined on a WRS-1B (Shanghai Jingke Industrial Co. Ltd., China) digital m.p. apparatus and were not calibrated. Infrared (IR) spectra were recorded on a (Nicolet Avatar 330 FT-IR, Thermo Scientific, Waltham, MA). All starting materials and reagents were purchased from commercial suppliers and used directly without further purification. The syntheses of the compounds are outlined in scheme 1.
Scheme 1.

Synthesis of PDE9 inhibitors (PDE9 I).
Compounds P1–P3 were synthesized according to the protocol previously reported in Shao et al. (2014). The syntheses of the target compounds are briefly described as follows: i-PrOH (2 ml), pyrimidinone (0.3 mmol), amine (1.0 mmol), and Et3N (1.0 mmol) were added to a 10-ml sealed vial. The reaction mixture was stirred in an oil bath preheated to 100°C. After pyrimidinone was consumed, as indicated by thin-layer chromatography, the reaction mixture was cooled to room temperature and concentrated in vacuo. The residue was purified by flash column chromatography on silica gel to provide the target compounds. The following sections describe the targeted compounds.
(±)-1-Isopropyl-6-((1-Phenylethyl)Amino)-1,5-Dihydro-4H-Pyrazolo[3,4-Day]Pyrimidin-4-One.
White solids (83 mg, yield: 93%); m.p. 67–68°C; MS (ESI−): m/z 296 ([M-H]−); 1H NMR (400 MHz, CDCl3) 10.70 (brs, 1H), 7.59 (s, 1H), 7.47–7.42 (m, 2H), 7.35 (t, J = 7.5 Hz, 2H), 7.30–7.26 (m, 1H), 7.20 (d, J = 7.3 Hz, 1H), 5.26 (q, J = 7.0 Hz, 1H), 4.89–4.79 (m, 1H), 1.66 (d, J = 6.8 Hz, 3H), 1.51 (d, J = 6.8 Hz, 3H), and 1.46 (d, J = 6.8 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 160.3, 154.0, 152.1, 143.7, 133.9, 128.6, 127.3, 126.1, 99.9, 50.6, 49.0, 22.60, 21.6, and 21.5; and IR (KBr, cm−1): 3327, 2977, 1681, 1615, 1553, 1507, 1442, 1259, 1120, 782, and 670. Analysis calculated for C16H19N5O·0.5H2O: C, 62.73; H, 6.58; and N, 22.86; found: C, 62.90; H, 6.34; and N, 22.85.
6-(Benzyl(Methyl)amino)-1-Isopropyl-1,5-Dihydro-4H-Pyrazolo[3,4-Day]Pyrimidin-4-One.
White solids (72 mg, yield: 80%); m.p. 187–188°C; MS (ESI−), m/z 296 ([M-H]−); 1H NMR [500 MHz, dimethylsulfoxide (DMSO)-d6] δ 10.66 (brs, 1H), 7.75 (s, 1H), 7.36–7.32 (m, 2H), 7.30–7.25 (m, 3H), 4.82 (s, 2H), 4.74 (hept, J = 6.7 Hz, 1H), 3.08 (s, 3H), and 1.37 (d, J = 6.7 Hz, 6H); 13C NMR (126 MHz, DMSO-d6) δ 158.5, 153.3, 152.9, 137.3, 133.5, 128.4, 127.3, 127.1, 99.0, 52.4, 47.8, 35.7, and 21.5; and IR (KBr, cm−1): 3120, 3050, 2979, 2934, 1676, 1595, 1544, 1387, 1285, 1113, 1003, 974, 815, 779, 729, and 698. Analysis calculated for C16H19N5O: C, 64.63; H, 6.44; and N, 23.55; found: C, 64.59; H, 6.26; and N, 23.43.
(±)-6-((1-(4-Chlorophenyl)Ethyl)Amino)-1-Isopropyl-1,5-Dihydro-4H-Pyrazolo[3,4-Day]Pyrimidin-4-One.
White solids (69 mg, yield: 69%); m.p. 67–68°C; MS (ESI−): m/z 330 ([M-H]−); 1H NMR (400 MHz, CDCl3) δ 10.70 (brs, 1H), 7.65 (s, 1H), 7.34 (d, J = 8.5 Hz, 2H), 7.30–7.26 (m, 2H), 7.14 (d, J = 7.0 Hz, 1H), 5.18 (q, J = 6.9 Hz, 1H), 4.73–4.79 (m, 1H), 1.61 (d, J = 7.0 Hz, 3H), 1.48 (d, J = 6.7 Hz, 3H), and 1.41 (d, J = 6.7 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 160.3, 153.9, 152.0, 142.5, 133.7, 132.9, 128.7, 127.5, 99.9, 50.1, 49.0, 22.7, 21.6, and 21.5; and IR (KBr, cm−1): 3321, 2977, 1681, 1615, 1553, 1444, 1259, 1092, 1013, 827, 781, and 680. Analysis calculated for C16H18ClN5O: C, 57.92; H, 5.47; and N, 21.11; found: C, 58.20; H, 5.64; and N, 20.76.
(±)-1-Cyclopentyl-6-((1-Phenylethyl)Amino)-1,5-Dihydro-4H-Pyrazolo[3,4-Day]Pyrimidin-4-One.
White solids (78 mg, yield: 80%); m.p. 172–174°C; MS (ESI−): m/z 322 ([M-H]−); 1H NMR (400 MHz, CDCl3) δ 10.70 (brs, 1H), 7.58 (s, 1H), 7.45–7.39 (m, 2H), 7.36–7.31 (m, 2H), 7.29–7.24 (m, 1H), 7.13 (d, J = 7.0 Hz, 1H), 5.25 (q, J = 6.8 Hz, 1H), 4.96 (p, J = 7.6 Hz, 1H), 2.12–2.05 (m, 2H), 2.05–1.98 (m, 2H), 1.97–1.88 (m, 2H), 1.75–1.67 (m, 2H), and 1.65 (d, J = 6.9 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 160.3, 154.4, 152.1, 143.7, 133.9, 128.6, 127.3, 126.1, 99.9, 57.9, 50.6, 31.9, 31.7, 24.7, 24.7, and 22.6; and IR (KBr, cm−1): 3317, 2960, 2868, 1680, 1615, 1555, 1443, 1258, 1121, 1066, 1012, 782, 698, and 550. Analysis calculated for C18H21N5O·0.1 H2O: C, 66.48; H, 6.57; and N, 21.54; found: C, 66.47; H, 6.44; and N, 21.20.
6-(Benzyl(Methyl)Amino)-1-Cyclopentyl-1,5-Dihydro-4H-Pyrazolo[3,4-Day]Pyrimidin-4-One.
White solids (45 mg, yield: 46%); m.p. 169–170°C; MS (ESI−): m/z 322 ([M-H]−); 1H NMR (400 MHz, DMSO-d6) δ 7.76 (s, 1H), 7.39–7.23 (m, 5H), 4.96–4.86 (m, 1H), 4.82 (s, 2H), 3.08 (s, 3H), 2.03–1.76 (m, 6H), and 1.67–1.56 (m, 2H); 13C NMR (101 MHz, DMSO-d6) δ 158.6, 153.4, 137.4, 133.7, 128.5, 127.4, 127.2, 99.1, 56.8, 52.4, 35.8, 31.6, and 24.3; and IR (KBr, cm−1): 3112, 2953, 1865, 1593, 1566, 1546, 1388, 1358, 1293, 1055, 1004, 776, and 705. Analysis calculated for C18H21N5O·0.4 H2O: C, 65.39; H, 6.65; and N, 21.18; found: C, 65.79; H, 6.41; and N, 20.84.
(±)-6-((1-(4-Chlorophenyl)Ethyl)Amino)-1-Cyclopentyl-1,5,6,7-Tetrahydro-4H-Pyrazolo[3,4-Day]Pyrimidin-4-One.
White solids (49 mg, yield: 46%); m.p. 209–210°C; MS (ESI−): m/z 356 ([M-H]−); 1H NMR (400 MHz, CDCl3) δ 10.72 (brs, 1H), 7.64 (s, 1H), 7.36–7.32 (m, 2H), 7.30–7.26 (m, 2H), 7.07 (d, J = 7.1 Hz, 1H), 5.18 (q, J = 6.9 Hz, 1H), 4.90 (p, J = 7.5 Hz, 1H), 2.09–2.03 (m, 2H), 2.01–1.95 (m, 2H), 1.94–1.85 (m, 2H), 1.73–1.65 (m, 2H), and 1.61 (d, J = 7.0 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 160.3 154.3, 151.9, 142.5, 133.7, 132.9, 128.7, 127.4, 99.9, 57.9, 50.1, 31.9, 31.7, 24.7, and 22.9; and IR (KBr, cm−1): 3309, 2959, 2869, 1680, 1614, 1552, 1492, 1446, 1259, 1092, 1014, 825, and 780. Analysis calculated for C18H20ClN5O: C, 60.42; H, 5.63; and N, 19.57; found: C, 60.44; H, 5.84; and N, 19.28.
(R)-6-((1-(4-Chlorophenyl)Ethyl)Amino)-1-Cyclopentyl-1,5,6,7-Tetrahydro-4H-Pyrazolo[3,4-Day]Pyrimidin-4-One [(R)-C33].
White solids (56 mg, yield: 52%); MS (ESI−): m/z 356 ([M-H]−); 1H NMR (400 MHz, CDCl3) δ 10.74 (brs, 1H), 7.66 (s, 1H), 7.38–7.34 (m, 2H), 7.33–7.27 (m, 2H), 7.18 (d, J = 6.9 Hz, 1H), 5.21 (q, J = 6.9 Hz, 1H), 4.94 (p, J = 7.5 Hz, 1H), 2.12–1.91 (m, 6H), 1.77–1.68 (m, 2H), and 1.64 (d, J = 7.0 Hz, 3H); and 13C NMR (100 MHz, CDCl3) δ 160.3, 154.3, 151.9, 142.4, 133.7, 132.9, 128.7, 127.4, 99.9, 57.9, 50.1, 32.0, 31.7, 24.8, 24.7, and 22.8. Analysis calculated for C18H20ClN5O: C, 60.42; H, 5.63; and N, 19.57; found: C, 60.18; H, 5.68; and N, 19.39; 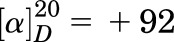 (c = 1 g/L, CHCl3).
(c = 1 g/L, CHCl3).
(S)-6-((1-(4-Chlorophenyl)Ethyl)Amino)-1-Cyclopentyl-1,5,6,7-Tetrahydro-4H-Pyrazolo[3,4-Day]Pyrimidin-4-One [(S)-C33].
White solids (52 mg, yield: 49%); MS (ESI−): m/z 356 ([M-H]−); 1H NMR (400 MHz, CDCl3) δ 10.71 (s, 1H), 7.66 (s, 1H), 7.40–7.34 (m, 2H), 7.34–7.29 (m, 2H), 7.24 (d, J = 7.0 Hz, 1H), 5.21 (q, J = 6.9 Hz, 1H), 4.94 (p, J = 7.5 Hz, 1H), 2.13–1.90 (m, 6H), 1.75–1.67 (m, 2H), and 1.64 (d, J = 7.0 Hz, 3H); and 13C NMR (100 MHz, CDCl3) δ 160.3, 154.3, 151.9, 142.5, 133.7, 132.9, 128.7, 127.4, 99.9, 57.9, 50.0, 32.0, 31.7, 24.8, 24.7, and 22.8. Analysis calculated for C18H20ClN5O: C, 60.42; H, 5.63; and N, 19.57; found: C, 60.54; H, 5.66; and N, 19.71; 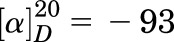 (c = 1 g/L, CHCl3).
(c = 1 g/L, CHCl3).
(±)-6-((1-(4-Chlorophenyl)-2-Methylpropyl)Amino)-1-Cyclopentyl-1,5,6,7-Tetrahydro-4H-Pyrazolo[3,4-Day]Pyrimidin-4-One (C40).
White solids (79 mg, yield: 68%); m.p. 210–212°C; MS (ESI−): m/z 386 ([M-H]−); 1H NMR (400 MHz, DMSO-d6) δ 10.23 (s, 1H), 7.72 (s, 1H), 7.37 (m, 4H), 7.08 (s, 1H), 4.88–4.77 (m, 1H), 4.70–4.65 (m, 1H), 2.07–2.03 (m, 1H), 2.01–1.73 (m, 6H), 1.70–1.49 (m, 2H), 0.95 (d, J = 6.7 Hz, 3H), and 0.82 (d, J = 6.7 Hz, 3H); 13C NMR (126 MHz, CDCl3) δ 160.3, 154.4, 152.4, 140.6, 133.8, 132.7, 128.4, 128.3, 99.8, 60.8, 57.7, 33.8, 32.0, 31.6, 24.7, 24.7, 19.9, and 19.2; and IR (KBr, cm−1): 3344, 2931, 2852, 1689, 1610, 1552, 1506, 1491, 1441, 1265, 1251, 1089, 1051, 1012, 989, 827, 816, 781, 621, 581, and 557. Analysis calculated for C20H24ClN5O·0.15H2O: C, 61.82; H, 6.30; and N, 18.02; found: C, 62.11; H, 6.12; and N, 17.74.
(±)-1-Isobutyl-6-((1-Phenylethyl)Amino)-1,5-Dihydro-4H-Pyrazolo[3,4-Day]Pyrimidin-4-One.
White solids (52 mg, yield: 54%); m.p. 84–85°C; MS (ESI−): m/z 310 ([M-H]−); 1H NMR (400 MHz, CDCl3) δ 10.66 (brs, H), 7.48 (s, H), 7.43–7.39 (m, 2H), 7.36–7.29 (m, 2H), 7.29–7.21 (m, 1H), 7.16 (d, J = 6.9 Hz, 1H), 5.20 (q, J = 6.8 Hz, 1H), 4.00–3.88 (m, 2H), 2.28–2.11 (m, 1H), 1.64 (d, J = 6.9 Hz, 3H), 0.88 (d, J = 6.7 Hz, 3H), and 0.81 (d, J = 6.7 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 160.3, 155.1, 152.3, 143.7, 134.1, 128.6, 127.3, 126.2, 99.4, 54.1, 50.7, 28.9, 22.4, 20.0, and 19.9; and IR (KBr, cm−1): 3489, 3268, 2962, 1680, 1611, 1566, 1520, 1457, 1277, 1094, 882, 756, 699, 554, and 476. Analysis calculated for C17H21N5O·0.3H2O: C, 64.45; H, 6.87; and N, 22.11; found: C, 64.71; H, 7.06; and N, 21.86.
6-(Benzyl(Methyl)Amino)-1-Isobutyl-1,5-Dihydro-4H-Pyrazolo[3,4-Day]Pyrimidin-4-One.
White solids (39 mg, yield: 42%); m.p. 187–188°C; MS (ESI−): m/z 310 ([M-H]−); 1H NMR (400 MHz, DMSO-d6) δ 7.78 (s, 1H), 7.37–7.22 (m, 5H), 4.80 (s, 2H), 3.88 (d, J = 7.1 Hz, 2H), 3.08 (s, 3H), 2.16–2.05 (m, 1H), and 0.79 (d, J = 6.7 Hz, 6H); 13C NMR (101 MHz, DMSO-d6) δ 159.2, 154.6, 153.9, 138.0, 134.4, 128.9, 127.9, 127.7, 99.0, 53.5, 53.0, 36.3, 29.0, and 20.2; and IR (KBr, cm−1): 3117, 2958, 1692, 1580, 1453, 1387, 1314, 1157, 1056, 1013, 884, 809, 780, 699, 627, 563, and 474. Analysis calculated for C17H21N5O: C, 65.57; H, 6.80; and N, 22.49; found: C, 65.56; H, 6.76; and N, 22.30.
(±)-6-((1-(4-Chlorophenyl)Ethyl)Amino)-1-Isobutyl-1,5-Dihydro-4H-Pyrazolo[3,4-day]Pyrimidin-4-One.
White solids (61 mg, yield: 58%); m.p. 88–89°C; MS (ESI−): m/z 344 ([M-H]−); 1H NMR (400 MHz, CDCl3) δ 10.79 (brs, 1H), 7.65 (s, 1H), 7.34–7.27 (m, 4H), 6.88 (d, J = 6.7 Hz, 1H), 5.13 (q, J = 6.8 Hz, 1H), 3.96–3.85 (m, 2H), 2.21–2.10 (m, 1H), 1.61 (d, J = 7.0 Hz, 3H), 0.87 (d, J = 6.7 Hz, 3H), and 0.79 (d, J = 6.7 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 160.4, 155.0, 152.0, 142.5, 134.0, 132.9, 128.7, 127.4, 99.5, 54.2, 50.3, 28.9, 22.7, 20.0, and 19.8; and IR (KBr, cm−1): 3429, 3270, 2964, 2930, 1678, 1608, 1560, 1516, 1458, 1401, 1318, 1270, 1171, 1094, 1014, 824, 780, 589, and 539. Analysis calculated for C17H20ClN5O·0.9H2O: C, 56.40; H, 6.07; and N, 19.34; found: C, 56.48; H, 6.14; and N, 19.12.
Protein Expression and Purification
The PDE9A2 catalytic domain (residues 181–506) was subcloned to vector pET15b and purified according to the protocols described by Huai et al. (2004). Briefly, the pET15-PDE9 plasmid was transferred into Escherichia coli strain BL21 (Codonplus, Stratagene, Santa Clara, CA) for overexpression. When the E. coli cell was grown in LB medium at 37°C to absorption (A600 = 0.7), 0.1 mM isopropyl β-d-thiogalactopyranoside was added to induce the expression at 15°C overnight. Recombinant PDE9A2 protein was purified by column chromatography of Ni-NTA (GE Healthcare, Pittsburgh, PA), Q-sepharose (GE Healthcare), and Superdex-200 (GE Healthcare). A typical batch of purification yielded 20–60 mg PDE9A2 from a liter of cell culture. The PDE9A2 protein had purity >90% as judged by SDS-PAGE.
The catalytic domains of PDE2A3 (222–904), PDE4D2 (86–413), PDE5A1 (535–860), PDE7A1 (130–482), PDE8A2 (480–820), and PDE10A2 (448–789) were purified by published protocols (Huai et al., 2003; Wang et al., 2005, 2006, 2007a, 2008a). PDE1B (10–516) was expressed and purified using a protocol similar to that for PDE9.
Enzymatic Assay
The enzymatic activities of PDE9A2 and other PDEs were assayed by using 3H-cGMP or 3H-cAMP as the substrate. The assay buffer was composed of 50 mM Tris-HCl pH 8.0, 10 mM MgCl2 or 4 mM MnCl2, 1 mM DTT, and 20 nM 3H-cGMP or 3H-cAMP (20,000–30,000 cpm/assay) (GE Healthcare). The reaction was carried out at room temperature for 15 minutes and then terminated by addition of 0.2 M ZnSO4. The reaction 3H-GMP or 3H-AMP products were precipitated by adding 0.2 N Ba(OH)2, whereas the unreacted 3H-cGMP or 3H-cAMP remained in the supernatant. Radioactivity in the supernatant was measured in 2.5 ml Ultima Gold liquid scintillation cocktails (Fisher Scientific, Pittsburgh, PA) by a liquid scintillation counter. The final concentrations of the enzymes used in the assay were 50–200 ng/ml, and they were hydrolyzed up to 70% of the substrates. A total of 9–12 concentrations of inhibitors were used for triplet measurement of the IC50 and S.D. values that were obtained by nonlinear regression.
Crystallization and Structure Determination
The crystals of the PDE9A2 catalytic domain (181–506) in the complex with racemic C33 and the (R)- and (S)-enantiomers were grown by the hanging drop vapor diffusion method. The PDE9A2 (8–10 mg/ml) in a buffer of 50 mM NaCl, 20 mM Tris·HCl, pH 7.5, 1 mM β-mercaptoethanol, and 1 mM EDTA was mixed with 2 mM racemic C33 or (R)-C33 and cocrystallized against a well buffer of 1.8–2.0 M sodium formate at 4°C. The PDE9-(S)-C33 crystals were prepared by soaking the PDE9/3-isobutyl-1-methylxanthine (IBMX) crystals in the crystallization buffer plus 2 mM (S)-C33 at 25°C for 1 day. The PDE9-IBMX crystals were grown by the hanging drop method against a well buffer of 0.1 M HEPES pH 7.5, 3.0 M sodium formate at 4°C. The crystallization buffer, plus 20% glycerol, was used as the cryosolvent to freeze the crystals. X-ray diffraction data were collected on Beamline X29 at Brookhaven National Laboratory (Upton, NY) (for racemic C33) or BM22 of SER-CAT at Advanced Photon Source in Chicago (for (R)-C33 and (S)-C33), and processed by HKL 2000 (Otwinowski and Minor, 1997). The structures were solved by molecular replacement, using the PDE9A2 catalytic domain as the initial model. The resulting models were rebuilt by the program COOT (Emsley et al., 2010) and refined by REFMAC (Winn et al., 2003). The statistics of the data collection and structure refinement are listed in Table 1.
TABLE 1.
Statistics on the diffraction data and structure refinement
| PDE9-C33 | PDE9-(S)-C33 | PDE9-(R)-C33 | |
|---|---|---|---|
| Data collection | |||
| Space group | P41212 | P41212 | P41212 |
| Unit cell (a, c, Å) | 103.6, 268.7 | 104.7, 270.3 | 105.4, 270.1 |
| Resolution (Å) | 2.0 | 2.7 | 3.1 |
| Total measurements | 1,215,951 | 688,294 | 161,116 |
| Unique reflections | 96,827 | 42,348 | 24,552 |
| Completeness (%) | 97.2 (91.9)a | 100.0 (100.0) | 84.9 (85.4) |
| Average I/σ | 20.8 (3.5)a | 7.2 (5.1) | 7.6 (2.0) |
| Rmerge | 0.074 (0.274)a | 0.125 (0.668) | 0.156 (0.717) |
| Structure refinement | |||
| R-work | 0.224 (0.457)a | 0.205(0.291) | 0.215 (0.301) |
| R-free | 0.247 (0.470)a | 0.231 (0.326) | 0.244 (0.364) |
| Resolution (Å) | 30.0-2.0 | 50.0-2.7 | 50.0-3.1 |
| Reflections | 94,640 (5%)b | 40,241 (5%) | 23,302 (5%) |
| RMSD for bond length (Å) | 0.007 | 0.008 | 0.007 |
| RMSD for bond angle (o) | 1.1 | 1.1 | 1.1 |
| Average B-factor (Å2) | |||
| Protein | 40.4 (5357)c | 41.6 (5337) | 62.1 (5357) |
| Inhibitor | 37.9 (50) | 37.8 (50) | 61.3 (50) |
| Zn2+ | 48.1 (2) | 51.7 (2) | 71.8 (2) |
| Mg2+ | 31.8 (2) | 32.5(2) | 50.6 (2) |
| Water | 39.0 (255) | 29.1 (12) | 42.6 (12) |
| Ramachandran plot (%) | |||
| Favored | 93.8 | 93.0 | 91.5 |
| Allowed | 5.9 | 6.3 | 8.3 |
| Generally allowed | 0.3 | 0.7 | 0.2 |
| Disallowed | 0.0 | 0.0 | 0.0 |
The numbers in parentheses are for the highest resolution shells of 2.0–2.05, 2.7–2.77, and 3.1–3.17 Å, respectively, for the PDE9 structures in the complex with racemic C33, (S)-C33, and (R)-C33.
The percentage of reflections omitted for calculation of R-free.
The number of atoms in the structure refinement.
Pharmacokinetics Analysis
Six male Sprague Dawley rats with body weights of 240–275 g were used for the pharmacokinetic experiments. Compound C33 was dissolved in 5% DMSO, 5% solutol, and 90% saline to make a 5 mg/ml stock for i.v. administration and 1 mg/ml for oral administration (by mouth). A final dosage of the 5 mg/kg formulated compound was given in both administration modes. The blood samples were taken at various time points over 24 hours. The concentration of the compounds in blood was analyzed by liquid chromatography–tandem MS (Shimadzu liquid chromatographic system, and API4000 mass spectrometer, Applied Biosystems, Ontario, Canada).
Stability in Liver Microsomes
Warfarin and testosterone was purchased from Sigma (St. Louis, MO) and Acros (Geel, Belgium), respectively. Human and mouse (CD-1) liver microsomes (0.5 ml) (BD Gentest Corporation, Woburn, MA) were preincubated with 0.75 µM sample in a buffer of 0.1 M potassium phosphate pH 7.4, and 2 mM MgCl2 in a 96-well plate at 37°C for 5 minutes. The enzymatic reaction was initiated by adding 3 mM NADPH and terminated by adding 90 µl acetonitrile at time points of 0, 5, 10, 20, and 30 minutes. Parallel incubations were performed for the positive (testosterone) and negative (warfarin) controls. The supernatant was injected into liquid chromatography–tandem MS for analysis.
Results
Structure-Based Design and Enzymatic Activity of PDE9 Inhibitors.
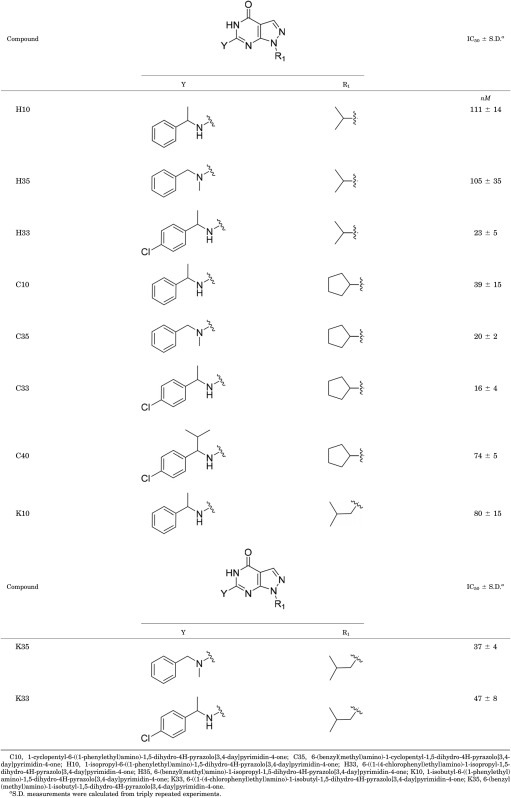
We previously reported a potent PDE9 inhibitor, 3r (Fig. 1), which has an IC50 value of 0.6 nM against PDE9, but only moderate metabolic stability and 10% bioavailability (Shao et al., 2014). To improve the in vivo stability, we designed a series of analogs of 3r and docked them to the PDE9 crystal structures for confirmation. Since 3r contains an amide bond that is often labile to acids and proteinases and may thus account for its moderate bioavailability, we removed the amide unit of 3r and also optimized the tail group with hydrocarbon fragments. Removal of the amide group would be expected to lead to a loss of the hydrogen bond between the amide nitrogen of the inhibitor and OH of Tyr424, and thus somewhat sacrifice the affinity and selectivity because Tyr424 is a unique residue for PDE8 and PDE9 (phenylalanine in other PDE families). However, if the pyrazolopyrimidinone and cyclic pentanyl groups that contribute the hydrogen bonds with the invariant glutamine and hydrophobic stack against the phenylalanine are retained as the scaffold of pharmacophore, optimization of the remaining moiety may lead to finding new compounds with nearly reasonable affinity and selectivity. In addition, since the benzenylamine link is usually sensitive to P450 metabolic enzymes in vivo, we hoped that introduction of the N-alpha substitutent would protect oxidation of benzenylamine to benzamide. Thus, we rationally designed a series of new compounds on the basis of the crystal structures of the PDEs and performed molecular docking to confirm the design. Compounds from this rational design and docking were then chemically synthesized and assayed for their enzymatic properties. The inhibitions of some representative compounds on PDE9 are listed in Table 2, among which racemic C33 shows the best IC50 value of 16 nM against PDE9. Overall, the IC50 values of the listed compounds are not dramatically different, perhaps due to the modified groups targeting similar protein regions and interacting with similar residues.
Fig. 1.

Chemical structures of PDE9 inhibitors assessed in this study (*, chiral carbon).
TABLE 2.
Inhibition of synthetic compounds on PDE9
 |
|
|---|---|
Based on the observation that the methyl group on the chiral carbon is neighboring a small hydrophobic pocket in the PDE9-r3 crystal structure (Shao et al., 2014), we hoped that the tail of (R)- or (S)-C33 may interact with the hydrophobic pocket and further improve the affinity and selectivity. Therefore, racemic and enantiomeric C33s were chemically synthesized and their inhibitions on the PDE families were evaluated (Table 3). The enzymatic assay revealed that racemic, (S)-C33, and (R)-C33 have IC50 values of 16, 11, and 56 nM, respectively, for inhibition on the PDE9 catalytic domain, and (S)-C33 has reasonably good selectivity over the other PDE families (Table 3), indicating its potential for further pharmacological studies.
TABLE 3.
Affinity of C33 with PDE families
The numbers in parentheses are the fold of selectivity of inhibitors against PDE9 over other PDEs (IC50, nM).
| PDE | (S)-C33 | C33 | (R)-C33 | 3ra |
|---|---|---|---|---|
| PDE9A (181–506) | 11 ± 4 | 16 ± 4 | 58 ± 2 | 0.60 ± 0.02 |
| PDE1B2 (10–487) | 554 ± 64 (46) | 152 ± 15 (9) | 80 ± 4 (1.4) | 473 ± 14 (800) |
| PDE2A3 (222–904) | 2.4 ± 0.5 x 103 (200) | 437 ± 77 (27) | 479 ± 79 (8) | 13 ± 0.2 × 103 (22,000) |
| PDE4D2 (86–413) | 1.3 ± 0.1 x 103 (108) | 1.8 ± 0.2 × 103 (110) | 3.2 ± 0.6 × 103 (56) | 21 ± 0.7 × 103 (35,000) |
| PDE5A1 (535–860) | 366 ± 38 (31) | 197 ± 36 (12) | 787 ± 38 (14) | 91 ± 23 (151) |
| PDE7A1 (130–482) | 2.5 ± 0.4 × 103 (208) | 1.7 ± 0.1 × 103 (104) | 1.3 ± 0.1 × 103 (23) | 1.8 ± 0.7 × 103 (3000) |
| PDE8A1 (480–820) | 1.0 ± 0.2 × 103 (83) | 1.6 ± 0.2 × 103 (98) | 4.4 ± 0.4 × 103 (76) | >100,000 |
| PDE10A2 (448–789) | 2.3 ± 0.3 × 103 (192) | 1.2 ± 0.1 × 103 (73) | 774 ± 11 (13) | 6.9 ± 0.1 × 103 (11,000) |
The data are cited from Shao et al. (2014).
Improvement in Bioavailability and Stability in Live Microsomes.
While an IC50 value of 11 nM for (S)-C33 is in the IC50 range of most drugs, it is significantly worse than 0.6 nM for 3r, and also the enzymatic selectivity of (S)-C33 over other PDE families significantly decreases. However, in terms of bioavailability and in vivo stability, C33 shows significant improvement over 3r. The oral administration of racemic C33 yielded a Cmax value of 373 ng/ml and bioavailability of 56.5% (Table 4), in comparison with Cmax values of 16 and 217 ng/ml and bioavailability of 1.3% and 9.8% for 28s (Meng et al., 2012) and 3r (Shao et al., 2014), respectively. The blood concentration of C33 in the oral administration mode was 113 ng/ml after 6 hours, which is about 19-fold higher than the IC50 value of 6 ng/ml, indicating practical usability and the potential for C33 as a drug lead. In addition, the replacement of the amide bond with a hydrocarbon fragment in C33 yields stronger hydrophobicity than 3r, as shown by a log P value of 3.59 C33 in comparison with a log P value of 1.86 for 3r, implying a strong ability to penetrate the cell membrane.
TABLE 4.
Pharmacokinetics profile of PDE9 inhibitor C33
| Dosing Route |
T1/2 |
Tmax |
Cmax |
AUC0-t |
AUC0-inf |
MRTinf |
Vz |
CLz |
F |
|---|---|---|---|---|---|---|---|---|---|
| hour | hour | ng/ml | ng/h/ml | ng/h/ml | hour | l/kg | l/h/kg | % | |
| By mouth | 1.5 ± 0.2 | 2.0 ± 0.0 | 372 ± 6 | 1600 ± 300 | 1700 ± 300 | 3.5 ± 0.3 | NA | NA | 56.5 |
| i.v. | 1.4 ± 0.1 | 0.02 ± 0.00 | 5300 ± 900 | 3000 ± 200 | 3100 ± 200 | 1.6 ± 0.2 | 3.2 ± 0.1 | 1.6 ± 0.1 |
AUC, area under the curve; Cl, clearance; MRT, mean residence time; NA, not applicable.
In addition, the metabolic stability of C33 in human and mouse liver microsomes suggests that C33 may have better in vivo stability than 3r. Apparently, the removal of the amide group from 3r significantly improved the stability, as shown by the T1/2 values of 51 and 12 minutes for racemic C33 and 3r, respectively, in the human liver microsome, and of 33 and 22 minutes, respectively, in the mouse liver microsome (Table 5). In the enantiomeric forms of C33, (S)-C33 is apparently more stable than (R)-C33, as shown by the T1/2 values of 105 and 54 minutes in human and mouse microsomes for (S)-C33, in comparison with 44 and 22 minutes for (R)-C33. The explanation is not clear, but may be related to their different conformations and interactions.
TABLE 5.
Metabolic stability of PDE9 inhibitors in human liver microsomes
| Compound |
k |
T1/2 |
CLint |
CLapp |
CLh |
Eh |
|---|---|---|---|---|---|---|
| minute | ml/min/mg | ml/min/mg | ml/min/mg | % | ||
| Testosterone | 0.035 | 20.0 | 0.069 | 66.9 | 15.4 | 78.0 |
| 0.165 | 4.2 | 0.329 | 1296 | 84.2 | 93.5 | |
| Warfarin | −0.001 | Stable | Stable | Stable | Stable | |
| −0.003 | Stable | Stable | Stable | Stable | ||
| (S)-C33 | 0.007 | 105 | 0.013 | 12.7 | 7.8 | 38.9 |
| 0.013 | 53.5 | 0.026 | 102 | 47.8 | 53.1 | |
| (R)-C33 | 0.016 | 44.0 | 0.032 | 30.4 | 12.1 | 60.3 |
| 0.034 | 20.2 | 0.069 | 270 | 67.5 | 75.0 | |
| Racemic C33 | 0.013 | 51.4 | 0.027 | 26.0 | 11.3 | 56.5 |
| 0.022 | 31.3 | 0.044 | 174 | 59.4 | 66.0 | |
| C40 | 0.011 | 62.2 | 0.022 | 21.5 | 10.4 | 51.8 |
| 0.002 | 291 | 0.008 | 19 | 15.5 | 17.2 | |
| 3ra | 0.058 | 11.9 | 0.116 | 112 | 17.0 | 84.8 |
| 0.031 | 22.3 | 0.062 | 244 | 65.8 | 73.1 |
The data are cited from Shao et al. (2014). Cl, clearance.
On the other hand, we replaced the methyl group of C33 with an isopropanyl group to seek a spatial obstacle for protection of the C–N bond, and thus lead to the design of C40 (Fig. 1). Indeed, the stability of C40 was significantly improved, as shown by the T1/2 values of 62 and 291 minutes in the human and mouse liver microsomes, respectively (Table 5), although the IC50 value of C40 against PDE9 was increased to 73.5 nM.
Asymmetry of PDE9 Dimers.
The crystallographic asymmetric units of PDE9 in complex with racemic or enantiomeric C33 contain two molecules of the PDE9 catalytic domain (Fig. 2). Superposition of subunit A over B in the PDE9-C33 dimer yielded a root-mean-square deviation (RMSD) of 0.62 Å for the Cα atoms of the whole domain (residues 185–506). A systematic superposition of subunit A over B of the other PDE9 structures in the RCSB Protein Data Bank resulted in RMSDs in a range of 0.55 and 0.78 Å for the Cα atoms of residues 185–506. These small RMSDs indicate an overall similarity between the two subunits of the PDE9 dimers. On the other hand, superposition of PDE9-C33 over the other PDE9 structures yielded RMSDs of 0.27–0.49 and 0.22–0.42 Å for the comparisons of A versus A and B versus B, respectively. Similar RMSDs were obtained from the cross-superposition among other PDE9 structures. Since superposition between structures reported from different laboratories often produces higher RMSDs than comparison between subunits within the same structure, the consistently and significantly larger RMSDs for A versus B in the same dimers than for A versus A or B versus B between different structures suggest asymmetry of the PDE9 dimers.
Fig. 2.

Ribbon diagram of PDE9-C33. (A) The PDE9A dimer observed in all PDE9 structures. Inhibitor C33 is shown as yellow sticks. (B) Superposition of subunit A (cyan, residues 185–506) over subunit B (green).
When the N- and C-terminal residues were excluded, the superposition between residues 192–486 of the PDE9-C33 dimer yielded a RMSD of 0.17 Å for the Cα atoms, suggesting flexibility of the N- and C-termini. A careful examination of the superposition revealed RMSDs of 0.25–0.62 Å for residues 432–440 (the M-loop), which are 1.5- to 4-fold of the overall average of 0.17 Å for residues 192–486, suggesting significant conformational differences between two M-loops in the PDE9 dimer. A systematic comparison between subunits A and B of the PDE9 structures in the Protein Data Bank reveals the average difference between the Cα atoms of the M-loop in the dimer is about twice the overall average of the PDE9 catalytic domain (Table 6), thus confirming the asymmetry of the M-loop. While the N- and C-termini have the largest RMSD differences, their biologic relevance is unclear because terminal flexibility is often due to lack of lattice packing in many crystal structures. However, the difference between the M-loops of the PDE9 dimers is statistically significant and appears to be biologically relevant.
TABLE 6.
The average difference between the Cα atoms of subunits A and B of the PDE9 dimers (Å)
| C33 | 4QGE (3r) | 4GH6 (28s) | 3JSI | 3JSW | 4G2J | 4G2L | 3K3E | 3QI3 | 2HD1 | 3DYN (cGMP) | 3DY8 (GMP) | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Residues 181-506 | 0.32 | 0.33 | 0.36 | 0.32 | 0.35 | 0.33 | 0.31 | 0.41 | 0.43 | 0.39 | 0.31 | 0.30 |
| M-loop (S433-K446) | 0.59 | 0.65 | 0.58 | 0.48 | 0.74 | 0.74 | 0.67 | 1.11 | 1.34 | 0.80 | 0.92 | 0.70 |
The asymmetry of the PDE9 dimer was first observed in the crystal structure of the PDE9 catalytic domain in the complex with the IBMX inhibitor, in which two IBMX molecules bind the active sites of the PDE9 dimer with different orientations (Huai et al., 2004). Later, it was shown that the substrate cGMP or product GMP easily replaced IBMX in subunit A of the PDE9-IBMX dimer in the soaking experiments, but not IBMX in subunit B (Liu et al., 2008). Finally, the asymmetry was reported for the binding of inhibitor BAY73-6691 to the Q453E mutant (Hou et al., 2011). The subtle conformational asymmetry of the M-loop, which is revealed by this study, may respond to the asymmetric binding/replacement of the inhibitors.
Different Conformations of Bound C33 Enantiomers.
In the PDE9 structure in the complex with racemic C33, the electron density in the maps of (2Fo − Fc) and (Fo − Fc) revealed binding of the (S)-C33 enantiomer to subunits A and (R)-C33 to subunit B of the PDE9 dimer (Fig. 3). In the structures of PDE9 in the complex with enantiomerically pure (S)-C33 or (R)-C33, the maps showed unique conformations of (S)-C33 and (R)-C33 bound to both subunits A and B of their PDE9 dimers. These conformations are identical to that of the (S)- or (R)-enantiomer, respectively, in the racemic C33-PDE9 complex.
Fig. 3.

Binding of C33 to PDE9A. (A) Surface presentation on binding of (R)-C33 (orange sticks) and (S)-C33 (yellow) to PDE9 dimers. (R)-C33 and (S)-C33 showed unique conformation in the PDE9 dimer structures, but in the PDE9 dimer in the complex with racemic C33, (R)-C33 binds subunit B and (S)-C33 binds subunit A. The gray and cyan sticks represent the key residues involved in binding. Dotted lines represent hydrogen bonds. (B) Ribbon model for (S)-C33 binding to the PDE9 dimer. The red mesh is the electron density in the difference (Fo − Fc) map that was calculated from the structure with omission of (S)-C33 and contoured at 3σ. (C) Binding of (R)-C33 to subunit B of the PDE9 dimer.
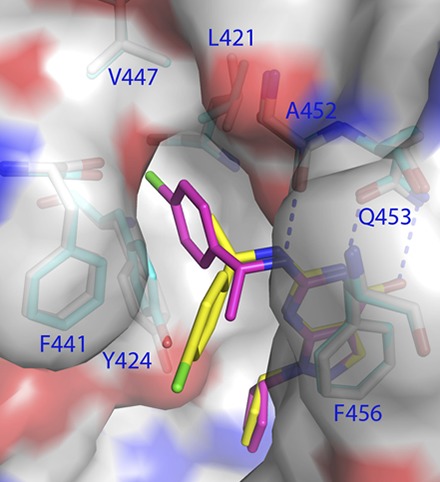
The pyrazolopyrimidinone and cyclic pentanyl rings of C33 in all three structures had the same conformation and were well superimposable. The pyrazolopyrimidinone rings of C33 formed two hydrogen bonds with invariant Gln453 and aromatic π-stack against Phe456, in addition to the hydrophobic interactions with Leu420 and Ala452. The nitrogen linked to pyrazolopyrimidinone formed a hydrogen bond with the carbonyl oxygen of Ala452. The cyclic pentanyl rings of racemic or enantiomeric C33 occupied the same location near the metal binding pocket and contacted Met365 and Tyr424. However, the chlorophenyl tails of (S)-C33 and (R)-C33 oriented to different directions and defined the different bound conformations of the two enantiomers in the PDE9 dimers (Fig. 3). The chlorophenyl tail of (S)-C33 interacted with a small hydrophobic pocket, which we tentatively name the M-pocket (see Discussion), and contacted residues of Leu420, Leu421, Tyr424, Phe441, Met442, Ala452, and Phe456. In comparison, the chlorophenyl tail of (R)-C33 pointed out the molecular surface and contacted residues of Leu420, Tyr424, Phe441, Ala452, and Phe456 (Fig. 3C). Apparently, the (S)-C33 tail makes more van der Waals interactions with PDE9 than with (R)-C33, thus explaining the slightly better binding affinity.
Since the link between chiral carbon and nitrogen (Fig. 1) is a single bond, free rotation of the bond might produce an orientation of the (R)-C33 tail in a similar conformation as (S)-C33 when interacting with the M-pocket. However, the crystal structure revealed that the chlorobenzyl tail of (R)-C33 oriented to the molecular surface of the PDE9 dimer, instead of interacting with the M-pocket. The different conformations and interactions of the tails of the (S)- and (R)-C33 enantiomers might be impacted by the subtle asymmetry of the M-pocket in the PDE9 dimer. Therefore, the structural information suggests that both subunits of the PDE9 dimer need to be considered for design of PDE9 inhibitors.
Discussion
An important Subpocket for Selectivity of PDE Inhibitors.
The early studies on the crystal structures of PDE9 in the complex with BAY73-6691 (Wang et al., 2010) and 28s (Meng et al., 2012) showed a small pocket neighboring the inhibitor binding site. This study revealed that the chlorophenyl tail of inhibitor (S)-C33 interacts with the small subpocket that is composed of a portion of helices H14 and H15 and the M-loop, which we thus tentatively named the M-pocket (Fig. 4). Helices H14 (Leu420, Leu421, and Phe425) and H15 contribute two walls of the pocket, while Val447 and the backbone of the M-loop form the bottom of the pocket. This small pocket is gated by Ala452 of H15 and Phe441 of the M-loop and occupies a similar location as the P-pocket in parasite PDEs. Sequence alignment reveals that the gating residues vary significantly across PDE families (Fig. 4C), thus making the pocket inaccessible in many human PDEs (Wang et al., 2007b, 2012).
Fig. 4.

The M-pocket for inhibitor binding. (A) Surface model of the M-pocket of PDE9. (B) Superposition of PDE9 (green ribbons) over PDE5 (pale) and PDE8A (salmon). Dotted lines represent hydrogen bonds. (C) Sequence alignment for region around the M-pocket of PDEs. The green color highlights helices in the crystal structures. The two residues in the red color gate the pocket.
In addition, the M-loop shows significant differences in sequence and conformations (Fig. 4) and is disordered in some PDE families. Therefore, we believe that the M-pocket may serve as a selectivity determinant in inhibitor binding, and is useful in improvement of inhibitor affinity and selectivity. For example, the M-pocket in PDE5, which is composed of Val782, Ala783, Phe786, Phe787, and Ile813 (Fig. 5A), is slightly deeper than that of PDE9. However, two large gating residues of Leu804 and Met816 would allow a small group to penetrate into the pocket, such as an ethoxyl fragment in the PDE5-sildenafil structure (Wang et al., 2008b). This might explain why (S)-C33 has better selectivity than (R)-C33 against PDE5 (Table 2). The M-pocket in the PDE8A1 structure, which is made up of residues Ser745, Tyr748, Phe749, Phe767, and Cys772, is much shallower and smaller than that of PDE9 and might not well accommodate the C33 inhibitors (Fig. 5B), thus explaining the poor binding of C33 to PDE8. In the PDE1 structure, the M-loop has good conservation of amino acids, as shown by the correspondence of Leu388, Met389, and Phe392 of PDE1B to Leu420, Leu421, and Y424 of PDE9, respectively. Since part of the M-pocket is disordered in PDE1, it might reasonably predict poor selectivity of PDE9 inhibitors against PDE1, as observed in the PDE9 structure (Wunder et al., 2005).
Fig. 5.

The M-pockets in other PDE families. (A) Surface presentation of the M-pocket of PDE5. (B) Surface presentation of the M-pocket of PDE8A. IBMX is a nonselective inhibitor of PDEs. (C) Ribbon presentation of the superposition of PDE9 (green) over PDE1B (cyan). The M-loop of PDE1B is partially disordered. The corresponding residues between PDE9A2 and PDE1B are M365/M336, N405/H373, L420/L388, L421/M389, Y424/F392, Q453/Q421, and F456/F424.
Supplementary Material
Acknowledgments
Diffraction data were collected at Beamline X29A at Brookhaven National Laboratory (Upton, NY) and the Southeast Regional Collaborative Access Team (SER-CAT) 22-BM beamline at the Advanced Photon Source (Argonne National Laboratory, Chicago, IL). Supporting institutions may be found at www.ser-cat.org/members.html. Use of the Advanced Photon Source (Chicago) was supported by the Office of Basic Energy Sciences, Office of Science, U.S. Department of Energy (Washington, DC), under Contract No. W-31-109-Eng-38.
Abbreviations
- brs
broad singlet
- C33
6-((1-(4-chlorophenyl)ethyl)amino)-1-cyclopentyl-1,5,6,7-tetrahydro-4H-pyrazolo[3,4-day]pyrimidin-4-one
- C40
6-((1-(4-chlorophenyl)-2-methylpropyl)amino)-1-cyclopentyl-1,5,6,7-tetrahydro-4H-pyrazolo[3,4-day]pyrimidin-4-one
- d
doublet
- DMSO
dimethylsulfoxide
- hept
heptat
- IBMX
3-isobutyl-1-methylxanthine
- IR
infrared
- m
multiplet
- MS
mass spectrometry
- p
pentet
- PDE
phosphodiesterase
- q
quartet
- RMSD
root-mean-square deviation
- s
singlet
- t
triplet
Authorship Contributions
Participated in research design: Wan, Liu, Ke, Luo.
Conducted experiments: M. Huang, Hou, Shao, Wu, Cui, Liang.
Performed data analysis: M. Huang, Shao, Cui, Liang, Li, Zhu, Y. Huang.
Wrote or contributed to the writing of the manuscript: Wan, Ke, Luo.
Footnotes
This work is partially supported by the National Institutes of Health [Grant GM59791] to H.K.; Natural Science Foundation of China [21272287, 81373258]; Major Project of Guangdong Province [2012A080201007]; Guangdong Natural Science Foundation [S2011030003190, S2013010014867]; Guangzhou Science Foundation [2014J4100165]; and the Research Fund for the Doctoral Program of Higher Education of China [20130171110096].
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.
References
- Agranat I, Caner H, Caldwell J. (2002) Putting chirality to work: the strategy of chiral switches. Nat Rev Drug Discov 1:753–768. [DOI] [PubMed] [Google Scholar]
- Claffey MM, Helal CJ, Verhoest PR, Kang Z, Fors KS, Jung S, Zhong J, Bundesmann MW, Hou X, Lui S, et al. (2012) Application of structure-based drug design and parallel chemistry to identify selective, brain penetrant, in vivo active phosphodiesterase 9A inhibitors. J Med Chem 55:9055–9068. [DOI] [PubMed] [Google Scholar]
- Conti M, Beavo J. (2007) Biochemistry and physiology of cyclic nucleotide phosphodiesterases: essential components in cyclic nucleotide signaling. Annu Rev Biochem 76:481–511. [DOI] [PubMed] [Google Scholar]
- Deninno MP, Andrews M, Bell AS, Chen Y, Eller-Zarbo C, Eshelby N, Etienne JB, Moore DE, Palmer MJ, Visser MS, et al. (2009) The discovery of potent, selective, and orally bioavailable PDE9 inhibitors as potential hypoglycemic agents. Bioorg Med Chem Lett 19:2537–2541. [DOI] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K. (2010) Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66:486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heckman PRA, Wouters C, Prickaerts J. (2015) Phosphodiesterase inhibitors as a target for cognition enhancement in aging and Alzheimer’s disease: a translational overview. Curr Pharm Des 21:317–331. [DOI] [PubMed] [Google Scholar]
- Hou J, Xu J, Liu M, Zhao R, Luo HB, Ke H. (2011) Structural asymmetry of phosphodiesterase-9, potential protonation of a glutamic acid, and role of the invariant glutamine. PLoS One 6:e18092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huai Q, Wang H, Sun Y, Kim HY, Liu Y, Ke H. (2003) Three-dimensional structures of PDE4D in complex with roliprams and implication on inhibitor selectivity. Structure 11:865–873. [DOI] [PubMed] [Google Scholar]
- Huai Q, Wang H, Zhang W, Colman RW, Robinson H, Ke H. (2004) Crystal structure of phosphodiesterase 9 shows orientation variation of inhibitor 3-isobutyl-1-methylxanthine binding. Proc Natl Acad Sci USA 101:9624–9629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutson PH, Finger EN, Magliaro BC, Smith SM, Converso A, Sanderson PE, Mullins D, Hyde LA, Eschle BK, Turnbull Z, et al. (2011) The selective phosphodiesterase 9 (PDE9) inhibitor PF-04447943 (6-[(3S,4S)-4-methyl-1-(pyrimidin-2-ylmethyl)pyrrolidin-3-yl]-1-(tetrahydro-2H-pyran-4-yl)-1,5-dihydro-4H-pyrazolo[3,4-d]pyrimidin-4-one) enhances synaptic plasticity and cognitive function in rodents. Neuropharmacology 61:665–676. [DOI] [PubMed] [Google Scholar]
- Kleiman RJ, Chapin DS, Christoffersen C, Freeman J, Fonseca KR, Geoghegan KF, Grimwood S, Guanowsky V, Hajós M, Harms JF, et al. (2012) Phosphodiesterase 9A regulates central cGMP and modulates responses to cholinergic and monoaminergic perturbation in vivo. J Pharmacol Exp Ther 341:396–409. [DOI] [PubMed] [Google Scholar]
- Kroker KS, Mathis C, Marti A, Cassel JC, Rosenbrock H, Dorner-Ciossek C. (2014) PDE9A inhibition rescues amyloid beta-induced deficits in synaptic plasticity and cognition. Neurobiol Aging 35:2072–2078. [DOI] [PubMed] [Google Scholar]
- Kroker KS, Rast G, Giovannini R, Marti A, Dorner-Ciossek C, Rosenbrock H. (2012) Inhibition of acetylcholinesterase and phosphodiesterase-9A has differential effects on hippocampal early and late LTP. Neuropharmacology 62:1964–1974. [DOI] [PubMed] [Google Scholar]
- Liddie S, Anderson KL, Paz A, Itzhak Y. (2012) The effect of phosphodiesterase inhibitors on the extinction of cocaine-induced conditioned place preference in mice. J Psychopharmacol 26:1375–1382. [DOI] [PubMed] [Google Scholar]
- Liu S, Mansour MN, Dillman KS, Perez JR, Danley DE, Aeed PA, Simons SP, Lemotte PK, Menniti FS. (2008) Structural basis for the catalytic mechanism of human phosphodiesterase 9. Proc Natl Acad Sci USA 105:13309–13314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Londesborough J. (1985) Evidence that the peripheral cyclic AMP phosphodiesterase of rat liver plasma membranes is a metalloenzyme. Biochem J 225:143–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurice DH, Ke H, Ahmad F, Wang Y, Chung J, Manganiello VC. (2014) Advances in targeting cyclic nucleotide phosphodiesterases. Nat Rev Drug Discov 13:290–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng F, Hou J, Shao YX, Wu PY, Huang M, Zhu X, Cai Y, Li Z, Xu J, Liu P, et al. (2012) Structure-based discovery of highly selective phosphodiesterase-9A inhibitors and implications for inhibitor design. J Med Chem 55:8549–8558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mentel M, Blankenfeldt W, Breinbauer R. (2009) The active site of an enzyme can host both enantiomers of a racemic ligand simultaneously. Angew Chem Int Ed Engl 48:9084–9087. [DOI] [PubMed] [Google Scholar]
- Nagy D, Tingley FD, 3rd, Stoiljkovic M, Hajós M. (2015) Application of neurophysiological biomarkers for Huntington’s disease: evaluating a phosphodiesterase 9A inhibitor. Exp Neurol 263:122–131. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 276:307–326. [DOI] [PubMed] [Google Scholar]
- Schwam EM, Nicholas T, Chew R, Billing CB, Davidson W, Ambrose D, Altstiel LD. (2014) A multicenter, double-blind, placebo-controlled trial of the PDE9A inhibitor, PF-04447943, in Alzheimer’s disease. Curr Alzheimer Res 11:413–421. [DOI] [PubMed] [Google Scholar]
- Sekhon BS. (2013) Exploiting the power of stereochemistry in drugs: an overview of racemic and enantiopure drugs. J Mod Med Chem 1:10–36. [Google Scholar]
- Shaner NC, Steinbach PA, Tsien RY. (2005) A guide to choosing fluorescent proteins. Nat Methods 2:905–909. [DOI] [PubMed] [Google Scholar]
- Shao YX, Huang M, Cui W, Feng LJ, Wu Y, Cai Y, Li Z, Zhu X, Liu P, Wan Y, et al. (2014) Discovery of a phosphodiesterase 9A inhibitor as a potential hypoglycemic agent. J Med Chem 57:10304–10313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh N, Patra S. (2014) Phosphodiesterase 9: insights from protein structure and role in therapeutics. Life Sci 106:1–11. [DOI] [PubMed] [Google Scholar]
- van der Staay FJ, Rutten K, Bärfacker L, Devry J, Erb C, Heckroth H, Karthaus D, Tersteegen A, van Kampen M, Blokland A, et al. (2008) The novel selective PDE9 inhibitor BAY 73-6691 improves learning and memory in rodents. Neuropharmacology 55:908–918. [DOI] [PubMed] [Google Scholar]
- Vardigan JD, Converso A, Hutson PH, Uslaner JM. (2011) The selective phosphodiesterase 9 (PDE9) inhibitor PF-04447943 attenuates a scopolamine-induced deficit in a novel rodent attention task. J Neurogenet 25:120–126. [DOI] [PubMed] [Google Scholar]
- Venkatachalam CM, Jiang X, Oldfield T, Waldman M. (2003) LigandFit: a novel method for the shape-directed rapid docking of ligands to protein active sites. J Mol Graph Model 21:289–307. [DOI] [PubMed] [Google Scholar]
- Verhoest PR, Fonseca KR, Hou X, Proulx-Lafrance C, Corman M, Helal CJ, Claffey MM, Tuttle JB, Coffman KJ, Liu S, et al. (2012) Design and discovery of 6-[(3S,4S)-4-methyl-1-(pyrimidin-2-ylmethyl)pyrrolidin-3-yl]-1-(tetrahydro-2H-pyran-4-yl)-1,5-dihydro-4H-pyrazolo[3,4-d]pyrimidin-4-one (PF-04447943), a selective brain penetrant PDE9A inhibitor for the treatment of cognitive disorders. J Med Chem 55:9045–9054. [DOI] [PubMed] [Google Scholar]
- Verhoest PR, Proulx-Lafrance C, Corman M, Chenard L, Helal CJ, Hou X, Kleiman R, Liu S, Marr E, Menniti FS, et al. (2009) Identification of a brain penetrant PDE9A inhibitor utilizing prospective design and chemical enablement as a rapid lead optimization strategy. J Med Chem 52:7946–7949. [DOI] [PubMed] [Google Scholar]
- Wang H, Kunz S, Chen G, Seebeck T, Wan Y, Robinson H, Martinelli S, Ke H. (2012) Biological and structural characterization of Trypanosoma cruzi phosphodiesterase C and implications for design of parasite selective inhibitors. J Biol Chem 287:11788–11797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Liu Y, Chen Y, Robinson H, Ke H. (2005) Multiple elements jointly determine inhibitor selectivity of cyclic nucleotide phosphodiesterases 4 and 7. J Biol Chem 280:30949–30955. [DOI] [PubMed] [Google Scholar]
- Wang H, Liu Y, Hou J, Zheng M, Robinson H, Ke H. (2007a) Structural insight into substrate specificity of phosphodiesterase 10. Proc Natl Acad Sci USA 104:5782–5787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Liu Y, Huai Q, Cai J, Zoraghi R, Francis SH, Corbin JD, Robinson H, Xin Z, Lin G, et al. (2006) Multiple conformations of phosphodiesterase-5: implications for enzyme function and drug development. J Biol Chem 281:21469–21479. [DOI] [PubMed] [Google Scholar]
- Wang H, Luo X, Ye M, Hou J, Robinson H, Ke H. (2010) Insight into binding of phosphodiesterase-9A selective inhibitors by crystal structures and mutagenesis. J Med Chem 53:1726–1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Yan Z, Geng J, Kunz S, Seebeck T, Ke H. (2007b) Crystal structure of the Leishmania major phosphodiesterase LmjPDEB1 and insight into the design of the parasite-selective inhibitors. Mol Microbiol 66:1029–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Yan Z, Yang S, Cai J, Robinson H, Ke H. (2008a) Kinetic and structural studies of phosphodiesterase-8A and implication on the inhibitor selectivity. Biochemistry 47:12760–12768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Ye M, Robinson H, Francis SH, Ke H. (2008b) Conformational variations of both phosphodiesterase-5 and inhibitors provide the structural basis for the physiological effects of vardenafil and sildenafil. Mol Pharmacol 73:104–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn MD, Murshudov GN, Papiz MZ. (2003) Macromolecular TLS refinement in REFMAC at moderate resolutions. Methods Enzymol 374:300–321. [DOI] [PubMed] [Google Scholar]
- Wu G, Robertson DH, Brooks CL, 3rd, Vieth M. (2003) Detailed analysis of grid-based molecular docking: A case study of CDOCKER-A CHARMm-based MD docking algorithm. J Comput Chem 24:1549–1562. [DOI] [PubMed] [Google Scholar]
- Wunder F, Tersteegen A, Rebmann A, Erb C, Fahrig T, Hendrix M. (2005) Characterization of the first potent and selective PDE9 inhibitor using a cGMP reporter cell line. Mol Pharmacol 68:1775–1781. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.