

Abstract
Doxorubicin (DOX), an effective cancer chemotherapeutic agent, induces dose-dependent cardiotoxicity, in part due to its ability to cause oxidative stress. We investigated the role of multidrug resistance–associated protein 1 (Mrp1/Abcc1) in DOX-induced cardiotoxicity in C57BL wild-type (WT) mice and their Mrp1 null (Mrp1−/−) littermates. Male mice were administered intraperitoneal DOX (3 or 2 mg/kg body weight) or saline twice a week for 3 weeks and examined 2 weeks after the last dose (protocol A total dose: 18 mg/kg) or for 5 weeks, and mice were examined 48 hours and 2 weeks after the last dose (protocol B total dose: 20 mg/kg). Chronic DOX induced body weight loss and hemotoxicity, adverse effects significantly exacerbated in Mrp1−/− versus WT mice. In the heart, significantly higher basal levels of glutathione (1.41-fold ± 0.27-fold) and glutathione disulfide (1.35-fold ± 0.16-fold) were detected in Mrp1−/− versus WT mice, and there were comparable decreases in the glutathione/glutathione disulfide ratio in WT and Mrp1−/− mice after DOX administration. Surprisingly, DOX induced comparable increases in 4-hydroxynonenal glutathione conjugate concentration in hearts from WT and Mrp1−/− mice. However, more DOX-induced apoptosis was detected in Mrp1−/− versus WT hearts (P < 0.05) (protocol A), and cardiac function, assessed by measurement of fractional shortening and ejection fraction with echocardiography, was significantly decreased by DOX in Mrp1−/− versus WT mice (P < 0.05; 95% confidence intervals of 20.0%–24.3% versus 23.7%–29.5% for fractional shortening, and 41.5%–48.4% versus 47.7%–56.7% for ejection fraction; protocol B). Together, these data indicate that Mrp1 protects the mouse heart against chronic DOX-induced cardiotoxicity.
Introduction
Doxorubicin (DOX) is an effective chemotherapeutic anthracycline used for a variety of solid tumors and hematologic malignancies; however, its clinical use is significantly limited by its dose-dependent cardiotoxicity (Minotti et al., 2004; Octavia et al., 2012). Cardiomyopathy is the dose-limiting toxicity in cancer patients treated with DOX, with an incidence 2.5 times higher than in untreated patients (Doyle et al., 2005). It is well established that the reactive oxygen species (ROS) generated by DOX, which cycles between the quinone and semiquinone in cardiac mitochondria, contribute significantly to cardiac pathology (Yen et al., 1996; Gewirtz, 1999; Minotti et al., 2004). However, the self-regulation and defense mechanisms of heart tissue in this process remain unclear.
Multidrug resistance–associated protein 1 (MRP1/ABCC1), a member of the ATP-binding cassette (ABC) transporter protein superfamily, is ubiquitously expressed, especially in the heart, skin, lung, brain capillary endothelial cells, and small intestine (Flens et al., 1996; Nies et al., 2004). Mrp1 mediates the efflux of glutathione (GSH) and glutathione disulfide (GSSG) as well as GSH-, glucuronate-, and sulfate-conjugated organic anions, including leukotriene C4 and the 4-hydroxy-2-nonenal glutathione conjugate (GS-HNE) (Leier et al., 1994; Cole and Deeley, 1998; Renes et al., 2000; Leslie et al., 2001; Jungsuwadee et al., 2012). Although Mrp1−/− mice have normal fertility and viability, their ability to transport key endo- and xenobiotics is compromised. Thus, Mrp1−/− mice are hypersensitive to the anticancer drug etoposide and exhibit reduced inflammatory responses attributed to disrupted leukotriene homeostasis caused by the inability to efflux leukotriene C4 (Wijnholds et al., 1997; Yoshioka et al., 2009).
Accumulating case-control cohort clinical studies have shown that several MRP1 single nucleotide polymorphisms are related to susceptibility to cardiotoxicity observed in many cancer patients treated with anthracyclines such as DOX (Wojnowski et al., 2005; Semsei et al., 2012; Visscher et al., 2012). These single nucleotide polymorphisms may affect efflux of not only DOX but also other important endobiotics (Jungsuwadee et al., 2012). Thus, MRP1 could play an important role in the regulation of oxidative stress by effluxing GSH and GSSG as well as GS-HNE (Jungsuwadee et al., 2012; Cole, 2014a,b).
4-Hydroxy-2-nonenal (HNE) is an α,β-unsaturated aldehyde derived from peroxidation of ω-6 polyunsaturated fatty acids and is a major toxic product of oxidative stress (Esterbauer et al., 1991; Butterfield and Stadtman 1997). As a potent electrophile, HNE reacts with cysteine, histidine, and lysine residues of proteins, likely resulting in functional impairment (Esterbauer et al., 1991); HNE can also form adducts with DNA, inducing mutations (Feng et al., 2004). HNE is partially detoxified by conjugation with GSH, either spontaneously or through GSH-S-transferases to form GS-HNE, which must be effluxed to alleviate intracellular toxicity (Völkel et al., 2005).
GSH is the most abundant cellular nonprotein thiol and is a critical factor responsible for the maintenance of cellular redox balance and antioxidant defense during oxidative stress. In the presence of ROS, GSH is rapidly oxidized to GSSG, resulting in a decreased intracellular GSH/GSSG ratio, an indicator of oxidative stress. Maintenance of GSH homeostasis plays a vital role in a multitude of cellular processes, including drug and free radical detoxification, cell differentiation, proliferation, and apoptosis (Jones, 2008). Mrp1−/− mice have increased basal levels of GSH attributed to its decreased efflux, whereas overexpression of Mrp1 decreases intracellular levels of GSH (Cole et al., 1990; Rappa et al., 1997). Mrp1 also mediates GSSG efflux, so that loss of Mrp1 could potentially disrupt the balance of the GSH/GSSG redox couple, impairing a normal tissue’s ability to protect itself against oxidative stress–induced injury.
In this study, we explored the role of Mrp1 in the regulation of DOX-induced cardiotoxicity in mice. Although Mrp1 is highly conserved between humans and rodents (with 88% sequence homology), the human isoform (MRP1) is able to transport DOX, whereas murine Mrp1 has only a negligible ability to transport this anthracycline (Stride et al., 1997). Therefore, the Mrp1−/− mouse provides a good model to study the role of Mrp1 in DOX-induced cardiotoxicity independently from the effects on DOX retention. Here, we present evidence for the first time that loss of Mrp1 potentiated the cardiac dysfunction induced by chronic DOX treatment and changed the intracellular GSH and GSSG levels in heart tissue. These results provide novel insights into the role of Mrp1 in cardiac protection beyond the ability to transport DOX.
Materials and Methods
Animals and Treatment.
C57BL/6 wild-type (WT) mice and Mrp1-disrupted C57BL/6 (Mrp1−/−) mice, initially a gift from Dr. Gary Kruh, were backcrossed for more than 10 generations (Mrp1−/− mice will be available at The Jackson Laboratory Repository, JAX Stock No. 028129); littermates were bred in-house and maintained in the Division of Laboratory Animal Resources (University of Kentucky, Lexington, KY). All experiments complied with the requirements of the University of Kentucky Institutional Animal Care and Use Committee. The animal treatment protocols were based on studies demonstrating decreased cardiac function after chronic treatment with DOX (Zhang et al., 2009). All experiments used male mice aged 10–12 weeks weighing 25–35 g. Mice were administered intraperitoneal DOX (Pfizer, NY) at a dose of 3 mg/kg body weight (protocol A) or 2 mg/kg body weight (protocol B), or an equivalent volume of saline, twice a week for 3 or 5 weeks, resulting in a cumulative DOX dose of 18 mg/kg (protocol A) or 20 mg/kg (protocol B) (Fig. 1). Hydration and nutritional gel (72-07-5022; ClearH2O, Portland, ME) were provided as supplements (30 ml per five mice and changed every 2 days), in addition to pelleted food and water provided throughout the treatment period. Animal body weight was recorded throughout the experimental period. Animals were euthanized and examined 48 hours or 2 weeks after the last DOX treatment.
Fig. 1.

Chronic DOX treatment protocols. In protocol A, WT and Mrp1−/− mice were administered intraperitoneal DOX (3 mg/kg body weight) or an equivalent volume of saline, twice a week for 3 weeks, resulting in a cumulative dose of 18 mg/kg DOX. In protocol B, WT and Mrp1−/− mice were administered intraperitoneal DOX (2 mg/kg body weight) or an equivalent volume of saline, twice a week for 5 weeks, resulting in a cumulative dose of 20 mg/kg DOX.
Transthoracic Echocardiography.
Transthoracic echocardiography was performed with a Vevo 2100 High-Resolution In Vivo Imaging System (Visual Sonics Inc., Toronto, ON, Canada). The mice were lightly anesthetized with isoflurane (0.5%–1.5%) until the heart rate stabilized at approximately 500 beats per minute. Using the M-mode from parasternal short-axis images, the left ventricular end-diastolic dimension (LVEDD) and left ventricular end-systolic dimension (LVESD) were measured. The percentage of left ventricular fractional shortening (LVFS) was calculated as 100 × [(LVEDD − LVESD)/LVEDD]. Left ventricular end-diastolic volume (LVEDV) was estimated as [7.0/(2.4 + LVEDD)] × LVEDD3. Left ventricular end-systolic volume (LVESV) was calculated as [7.0/(2.4 + LVESD)] × LVESD3. Left ventricular ejection fraction (LVEF) was determined using LVEDV and LVESV as follows: [(LVEDV − LVESV)/LVEDV] × 100%. Echocardiography was conducted by investigators who were blinded to treatment group assignments and genotype.
Complete Blood Count.
Peripheral blood (approximately 20 µl) was collected from WT and Mrp1−/− mice by submandibular bleeding 48 hours and 2 weeks after the last DOX treatment (protocol B). Blood cell parameters were obtained on the Hemavet 950FS automated hematology analyzer (Drew Scientific, Dallas, TX).
Terminal Deoxynucleotidyl Transferase–Mediated Digoxigenin-Deoxyuridine Nick-End Labeling Assay.
The tip of the mouse heart ventricle was fixed in 10% formalin and embedded in paraffin, and 4-μm sections were cut and stained using the ApopTag Peroxidase In Situ Apoptosis Detection Kit (S7100; Millipore, Billerica, MA) according to the manufacturer’s instructions. Counterstaining with Mayer’s hematoxylin aided in morphologic evaluation in which normal nuclei were stained blue and apoptotic nuclei were stained brown. The number of terminal deoxynucleotidyl transferase–mediated digoxigenin-deoxyuridine nick-end labeling (TUNEL)–positive cells was quantitated using Aperio (Leica Biosystems Inc., Buffalo Grove, IL) scanning image analysis of sections. DNase I treatment was carried out as the positive control.
Measurement of GS-HNE in Mice Hearts by Liquid Chromatography–Tandem Mass Spectrometry.
The heart ventricle was prepared by removal of the atrium and attached fat tissue and vessels. GS-HNE measurement by liquid chromatography–tandem mass spectrometry was performed exactly as described (Deng et al., 2015).
Heart Ventricle Homogenate and High-Performance Liquid Chromatography Assay of GSH and GSSG.
The heart ventricle was prepared as above and then homogenized in 10 volumes of ice-cold buffer consisting of 0.225 M mannitol, 0.075 M sucrose, 1 mM EGTA, and protease inhibitors (1 mM phenylmethyl sulfonyl fluoride, 1 μg/ml leupeptin, 1μg/ml aprotinin, and 1 μg/ml pepstatin). The protein concentrations of heart ventricle homogenates were determined with the bicinchoninic acid protein assay. The measurement of GSH and GSSG by high-performance liquid chromatography was performed exactly as described (Deng et al., 2015) and used by others (Yamazaki et al., 2008, 2012; Yamazaki and Villarreal, 2011; Kennedy et al., 2013; Barajas-Espinosa et al., 2014). The use of authentic GSH and GSSG to develop the standard curves ensured quantitation of the correct entities by high-performance liquid chromatography, thus validating the assay. Furthermore, the GSH/GSSG ratios obtained in treatment-naïve and saline-treated WT hearts are consistent with those reported for the normal mouse heart in the literature (Han et al., 2009; Watanabe et al., 2013), supporting the validity of the assay.
RNA Extraction and Real-Time Quantitative Polymerase Chain Reaction.
Total RNA was isolated from heart ventricles using TRIzol reagent (Sigma-Aldrich, St. Louis, MO) according to the manufacturer’s instructions. RNA concentrations were determined using a NanoPhotometer (Implen GmbH, Munich, Germany). Total RNA (2 μg) was converted into cDNA with the SuperScript III First-Strand Synthesis System for real-time polymerase chain reaction (RT-PCR) (Invitrogen, Carlsbad, CA), and the mixture was diluted without purification in sterile water and used for quantitative RT-PCR analysis. mRNA expression of specific genes was quantified using the LightCycler 480 Real-Time PCR System (Roche Applied Science, Mannheim, Germany). The following forward and reverse primers were used: brain natriuretic peptide (BNP), 5′-GTCAGTCGTTTGGGCTGTAAC-3′ (forward) and 5′-AGACCCAGGCAGAGTCAGA-3′ (reverse); and 18S rRNA, 5′-GCAATTATTCCCCATGAACG-3′ (forward) and 5′-GGGACTTAATCAACGCAAGC-3′ (reverse). Primers were obtained from Integrated DNA Technologies (Coralville, IA), and Universal Probe Library probes 71 (BNP) and 48 (18S rRNA) were from Roche Applied Science. The quantitative RT-PCR reactions were performed in duplicate in 15 μl reaction volume containing 2 μl 1:10 diluted cDNA and 1× LightCycler 480 Probes Master Mix. 18S rRNA was selected as the reference gene, for which cDNA samples were diluted 1:5000. Data were evaluated by calibrator-normalized relative quantification with efficiency correction using Light Cycler 480 software (version 1.5; Roche Applied Science).
Statistical Analysis.
All data are expressed as the mean ± S.E. for 5–12 mice per group, as detailed in the figure legends. For body weight data, a linear mixed model was considered with fixed effects of treatment group, day, and their interaction and random effects of both the intercept and slope. The slope of weight loss and the weight change at the end of the treatment were compared between two treatment groups based on the linear mixed model. For data from other experiments, we first used a Bartlett test to test homogeneity of variance across all groups. If the Bartlett test results were not significant, further statistical analysis was performed using a one-way analysis of variance with post hoc analysis by the Newman–Keuls method. If the Bartlett test results were significant, further statistical analysis was performed using Welch t tests for pairwise comparisons between groups of interest with the Bonferroni correction for multiple comparison adjustment.
Results
Effects of Chronic DOX Administration on Body Weight and Blood Counts.
We treated Mrp1−/− mice and their WT littermates with either 3 mg/kg body weight, twice a week for 3 weeks (Fig. 2A), or 2 mg/kg body weight, twice a week for 5 weeks (Fig. 2B). DOX markedly decreased body weight in WT and Mrp1−/− mice; after DOX treatment was discontinued and during the 2-week recovery period, body weight stabilized and began to recover in all DOX-treated animals. Comparison of the slope of weight loss for DOX-treated Mrp1−/− versus WT mice and the weight change in the two groups at the end of the treatment (days 21 and 35 for the 3 and 2 mg/kg DOX treatment groups, respectively) showed that the magnitude of the decrease was significantly larger in Mrp1−/− mice compared with WT mice (P < 0.001 for both comparisons). With the 3 mg/kg DOX treatment, 4 of 13 Mrp1−/− mice were euthanized due to body weight loss (22%–25% of initial body weight) before the last day of the experimental period. Thus, for subsequent experiments, except for the TUNEL assay, data were acquired using the 2 mg/kg DOX dose (protocol B). DOX treatment also significantly decreased heart weight in both genotypes (Supplemental Fig. 1).
Fig. 2.

Body weight changes in WT and Mrp1−/− mice during chronic DOX treatment. WT and Mrp1−/− mice were administered intraperitoneal DOX with protocol A (A) or protocol B (B) and maintained for an additional 2 weeks (“recovery”); animal weight was monitored throughout. The arrow indicates that one mouse was euthanized on each of the indicated days due to body weight loss (loss of 22%–25% of initial body weight). Values are means ± S.E. n = 12–14 for (A) and n = 12 before day 35 and n = 6 after day 35 for (B).
Chronic DOX treatment (2 mg/kg) significantly decreased white blood cell (WBC) and lymphocyte counts in both genotypes 48 hours after the last DOX treatment (Fig. 3, A and B). Two weeks later, the WBC, including lymphocyte counts, had recovered in WT mice but remained significantly decreased in Mrp1−/− mice (P < 0.05) (Fig. 3, C and D).
Fig. 3.

The effects of DOX on WBC and lymphocyte counts. Mice were treated with protocol B, and WBC and lymphocyte counts were measured 48 hours (A and B) and 2 weeks (C and D) after the last DOX dose. Each bar represents the mean ± S.E. (n = 8 for the saline-treated group; n = 12 for the DOX-treated group). *P < 0.05 DOX versus saline of the same genotype; #P < 0.05 Mrp1−/− versus respective WT mice by Newman–Keuls multiple comparison test after one-way analysis of variance. K/µl, 1000 cells per microliter; LY, lymphocyte.
Cardiac Function after Chronic DOX Administration.
To determine whether cardiac contractile function was affected by chronic DOX administration, LVEDD and LVESD were assessed by in vivo echocardiography 2 weeks after the last DOX treatment (2 mg/kg), and LVFS and LVEF were calculated. Figure 4A shows representative echocardiograms of WT and Mrp1−/− mice after saline or DOX administration. There were no significant differences in LVFS and LVEF between WT and Mrp1−/− saline-treated mice (Fig. 4B), indicating that Mrp1−/− mice had normal basal contractile function. DOX treatment increased both LVEDD and LVESD in WT and Mrp1−/− mice, and the increase in LVESD was statistically significant in Mrp1−/− mice (data not shown). Importantly, DOX significantly reduced LVFS and LVEF in Mrp1−/− mice, whereas there were no significant changes in these values in WT mice (Fig. 4, B and C). This cardiac dysfunction in Mrp1−/− mice after DOX treatment coincided with increased mRNA expression of BNP, which is a marker of systolic left ventricular dysfunction in the heart (Fig. 4D). These data clearly demonstrate that loss of Mrp1 exacerbated DOX-induced cardiac dysfunction.
Fig. 4.

Chronic DOX treatment leads to more severe systolic dysfunction in Mrp1−/− versus WT mice. Mice were treated with protocol B, and cardiac function was assessed through M-mode transthoracic echocardiography. (A) Representative M-mode echocardiogram of left ventricular wall motion. (B) Fractional shortening. (C) Ejection fraction. (D) mRNA expression of BNP analyzed by quantitative RT-PCR 2 weeks after chronic saline or DOX treatment. Each bar represents the mean ± S.E. (n = 8 for the saline-treated group; n = 12 for the DOX-treated group). *P < 0.05 DOX versus saline of the same genotype; #P < 0.05 Mrp1−/− versus respective WT mice by the Newman–Keuls multiple comparison test after one-way analysis of variance.
DOX-Induced Apoptosis in the Mouse Heart.
We investigated whether loss of Mrp1 potentiated DOX-induced cell apoptosis in the heart, using TUNEL staining to evaluate heart sections. Cells containing intensive terminal deoxynucleotidyl transferase–positive staining in the nuclei were considered apoptotic. DOX treatment (3 mg/kg) increased apoptosis in the mouse heart of both genotypes; however, Mrp1−/− hearts showed significantly more extensive (approximately 3.5-fold higher) TUNEL staining in the nuclei compared with WT hearts (P < 0.05) (Fig. 5), consistent with the more severe cardiotoxicity seen with measurements of systolic function.
Fig. 5.

Apoptosis in mouse myocardium. Mice were treated using protocol A, and apoptosis from mouse myocardium was detected by TUNEL staining. Representative photomicrographs are shown, demonstrating TUNEL staining of heart sections from saline-treated WT (A), DOX-treated WT (B), saline-treated Mrp1−/− (C), and DOX-treated Mrp1−/− (D) mice. TUNEL-positive cells are indicated by brown staining, and the TUNEL-positive nuclei are indicated by arrows. (E) Histogram showing the quantitative analysis of TUNEL-positive cells. Each bar represents the mean ± S.E. (n = 6 for the saline-treated group; n = 10 for DOX-treated WT mice; n = 8 for DOX-treated Mrp1−/− mice). *P < 0.05 DOX versus saline of the same genotype; #P < 0.05 Mrp1−/− versus respective WT mice by Welch t test.
DOX Treatment Increases GS-HNE in the Mouse Heart.
DOX initiates ROS and causes lipid peroxidation, with HNE as the major toxic lipid metabolite. The highly electrophilic HNE reacts rapidly with nucleophiles, particularly GSH, to form GS-HNE. We therefore characterized the retention of GS-HNE in WT and Mrp1−/− mouse heart tissue after chronic DOX treatment (2 mg/kg). As shown in Fig. 6, DOX increased GS-HNE in WT and Mrp1−/− mouse hearts measured 48 hours (Fig. 6A) and 2 weeks (Fig. 6B) after the last DOX treatment; the increase was statistically significant at 2 weeks. However, in contrast with our expectations, there was no significant difference in GS-HNE levels between genotypes.
Fig. 6.

Concentration of GS-HNE in the heart after saline and DOX treatment. Mice were treated with protocol B, and the GS-HNE concentration in the heart was measured by liquid chromatography–tandem mass spectrometry 48 hours (A) or 2 weeks (B) after the last dose of saline or DOX. Each bar represents the mean ± S.E. (n = 5 for the saline-treated group; n = 6 for the DOX-treated group). *P < 0.05 DOX versus saline of the same genotype by the Newman–Keuls multiple comparison test after one-way analysis of variance.
GSH and GSSG Levels in the Mouse Heart.
Because GSH and GSSG are known substrates for Mrp1, we investigated whether loss of Mrp1 would alter GSH and GSSG cellular levels, thus further compromising the antioxidant capacity of cells. We examined GSH and GSSG 48 hours and 2 weeks after the last DOX dose. DOX treatment (2 mg/kg) significantly decreased both GSH and the GSH/GSSG ratio at 48 hours, indicating oxidative stress (Fig. 7A). GSH and GSH/GSSG had returned to control levels 2 weeks later, at the end of the recovery period (Fig. 7B). Although there was no significant difference in the GSH/GSSG ratio between WT and Mrp1−/− mice at either time point, it is interesting to note the significantly higher basal (saline treatment) levels of GSH and GSSG in the Mrp1−/− mouse heart compared with the WT mouse heart. GSSG levels were also significantly higher in Mrp1−/− versus WT mice after DOX treatment at both time points. These observations are consistent with the function of Mrp1 to efflux GSH and GSSG.
Fig. 7.
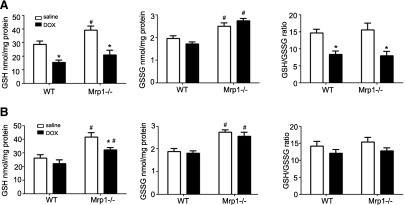
Assessment of GSH, GSSG, and the GSH/GSSG ratio in the mouse heart. Mice were treated with protocol B, and GSH and GSSG concentrations were measured by high-performance liquid chromatography at 48 hours (A) or 2 weeks (B) after the last dose of DOX. Each bar represents the mean ± S.E (n = 6). *P < 0.05 DOX versus saline of the same genotype; #P < 0.05 versus respective WT mice by Newman–Keuls multiple comparison test after one-way analysis of variance.
Discussion
The key finding in this study is that global deletion of Mrp1 potentiates DOX-induced cardiac toxicity in mice, as measured by echocardiography, apoptosis, and the ventricular dysfunction marker, BNP. DOX is an antitumor anthracycline that is effective in treating a wide variety of cancers, but it produces dose-limiting cardiac toxicity. Although the mechanism of DOX-induced cardiotoxicity is complex and multifactorial, it is well accepted that oxidative stress contributes significantly to DOX-induced heart failure (Yen et al., 1996; Minotti et al., 2004; Kang, 2007). We and others have shown that adduction of cardiac proteins with HNE, a toxic product of lipid peroxidation induced by oxidative stress, is increased in DOX-treated mice (Renes et al., 2000; Jungsuwadee et al., 2006). In addition, sarcolemmal membrane vesicles from WT mouse heart transport GS-HNE, but this activity is absent in such vesicles from Mrp1−/− mice (Jungsuwadee et al., 2012). We recently found that 72 hours after a single dose of DOX (15 mg/kg body weight, i.v.), there is significantly more nuclear injury in the Mrp1−/− heart compared with the WT heart (Deng et al.; 2015). On the basis of these findings, we hypothesized that Mrp1 protects against DOX-induced cardiac dysfunction.
Here, we extended these findings to assess whether loss of Mrp1 affected cardiac function after DOX treatment and we found that chronic DOX treatment caused more severe left ventricle dysfunction in Mrp1−/− mice, presenting as decreased LVFS and LVEF. These pathologic changes are also consistent with the higher BNP expression and more apoptotic nuclei observed in the Mrp1−/− mouse heart. Thus, despite the higher GSH levels present in hearts of Mrp1−/− mice due to loss of GSH efflux (Deng et al., 2015), loss of Mrp1 potentiated DOX-induced cardiac dysfunction and greater nuclear damage.
Several ABC transport mRNA/proteins have been reported to be present to various degrees in the heart (Couture et al., 2006) and could contribute to efflux of DOX. P-glycoprotein (Abcb1a, Abcb1b), breast cancer resistance protein (Abcg2), and Mrp2 (Abcc2) were demonstrated to contribute to DOX resistance by mediating its efflux (Couture et al., 2006; Vlaming et al., 2006; Natarajan et al., 2012). In addition, Mrp4 (Abcc4) is able to efflux GSH and GSSG (Ballatori et al., 2005). There is very low to no expression of Abcc2 in the heart (Couture et al., 2006), and we were unable to detect protein expression of Abcg2 in heart tissue (data not shown). To rule out potentially altered, compensatory expression of other transporters in the heart of Mrp1−/− mice, we measured the protein expression of Abcb1 and Abcc4 2 weeks after the last dose of DOX or saline and found no significant differences between WT and Mrp1−/− mice hearts after treatment with saline or DOX (Supplemental Fig. 2).
HNE is detected in heart tissues as early as 3 hours after DOX administration (Luo et al., 1997; Liu and Tan, 2003; Chaiswing et al., 2004). Furthermore, HNE-adducted protein is increased in the mouse heart after DOX treatment (20 mg/kg, i.p.) and Mrp1 is the sole transporter of GS-HNE in the mouse heart (Jungsuwadee et al., 2006, 2012). We anticipated that DOX treatment would increase HNE adduction of proteins, but we did not detect any differences in HNE-adducted proteins in heart homogenate between treatment or genotypes 48 hours and 2 weeks after DOX treatment. Similarly, we anticipated that the loss of Mrp1 would eliminate efflux of GS-HNE and cause its intracellular accumulation; however, we did not detect a significant difference in the GS-HNE concentration in the heart between WT and Mrp1−/− mice after chronic DOX treatment. GS-HNE was somewhat elevated at 48 hours after DOX and significantly increased 2 weeks after the last dose of DOX, but this increase was similar in WT and Mrp1−/− mice. By contrast, after a single dose of DOX, GS-HNE was significantly decreased in WT mice relative to saline treatment, whereas there was no change in GS-HNE in Mrp1−/− mice (Deng et al., 2015). We have shown that in WT mice, a single dose of DOX (20 mg/kg i.p.) induces expression of Mrp1 that is maximal at 6 hours (Jungsuwadee et al., 2006); this increased Mrp1 is a likely compensatory effect sufficient to efflux GS-HNE from the heart in WT mice. However, this increased Mrp1 is not sufficiently long lasting to sustain low GS-HNE levels in WT mice after 5 weeks of DOX treatment, because immunoblots of the heart from saline- and DOX-treated mice 48 hours and 2 weeks after chronic DOX treatment did not show any differences in expression of Mrp1 (data not shown).
In addition to conjugation with GSH to yield GS-HNE, HNE metabolism includes reduction to the corresponding alcohol (1,4-dihydroxy-2-nonene) or oxidation to the corresponding acid (4-hydroxy-2-nonenoic acid) (Alary et al., 2003; Völkel et al., 2005). The loss of GS-HNE efflux may saturate these metabolic pathways, but it can also lead to metabolism of GS-HNE via an NADH-dependent alcohol dehydrogenase-catalyzed reduction to glutathionyl-1,4-dihydroxy-2-nonene and/or an aldehyde dehydrogenase-catalyzed oxidation to glutathionyl-4-hydroxy-2-nonenoic acid. The biologic activities of these GSH conjugates are not yet well characterized nor has their cellular efflux by Mrp1 been described (Dalleau et al., 2013; Frohnert and Bernlohr, 2014). We did not measure these additional HNE metabolic products; thus, we cannot conclude whether loss of Mrp1 alters HNE metabolism in the mouse heart.
Because GSH/GSSG is the most important redox couple in the cell, we investigated how the loss of Mrp1-mediated efflux would affect maintenance of GSH and GSSG homeostasis after DOX treatment. Chronic DOX treatment decreased GSH and the GSH/GSSG ratio 48 hours after the last DOX treatment, indicating the presence of oxidative stress that was similar between the genotypes, but these values had recovered to control values 2 weeks later. These data are consistent with a decrease in GSH and GSH/GSSG upon oxidative stress that in turn triggers activation of the antioxidant stress system to increase GSH synthesis and restore a normal GSH/GSSG ratio. As an indicator of intracellular oxidative stress, the GSH/GSSG ratio was similar between WT and Mrp1−/− mice, whereas the levels of GSH and GSSG were higher in Mrp1−/− mice, regardless of whether they were treated with DOX or saline, consistent with loss of Mrp1-mediated efflux. In contrast with the effects of chronic DOX treatment, neither GSH nor the GSH/GSSG ratio was different from that in saline-treated mice 72 hours after a single intravenous dose of DOX (15 mg/kg) (Deng et al., 2015), implying that redox homeostasis was restored 72 hours after a single DOX dose and that although recovery had not occurred by 48 hours, recovery was complete 2 weeks after chronic DOX treatment.
Decreased recovery from hematopoietic toxicity was also seen in Mrp1−/− mice after DOX treatment. This transient myelosuppression, shown as decreased WBC counts and lymphocyte counts in DOX-treated mice, is probably caused by the suppression or apoptosis of hematopoietic progenitor cells. How Mrp1 inhibits the replenishment of these blood cells is not clear. Another key finding was that DOX treatment caused a more severe body weight loss in Mrp1−/− mice. This is likely associated with reduced food consumption, since DOX causes loss of appetite, a phenomenon commonly observed in DOX-treated cancer patients. It is also well known that DOX causes dose-limiting gastrointestinal injury due to its toxic effect on intestinal epithelium, including the rapidly dividing stem cells located at the base of the intestinal crypts. Apoptosis in the intestinal epithelium and the loss of villi throughout the small intestine could compromise digestive and absorptive capacities, finally causing severe body weight loss in mice (Ijiri and Potten, 1987). However, since these Mrp1−/− mice are constitutive knockout animals, we could not determine whether the intestines or other organs were primarily responsible for this differential extent of body weight loss between WT and Mrp1−/− mice.
In summary, the key important and novel finding provided by these studies is the demonstration that DOX causes a marked decrease in cardiac function, consistent with significantly greater apoptosis, in Mrp1−/− versus WT mice. These data are more surprising in view of the significantly higher concentrations of GSH in hearts of Mrp1−/− versus WT mice. Deletion of Mrp1 clearly perturbed the regulation of GSH due to retention of both GSH and GSSG in the cell, and this dysregulation may well contribute to the greater DOX cardiotoxicity in Mrp1−/− mice. These data suggest that Mrp1 has additional functions whose loss contributes to toxicity and provide critical information regarding the potential adverse sequelae of introduction of MRP1 inhibitors as adjuncts to clinical chemotherapy of multidrug-resistant tumors.
Supplementary Material
Acknowledgments
The authors thank Dr. John Seubert (University of Alberta, Edmonton, AB, Canada) for numerous helpful discussions regarding the chronic DOX treatment model (Zhang et al., 2009).
Abbreviations
- ABC
ATP-binding cassette
- BNP
brain natriuretic peptide
- DOX
doxorubicin
- GS-HNE
4-hydroxy-2-nonenal glutathione conjugate
- GSH
glutathione
- GSSG
glutathione disulfide
- HNE
4-hydroxy-2-nonenal
- LVEDD
left ventricular end-diastolic dimension
- LVEDV
left ventricular end-diastolic volume
- LVEF
left ventricular ejection fraction
- LVESD
left ventricular end-systolic dimension
- LVESV
left ventricular end-systolic volume
- LVFS
left ventricular fractional shortening
- ROS
reactive oxygen species
- RT-PCR
real-time polymerase chain reaction
- TUNEL
terminal deoxynucleotidyl transferase–mediated digoxigenin-deoxyuridine nick-end labeling
- WBC
white blood cell
- WT
wild type
Authorship Contributions
Participated in research design: Zhang, St. Clair, Vore.
Conducted experiments: Zhang.
Contributed new reagents or analytic tools: Sunkara, Morris.
Performed data analysis: Zhang, Vore.
Wrote or contributed to the writing of the manuscript: Zhang, Deng, Morris, Wang, St. Clair, Vore.
Footnotes
This research was supported by the National Institutes of Health National Cancer Institute [Grants R01CA139844 and P30CA177558 (to the Biostatistics and Bioinformatics, Biospecimen and Tissue Procurement and Redox Metabolism Shared Resources of the University of Kentucky Markey Cancer Center)] and by an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health [P20 GM103527]. W.Z. was supported by the American Heart Association [Predoctoral Fellowship 17060037].
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.
References
- Alary J, Guéraud F, Cravedi JP. (2003) Fate of 4-hydroxynonenal in vivo: disposition and metabolic pathways. Mol Aspects Med 24:177–187. [DOI] [PubMed] [Google Scholar]
- Ballatori N, Hammond CL, Cunningham JB, Krance SM, Marchan R. (2005) Molecular mechanisms of reduced glutathione transport: role of the MRP/CFTR/ABCC and OATP/SLC21A families of membrane proteins. Toxicol Appl Pharmacol 204:238–255. [DOI] [PubMed] [Google Scholar]
- Barajas-Espinosa A, Basye A, Jesse E, Yan H, Quan D, Chen CA. (2014) Redox activation of DUSP4 by N-acetylcysteine protects endothelial cells from Cd²⁺-induced apoptosis. Free Radic Biol Med 74:188–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butterfield DA, Stadtman ER. (1997) Protein oxidation processes in aging brain. Adv Cell Aging Gerontol 2:161–191. [Google Scholar]
- Chaiswing L, Cole MP, St Clair DK, Ittarat W, Szweda LI, Oberley TD. (2004) Oxidative damage precedes nitrative damage in adriamycin-induced cardiac mitochondrial injury. Toxicol Pathol 32:536–547. [DOI] [PubMed] [Google Scholar]
- Cole SP. (2014a) Multidrug resistance protein 1 (MRP1, ABCC1), a “multitasking” ATP-binding cassette (ABC) transporter. J Biol Chem 289:30880–30888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole SP. (2014b) Targeting multidrug resistance protein 1 (MRP1, ABCC1): past, present, and future. Annu Rev Pharmacol Toxicol 54:95–117. [DOI] [PubMed] [Google Scholar]
- Cole SP, Deeley RG. (1998) Multidrug resistance mediated by the ATP-binding cassette transporter protein MRP. BioEssays 20:931–940. [DOI] [PubMed] [Google Scholar]
- Cole SP, Downes HF, Mirski SE, Clements DJ. (1990) Alterations in glutathione and glutathione-related enzymes in a multidrug-resistant small cell lung cancer cell line. Mol Pharmacol 37:192–197. [PubMed] [Google Scholar]
- Couture L, Nash JA, Turgeon J. (2006) The ATP-binding cassette transporters and their implication in drug disposition: a special look at the heart. Pharmacol Rev 58:244–258. [DOI] [PubMed] [Google Scholar]
- Dalleau S, Baradat M, Guéraud F, Huc L. (2013) Cell death and diseases related to oxidative stress: 4-hydroxynonenal (HNE) in the balance. Cell Death Differ 20:1615–1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng J, Coy D, Zhang W, Sunkara M, Morris AJ, Wang C, Chaiswing L, St. Clair D, Vore M, Jungsuwadee P. (2015) Elevated glutathione is insufficient to protect against doxorubicin-induced nuclear damage in heart in multidrug resistance associated protein 1 (Mrp1/Abcc1) null mice. J Pharmacol Exp Ther DOI: 10.1124/jpet.115.225490 [published ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle JJ, Neugut AI, Jacobson JS, Grann VR, Hershman DL. (2005) Chemotherapy and cardiotoxicity in older breast cancer patients: a population-based study. J Clin Oncol 23:8597–8605. [DOI] [PubMed] [Google Scholar]
- Esterbauer H, Schaur RJ, Jr, Zollner H. (1991) Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic Biol Med 11:81–128. [DOI] [PubMed] [Google Scholar]
- Feng Z, Hu W, Tang MS. (2004) Trans-4-hydroxy-2-nonenal inhibits nucleotide excision repair in human cells: a possible mechanism for lipid peroxidation-induced carcinogenesis. Proc Natl Acad Sci USA 101:8598–8602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flens MJ, Zaman GJ, van der Valk P, Izquierdo MA, Schroeijers AB, Scheffer GL, van der Groep P, de Haas M, Meijer CJ, Scheper RJ. (1996) Tissue distribution of the multidrug resistance protein. Am J Pathol 148:1237–1247. [PMC free article] [PubMed] [Google Scholar]
- Frohnert BI, Bernlohr DA. (2014) Glutathionylated products of lipid peroxidation: a novel mechanism of adipocyte to macrophage signaling. Adipocyte 3:224–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gewirtz DA. (1999) A critical evaluation of the mechanisms of action proposed for the antitumor effects of the anthracycline antibiotics adriamycin and daunorubicin. Biochem Pharmacol 57:727–741. [DOI] [PubMed] [Google Scholar]
- Han B, Baliga R, Huang H, Giannone PJ, Bauer JA. (2009) Decreased cardiac expression of vascular endothelial growth factor and redox imbalance in murine diabetic cardiomyopathy. Am J Physiol Heart Circ Physiol 297:H829–H835. [DOI] [PubMed] [Google Scholar]
- Ijiri K, Potten CS. (1987) Further studies on the response of intestinal crypt cells of different hierarchical status to eighteen different cytotoxic agents. Br J Cancer 55:113–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones DP. (2008) Radical-free biology of oxidative stress. Am J Physiol Cell Physiol 295:C849–C868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jungsuwadee P, Cole MP, Sultana R, Joshi G, Tangpong J, Butterfield DA, St Clair DK, Vore M. (2006) Increase in Mrp1 expression and 4-hydroxy-2-nonenal adduction in heart tissue of Adriamycin-treated C57BL/6 mice. Mol Cancer Ther 5:2851–2860. [DOI] [PubMed] [Google Scholar]
- Jungsuwadee P, Zhao T, Stolarczyk EI, Paumi CM, Butterfield DA, St Clair DK, Vore M. (2012) The G671V variant of MRP1/ABCC1 links doxorubicin-induced acute cardiac toxicity to disposition of the glutathione conjugate of 4-hydroxy-2-trans-nonenal. Pharmacogenet Genomics 22:273–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang YJ. (2007) Antioxidant defense against anthracycline cardiotoxicity by metallothionein. Cardiovasc Toxicol 7:95–100. [DOI] [PubMed] [Google Scholar]
- Kennedy LH, Sutter CH, Leon Carrion S, Tran QT, Bodreddigari S, Kensicki E, Mohney RP, Sutter TR. (2013) 2,3,7,8-Tetrachlorodibenzo-p-dioxin-mediated production of reactive oxygen species is an essential step in the mechanism of action to accelerate human keratinocyte differentiation. Toxicol Sci 132:235–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leier I, Jedlitschky G, Buchholz U, Cole SP, Deeley RG, Keppler D. (1994) The MRP gene encodes an ATP-dependent export pump for leukotriene C4 and structurally related conjugates. J Biol Chem 269:27807–27810. [PubMed] [Google Scholar]
- Leslie EM, Ito K, Upadhyaya P, Hecht SS, Deeley RG, Cole SP. (2001) Transport of the β -O-glucuronide conjugate of the tobacco-specific carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol (NNAL) by the multidrug resistance protein 1 (MRP1). Requirement for glutathione or a non-sulfur-containing analog. J Biol Chem 276:27846–27854. [DOI] [PubMed] [Google Scholar]
- Liu QY, Tan BK. (2003) Relationship between anti-oxidant activities and doxorubicin-induced lipid peroxidation in P388 tumour cells and heart and liver in mice. Clin Exp Pharmacol Physiol 30:185–188. [DOI] [PubMed] [Google Scholar]
- Luo X, Evrovsky Y, Cole D, Trines J, Benson LN, Lehotay DC. (1997) Doxorubicin-induced acute changes in cytotoxic aldehydes, antioxidant status and cardiac function in the rat. Biochim Biophys Acta 1360:45–52. [DOI] [PubMed] [Google Scholar]
- Minotti G, Menna P, Salvatorelli E, Cairo G, Gianni L. (2004) Anthracyclines: molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol Rev 56:185–229. [DOI] [PubMed] [Google Scholar]
- Natarajan K, Xie Y, Baer MR, Ross DD. (2012) Role of breast cancer resistance protein (BCRP/ABCG2) in cancer drug resistance. Biochem Pharmacol 83:1084–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nies AT, Jedlitschky G, König J, Herold-Mende C, Steiner HH, Schmitt HP, Keppler D. (2004) Expression and immunolocalization of the multidrug resistance proteins, MRP1-MRP6 (ABCC1-ABCC6), in human brain. Neuroscience 129:349–360. [DOI] [PubMed] [Google Scholar]
- Octavia Y, Tocchetti CG, Gabrielson KL, Janssens S, Crijns HJ, Moens AL. (2012) Doxorubicin-induced cardiomyopathy: from molecular mechanisms to therapeutic strategies. J Mol Cell Cardiol 52:1213–1225. [DOI] [PubMed] [Google Scholar]
- Rappa G, Lorico A, Flavell RA, Sartorelli AC. (1997) Evidence that the multidrug resistance protein (MRP) functions as a co-transporter of glutathione and natural product toxins. Cancer Res 57:5232–5237. [PubMed] [Google Scholar]
- Renes J, de Vries EE, Hooiveld GJ, Krikken I, Jansen PL, Müller M. (2000) Multidrug resistance protein MRP1 protects against the toxicity of the major lipid peroxidation product 4-hydroxynonenal. Biochem J 350:555–561. [PMC free article] [PubMed] [Google Scholar]
- Semsei AF, Erdelyi DJ, Ungvari I, Csagoly E, Hegyi MZ, Kiszel PS, Lautner-Csorba O, Szabolcs J, Masat P, Fekete G, et al. (2012) ABCC1 polymorphisms in anthracycline-induced cardiotoxicity in childhood acute lymphoblastic leukaemia. Cell Biol Int 36:79–86. [DOI] [PubMed] [Google Scholar]
- Stride BD, Grant CE, Loe DW, Hipfner DR, Cole SP, Deeley RG. (1997) Pharmacological characterization of the murine and human orthologs of multidrug-resistance protein in transfected human embryonic kidney cells. Mol Pharmacol 52:344–353. [DOI] [PubMed] [Google Scholar]
- Vlaming ML, Mohrmann K, Wagenaar E, de Waart DR, Elferink RP, Lagas JS, van Tellingen O, Vainchtein LD, Rosing H, Beijnen JH, et al. (2006) Carcinogen and anticancer drug transport by Mrp2 in vivo: studies using Mrp2 (Abcc2) knockout mice. J Pharmacol Exp Ther 318:319–327. [DOI] [PubMed] [Google Scholar]
- Visscher H, Ross CJ, Rassekh SR, Barhdadi A, Dubé MP, Al-Saloos H, Sandor GS, Caron HN, van Dalen EC, Kremer LC, et al. Canadian Pharmacogenomics Network for Drug Safety Consortium (2012) Pharmacogenomic prediction of anthracycline-induced cardiotoxicity in children. J Clin Oncol 30:1422–1428. [DOI] [PubMed] [Google Scholar]
- Völkel W, Alvarez-Sánchez R, Weick I, Mally A, Dekant W, Pähler A. (2005) Glutathione conjugates of 4-hydroxy-2(E)-nonenal as biomarkers of hepatic oxidative stress-induced lipid peroxidation in rats. Free Radic Biol Med 38:1526–1536. [DOI] [PubMed] [Google Scholar]
- Watanabe Y, Watanabe K, Kobayashi T, Saito Y, Fujioka D, Nakamura T, Obata JE, Kawabata K, Mishina H, Kugiyama K. (2013) Chronic depletion of glutathione exacerbates ventricular remodelling and dysfunction in the pressure-overloaded heart. Cardiovasc Res 97:282–292. [DOI] [PubMed] [Google Scholar]
- Wijnholds J, Evers R, van Leusden MR, Mol CA, Zaman GJ, Mayer U, Beijnen JH, van der Valk M, Krimpenfort P, Borst P. (1997) Increased sensitivity to anticancer drugs and decreased inflammatory response in mice lacking the multidrug resistance-associated protein. Nat Med 3:1275–1279. [DOI] [PubMed] [Google Scholar]
- Wojnowski L, Kulle B, Schirmer M, Schlüter G, Schmidt A, Rosenberger A, Vonhof S, Bickeböller H, Toliat MR, Suk EK, et al. (2005) NAD(P)H oxidase and multidrug resistance protein genetic polymorphisms are associated with doxorubicin-induced cardiotoxicity. Circulation 112:3754–3762. [DOI] [PubMed] [Google Scholar]
- Yamazaki KG, Ihm SH, Thomas RL, Roth D, Villarreal F. (2012) Cell adhesion molecule mediation of myocardial inflammatory responses associated with ventricular pacing. Am J Physiol Heart Circ Physiol 302:H1387–H1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki KG, Romero-Perez D, Barraza-Hidalgo M, Cruz M, Rivas M, Cortez-Gomez B, Ceballos G, Villarreal F. (2008) Short- and long-term effects of (-)-epicatechin on myocardial ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol 295:H761–H767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki KG, Villarreal FJ. (2011) Ventricular pacing-induced loss of contractile function and development of epicardial inflammation. Am J Physiol Heart Circ Physiol 300:H1282–H1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen HC, Oberley TD, Vichitbandha S, Ho YS, St Clair DK. (1996) The protective role of manganese superoxide dismutase against adriamycin-induced acute cardiac toxicity in transgenic mice. J Clin Invest 98:1253–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshioka M, Sagara H, Takahashi F, Harada N, Nishio K, Mori A, Ushio H, Shimizu K, Okada T, Ota M, et al. (2009) Role of multidrug resistance-associated protein 1 in the pathogenesis of allergic airway inflammation. Am J Physiol Lung Cell Mol Physiol 296:L30–L36. [DOI] [PubMed] [Google Scholar]
- Zhang Y, El-Sikhry H, Chaudhary KR, Batchu SN, Shayeganpour A, Jukar TO, Bradbury JA, Graves JP, DeGraff LM, Myers P, et al. (2009) Overexpression of CYP2J2 provides protection against doxorubicin-induced cardiotoxicity. Am J Physiol Heart Circ Physiol 297:H37–H46. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.