

Abstract
Activation of kappa opioid receptors (KORs) expressed by peripheral sensory neurons that respond to noxious stimuli (nociceptors) can reduce neurotransmission of pain stimuli from the periphery to the central nervous system. We have previously shown that the antinociception dose-response curve for peripherally restricted doses of the KOR agonist (–)-(trans)-3,4-dichloro-N-methyl-N-[2-(1-pyrrolidinyl)cyclohexyl]benzeneacetamide (U50488) has an inverted U shape. Here, we found that the downward phase of the U50488 dose-response curve was blocked by an inhibitor of extracellular signal-regulated kinase (ERK) activation U0126. Local administration of the selective KOR agonist salvinorin A (Sal-A), also resulted in an inverted U-shaped curve; however, the downward phase was insensitive to U0126. By contrast, inhibition of c-Jun N-terminal kinase (JNK) partially blocked the downward phase of the dose-response curve to Sal-A, suggesting a role for JNK. In cultures of peripheral sensory neurons, U50488 and Sal-A inhibited adenylyl cyclase activity with similar efficacies; however, their ability to activate ERK and JNK differed. Whereas U50488 activated ERK but not JNK, Sal-A activated JNK but not ERK. Moreover, although both U50488 and Sal-A produced homologous desensitization, desensitization to U50488 was blocked by inhibition of ERK activation, whereas desensitization to Sal-A was blocked by inhibition of JNK. Substitution of an ethoxymethyl ether for the C2 position acetyl group of Sal-A reduced stimulation of JNK, prevented desensitization by ethoxymethyl ether for the C2 position acetyl group of Sal-A, and resulted in a monotonic antinociception dose-response curve. Collectively, these data demonstrate the functional selectivity of KOR ligands for signaling in peripheral sensory neurons, which results in differential effects on behavioral responses in vivo.
Introduction
Opioid receptors are key targets for drugs used for the treatment of pain. However, there are several serious adverse effects associated with opioid pharmacotherapy (e.g., respiratory depression, addiction, euphoria, and sedation), many of which are due to opioid actions at receptors located within the central nervous system (CNS). To avoid CNS-mediated adverse effects, it may be possible to target inhibitory opioid receptors expressed on peripheral pain-sensing neurons (nociceptors). µ-Opioid receptor (MOR), delta opioid receptor, and kappa opioid receptor (KOR) subtypes are expressed by peripheral nociceptors (Fields et al., 1980; Chen et al., 1997; Stein and Lang, 2009; Stein and Zollner, 2009). Both clinical and animal studies have shown that peripheral opioid receptors, when activated, can inhibit the transmission of nociceptive signals from the periphery to the CNS (Gupta et al., 2001; Kalso et al., 2002; Stein et al., 2003; Zollner et al., 2008). Moreover, recent evidence suggests that activation of these peripheral opioid receptors can contribute to analgesia produced by systemic opioid administration (Labuz et al., 2007; Weibel et al., 2013; Jagla et al., 2014). However, unlike their CNS counterparts, much less is known about the function and regulation of peripheral opioid receptor systems expressed by nociceptors.
Activation of KOR with peripherally restricted doses of KOR agonists produces antinociception in a variety of animal pain models, without demonstrable CNS-mediated adverse effects (for reviews, see Riviere, 2004; Kivell and Prisinzano, 2010; Vanderah, 2010; and Vadivelu et al., 2011). In a previous study using an inflammatory pain model system, we demonstrated that the KOR agonist (–)-(trans)-3,4-dichloro-N-methyl-N-[2-(1-pyrrolidinyl)cyclohexyl]benzeneacetamide (U50488) produces a strong inhibition of prostaglandin E2 (PGE2)-stimulated thermal allodynia when administered directly into the plantar surface of the rat hindpaw at doses that do not act systemically (Berg et al., 2011). However, the dose-response curve to U50488 is not monotonic; rather, it has an inverted U shape. At doses of U50488 above 0.1 µg, the antinociceptive effect decreases and returns to baseline levels at a dose of 10 µg. Inverted U-shaped dose-response curves reduce the therapeutic window of drugs and therefore limit their usefulness as therapeutic agents.
In this study, we examined the cellular mechanisms responsible for the downward phase of the inverted U-shaped dose-response curve to KOR agonists for the inhibition of thermal allodynia in vivo. We found that the downward phase of the dose-response curve to U50488 was KOR mediated and involved the activation of the extracellular signal-regulated kinase (ERK). Interestingly, the dose-response curve to a different KOR agonist, salvinorin A (Sal-A), also had an inverted U shape, but the downward phase was not mediated by ERK activation. Instead, activation of the c-Jun N-terminal kinase (JNK) played a role in the downward phase of the Sal-A dose-response curve. The addition of an ethoxymethyl ether to the C2 position acetyl group of Sal-A (EOM-Sal-B) reduced stimulation of JNK and resulted in a monotonic dose-response curve for the inhibition of thermal allodynia. These results indicate that in peripheral sensory neurons, ligands of different structures can differentially regulate cellular signaling mechanisms (i.e., functional selectivity), leading to differential effects on behavior in vivo. Further, these results suggest that modifications to a ligand can fine tune the signaling profile to enhance beneficial, and minimize adverse, physiologic effects.
Materials and Methods
Drugs and Chemicals.
The following compounds were purchased from Sigma-Aldrich (St. Louis, MO): bradykinin (BK) acetate salt, rolipram, nor-binaltorphimine dihydrochlorite (nor-BNI), (–)-trans-(1S, 2S)-U50488 hydrochloride hydrate (U50488), U0126 (mitogen/extracellular signal-regulated kinase 1/2 inhibitor), SP600125 (JNK inhibitor), (D-Ala2, N-MePhe4, Gly-ol)-enkephalin (DAMGO), and L-JNKi-1 trifluoroacetate salt (JNK-inhibitor peptide). SCH772984 (ERK inhibitor) and JNK-IN-8 (JNK inhibitor) were purchased from Selleckchem (Houston, TX). PGE2 was purchased from Cayman Chemical (Ann Arbor, MI). Sal-A was isolated from Salvia divinorum, and EOM-Sal-B was synthesized in the laboratory of Dr. Thomas Prisinzano (Lawrence, KS). 125I-cAMP was purchased from PerkinElmer Life and Analytical Sciences (Waltham, MA). Nerve growth factor was purchased from Harlan Laboratories (Indianapolis, IN), collagenase was purchased from Worthington Biochemicals (Freehold, NJ), and all other tissue culture reagents were purchased from Life Sciences (Carlsbad, CA).
Animals.
Adult male Sprague-Dawley rats (Charles River Laboratories, Inc., Wilmington, MA) weighing 250–300 g were used in this study. The animal study protocol was approved by the Institutional Animal Care and Use Committee of the University of Texas Health Science Center at San Antonio and conformed to the International Association for the Study of Pain and federal guidelines. Animals were housed for 1 week, with food and water available ad libitum, before experimentation.
Behavioral Assay of Nociceptor Function.
Agonist-mediated changes in paw withdrawal latency (PWL) to a thermal stimulus were measured with a plantar test apparatus as described previously (Berg et al., 2011). The radiant heat stimulus intensity was set to produce a baseline PWL of 10 ± 2 seconds, with a cutoff time of 25 seconds to prevent tissue damage. Because peripheral opioid receptor–mediated antinociception typically requires an inflammatory stimulus (Patwardhan et al., 2005; Berg et al., 2007, 2011; Obara et al., 2009; Rowan et al., 2009; Stein and Lang, 2009; Stein and Zollner, 2009; Sullivan et al., 2015), in all experiments, BK and PGE2 were used to enhance opioid receptor–mediated antinociception. After baseline PWL was determined, animals were pretreated (15 minutes) with BK (25 μg) via intraplantar (i.pl.) injection. BK injection produces a transient (<10 minutes) allodynia, such that PWL returns to baseline before opioid administration. Rats then received a coinjection (i.pl.) of PGE2 (0.3 µg) with U50488, Sal-A (doses indicated), or vehicle. Measurements of PWL were taken in duplicate at least 30 seconds apart at 5-minute intervals for 20 minutes after PGE2/opioid injections. Where indicated, antagonists or inhibitors were injected (i.pl.) with BK 15 minutes before injection of PGE2 with or without KOR agonists. Time-course data are expressed as the change (seconds) from individual PWL baseline values and represent mean ± S.E.M. Area under the curve was quantified by averaging the area under each individual time-course curve for rats in a group. Drugs were solubilized as follows: BK and nor-BNI were solubilized in phosphate buffered saline (PBS); U50488 was solubilized in double distilled H2O; U0126, SP600125, JNK inhibitor peptide, and Sal-A were solubilized in dimethyl sulfoxide (final dilution = 0.1%); and PGE2 was solubilized in ethanol (final dilution = 0.1%). All drugs were administered via i.pl. injection at a final volume of 50 μl. Observers were blinded to the treatment allocation.
Primary Sensory Neuron Cultures.
Primary cultures of rat trigeminal ganglion neurons were prepared as described previously (Berg et al., 2007, 2011). Cells were maintained in culture for 6 days before experimentation. For all experiments, cells were refed with serum-free Dulbecco’s modified Eagle’s medium without nerve growth factor on day 5 and used for experiments on the sixth day of culture (i.e., after a 24-hour serum/nerve growth factor free period).
Measurement of Cellular cAMP Accumulation.
Opioid receptor–mediated inhibition of PGE2-stimulated adenylyl cyclase activity was determined by measuring the amount of cellular cAMP accumulated in the presence of the phosphodiesterase inhibitor rolipram (100 µM) and PGE2 (1 µM) with or without the indicated κ-opioid receptor ligands as described previously (Berg et al., 2007, 2011; Sullivan et al., 2015). As in the behavior experiments, cells were pretreated for 15 minutes with BK (10 µM) for 15 minutes before the application of PGE2 ± opioid agonist. When tested, antagonists or inhibitors were administered 15 minutes before the opioid ligand (coadministered with BK). For experiments to assess Gαi protein mediation, cells were treated with pertussis toxin (PTx) (400 ng/ml) for 24 hours.
Desensitization of KOR-Mediated Inhibition of PGE2-Stimulated cAMP Accumulation.
To elicit desensitization, cells were pretreated with maximal concentrations of either U50488 (1 µM), Sal-A (1 µM), or EOM-Sal-B (1 µM) in the presence of BK for 15 minutes. After pretreatment, cells were washed several times and the ability of either U50488 (100 nM), Sal-A (100 nM), or EOM-Sal-B (100 nM) to inhibit PGE2-stimulated cAMP accumulation was determined. When tested, inhibitors were incubated with the cells 15 minutes before pretreatment KOR agonists.
Measurement of ERK1/2 Activation.
KOR agonist–mediated activation of ERK in peripheral sensory neurons was measured as described previously (Berg et al., 2011). ERK activity was determined by measuring the levels of phosphorylated ERK with the AlphaScreen SureFire Phospho-ERK1/2 Kit (PerkinElmer Life and Analytical Sciences) according to the manufacturer’s instructions and a FluoStar microplate reader equipped with AlphaScreen technology (BMG Labtech GMBH, Ortenberg, Germany).
Measurement of JNK Activation.
Peripheral sensory neurons were plated in six-well plates and maintained for 5 days as described above. Cells were washed with 4 ml per well of Hank’s balanced salt solution containing 20 mM HEPES (pH 7.4) and incubated for 15 minutes at 37°C before the addition of ligands. Cells were incubated with or without BK (10 µM, 15 minutes, 37°C), followed by a further incubation with maximal concentrations of U50488 (100 nM) or Sal-A (100 nM) for 2.5, 5, 15, 30, and 60 minutes in the presence of phosphatase inhibitor cocktail 3 (0.1%; Sigma-Aldrich) and okadaic acid (10 nM; Sigma-Aldrich). Incubation was terminated by aspiration of the buffer and the addition of 2 ml of ice-cold PBS to each well. Plates were placed on ice, PBS was aspirated, and cells were lysed by adding 50 μl of lysis buffer (Pierce, Thermo Scientific Rockford, IL), supplemented with 1% phosphatase inhibitor cocktail 3, 100 nM okadaic acid, and 1% protease inhibitor (Pierce, Thermo Scientific) per well. Cells were scraped and centrifuged at 2000 rpm at 4°C for 3 minutes, and the supernatant was collected and prepared for SDS-PAGE following the NuPAGE protocol (Novex, Life Technologies Grand Island, NY). Immunoblots were probed with anti–rat phospho-JNK rabbit monoclonal antibody (Cell Signaling Danvers, MA). Actin was measured as a loading control with an anti-rat actin goat polyclonal antibody (Santa Cruz Biotechnology, Santa Cruz, CA). Goat anti-rabbit IR800 and donkey anti-goat IR680 secondary antibodies (LI-COR Biosciences, Lincoln, NE) were used to detect phospho-JNK and actin, respectively. Immunoblots were imaged using a LI-COR Odyssey infrared imager, and relative band intensities were quantified using Odyssey software (LI-COR Biosciences).
Data Analysis.
For cell culture experiments, concentration-response data were fit to a logistic equation (eq. 1) using nonlinear regression analysis to provide estimates of maximal response (Rmax), potency (EC50), and slope factor (n).
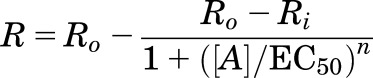 |
(1) |
where R is the measured response at a given agonist concentration (A), Ro is the response in the absence of the agonist, Ri is the response after maximal inhibition by the agonist, EC50 is the concentration of the agonist that produces a half-maximal response, and n is the slope index. Rmax (the maximal inhibition produced by the agonist) was calculated as Ro − Ri. Experiments were repeated at least three times. Statistical differences in concentration-response curve parameters between groups were analyzed with the Student’s paired t test. When only a single agonist concentration was used, statistical significance was assessed using one-way analysis of variance (ANOVA) followed by Bonferroni’s post hoc or Student’s t test (paired) using Prism software (GraphPad Software, Inc., San Diego, CA). P < 0.05 was considered statistically significant.
For behavioral experiments, time-course data were analyzed for statistical significance with two-way ANOVA followed by Bonferroni’s post hoc test. Area under the time-response curves were calculated and analyzed with one-way ANOVA followed by Bonferroni’s post hoc test. Data are presented as mean ± S.E.M., and P < 0.05 was considered statistically significant.
Results
Intraplantar injection of PGE2 (0.3 µg, 50 µl) elicited a mild (3–5 second reduction in PWL), prolonged (>20 minutes) thermal allodynia (Supplemental Fig. 1). As shown in Fig. 1, coinjection (i.pl.) of either U50488 or Sal-A at a dose of 0.1 µg not only completely abolished PGE2-induced thermal allodynia, but also produced robust antinociception (PWL was significantly above preinjection baseline). However, the antinociceptive effects of both U50488 and Sal-A were dose limited. As the dose of either agonist was increased above 0.1 µg, the magnitude of the antinociceptive effect diminished, resulting in an inverted U-shaped dose response curve (Fig. 1). Injection (i.pl.) of the selective KOR antagonist nor-BNI (0.3 ng, 10 × Ki) 15 minutes before either U50488 or Sal-A produced a rightward shift of both the ascending and descending limbs of the dose-response curve for each agonist (Fig. 1), suggesting that both phases of the dose-response curve were KOR mediated. nor-BNI alone did not alter PGE2-induced thermal allodynia (PWL: −4.7 ± 1.2 seconds versus −4.9 ± 1.6 seconds for Veh versus nor-BNI, respectively). Injection (i.pl.) of U50488 (Berg et al., 2011) or Sal-A (not shown) into the contralateral paw did not alter PGE2-induced thermal allodynia in the ipsilateral paw, indicating the actions of the KOR agonists were peripherally restricted.
Fig. 1.

Dose response curves for U50488 (A) and salvinorin A (B) to inhibit PGE2-induced thermal allodynia in the rat hindpaw. Rats received i.pl. injections of nor-BNI (0.3 ng) or vehicle (Veh) 15 minutes before induction of thermal allodynia by intraplantar injection of PGE2 (0.3 μg). U50488 (doses indicated), Sal-A (doses indicated), or vehicle was coinjected with PGE2. PWLs in response to radiant heat applied to the ventral surface of the hindpaw were measured in duplicate before nor-BNI/Veh injections and at 5-minute intervals for 20 minutes after the last injection. Data are expressed as the mean ± S.E.M. of the area under the time-course curves for individual rats. Each data point represents the mean ± S.E.M. of 4–10 animals.
ERK has been shown to have a role in regulating nociceptor function (Dai et al., 2002; Ji et al., 2009) and can be activated by KOR in the cells of the CNS (Bruchas and Chavkin, 2010). Previously, we have shown that activation of KOR with U50488 increases ERK activity in peripheral sensory neurons (Berg et al., 2011). To determine if activation of ERK was responsible for the descending phase of the dose-response curves for U50488- and Sal-A–mediated antinociception, we injected the selective mitogen-activated protein kinase kinase 1/2 inhibitor U0126 (50 µl, 10 µg, i.pl.) into the rat hindpaw 15 minutes before i.pl. injection of a dose of U50488 (10 µg) or Sal-A (10 µg). As shown in Fig. 2A, the response to U50488 was enhanced by U0126. By contrast, the response to Sal-A was insensitive to U0126 (Fig. 2B).
Fig. 2.

The descending limb of dose-response curves for U50488 and Sal-A is differentially regulated. Rats received intraplantar injections of the inhibitor of ERK activation, U0126 (10 µg), the inhibitors of JNK, SP600125 (1.0 µg) or L-JNKi-1 (30 µg), or vehicle 15 minutes before PGE2 (0.3 µg, i.pl.), with various doses of U50488 (A), Sal-A (B), or vehicle. PWL was measured before and at 5-minute intervals for 20 minutes following the last injection. Area under the curve values were calculated from the time-course data for each animal. The dose-response curve data for U50488 and Sal-A are reproduced from Fig. 1. Data represent the mean ± S.E.M. of four to eight rats. *P < 0.05 compared with the corresponding 10-µg dose of U50488 or Sal-A.
Previous studies have shown that JNK can be activated via KOR (Bruchas and Chavkin, 2010) and that activation of JNK is involved in pain responsiveness (Zhuang et al., 2006). Further, activation of JNK can disrupt KOR signaling (Bruchas et al., 2007b; Bruchas and Chavkin, 2010; Melief et al., 2010, 2011). To determine if the activation of JNK was responsible for the downward phase of the Sal-A dose-response curve for antinociception, we injected the selective JNK inhibitor SP600125 (50 µl, 1 µg, i.pl.) into the rat hindpaw 15 minutes before the injection of Sal-A (10 µg). SP600125 partially restored the antinociceptive response for Sal-A (Fig. 2B). Similar results were obtained with preinjection of L-JNKi-1 trifluoroacetate (50 µl, 30 µg, i.pl.) (Fig. 2B). Inhibition of JNK with SP600125 had no effect on the response to U50488 (Fig. 2A). Inhibitors of ERK or JNK did not alter PGE2-induced thermal allodynia (−5.85 ± 0.57 versus −5.97 ± 0.30 for PWL with ERK and JNK inhibitors, respectively).
As shown in Fig. 3A, both U50488 and Sal-A produced a concentration-dependent reduction in PGE2-stimulated cAMP levels, with similar maximal inhibition and potency values in peripheral sensory neurons in culture. Maximal inhibition was 76 ± 4 and 68 ± 5% for U50488 and Sal-A, respectively. The pEC50 values were 8.11 ± 0.33 (8 nM) and 8.29 ± 0.24 (5 nM) for U50488 and Sal-A, respectively. Similar to our previous findings with U50488 (Berg et al., 2011), pretreatment with nor-BNI (3 nM, 100 × Ki) for 15 minutes or with PTx (400 ng/ml) for 24 hours completely blocked Sal-A–mediated inhibition of cAMP accumulation (Fig. 3B), indicating that this signaling pathway is mediated by KOR activation of Gαi/o proteins. nor-BNI did not alter basal or PGE2-stimulated cAMP accumulation as reported previously (Berg et al., 2011, 2012).
Fig. 3.

U50488- or Sal-A–mediated inhibition of PGE2-stimulated cAMP accumulation in rat primary sensory neuron cultures. (A) Cells were treated with PGE2 (1 µM) with or without U50488 or Sal-A (doses indicated) for 15 minutes. (B) Cells were pretreated with nor-BNI (3 nM, 15 minutes) or PTx (400 ng/ml, 24 hours) and then incubated with PGE2 plus Sal-A (100 nM) for 15 minutes. Cellular levels of cAMP were measured with radioimmunoassay. Basal cAMP levels averaged 0.22 ± 0.03 pmol per well. Data are expressed as the percentage of PGE2-stimulated levels, which averaged 217 ± 22% above basal levels and represent the mean ± S.E.M. of six experiments. ***P < 0.001 compared with vehicle (VEH) pretreatment.
Results from the behavioral experiments (Fig. 2) indicated a role for the activation of ERK or JNK in regulating the antinociceptive effect of U50488 and Sal-A, respectively. Accordingly, incubation of peripheral sensory neurons with a maximal concentration of U50488 (100 nM) increased the activation of ERK (Fig. 4A) but did not activate JNK (Fig. 4B). By contrast, Sal-A (100 nM) did not increase ERK activation (Fig. 4A) but was effective at increasing the activation of JNK (Fig. 4B).
Fig. 4.

Activation of ERK and JNK by U50488 and Sal-A in primary cultures of peripheral sensory neurons. (A) Cells were treated with U50488 (100 nM) or Sal-A (100 nM) for the times indicated, and the level of phosphorylated ERK (pERK) was measured using the pERK surefire assay kit from PerkinElmer Life and Analytical Sciences, according to the manufacturer’s protocol. Data are expressed as the percentage increase in pERK over basal (no ligand) activity and represent the mean ± S.E.M. of four experiments. (B) Cells were treated with U50488 (100 nM) or Sal-A (100 nM) for the times indicated, and the level of phospho-JNK (pJNK) (54- and 46-kD bands) was measured using Western analysis. Immunoblots were analyzed using LI-COR Odyssey Imaging software. Data points represent pJNK levels (54- and 46-kD bands) normalized to actin as a percentage of basal levels and represent the mean ± S.E.M. of four experiments. Inset: representative immunoblots (54- and 46-kD bands) of cells treated with Sal-A (top) or U50488 (bottom). **P < 0.01 and ***P < 0.001 versus basal activity (time = 0).
As a possible mechanism for the downward phase of the KOR agonist dose-response curve for antinociception, we examined whether U50488 and/or Sal-A caused desensitization of KOR signaling in peripheral sensory neurons. Pretreatment of peripheral sensory neurons in culture with a maximal concentration of U50488 completely eliminated the subsequent inhibition of PGE2-stimulated cAMP accumulation in response to a second application of U50488 (Fig. 5A), but not to Sal-A or the MOR agonist DAMGO (Fig. 5C). Pretreatment with a maximal concentration of Sal-A likewise abolished the response to a second application of Sal-A (Fig. 5B) but not to U50488 or DAMGO (Fig. 5C). The desensitization produced by U50488 was blocked by the inhibition of ERK activation with U0126 or SCH772984 (Fig. 5A). Desensitization produced by Sal-A was blocked by the JNK inhibitors SP600125 or JK-IN-8 but not by U0126 (Fig. 5B). None of the inhibitors tested altered PGE2-stimulated activity nor the inhibition of cellular cAMP levels produced by either U50488 or Sal-A.
Fig. 5.

KOR agonist–induced desensitization of inhibition of PGE2-stimulated cAMP accumulation in peripheral sensory neuron cultures. (A) Cells were pretreated with U50488 (1 µM) or vehicle for 15 minutes (desensitizing stimulus), washed, and then incubated with PGE2 with U50488 (100 nM) for an additional 15 minutes (test stimulus). The ERK inhibitors U0126 (1 µM) or SCH777984 (10 nM), JNK inhibitors SP600125 (1 µM) or JNK-IN-8 (10 nM), or vehicle were added to cells 15 minutes before the desensitizing stimulus. (B) Cells were pretreated with Sal-A (1 µM) or vehicle for 15 minutes (desensitizing stimulus), washed, and then incubated with PGE2 with Sal-A (100 nM) for an additional 15 minutes (test stimulus). The ERK inhibitor U0126 (1 µM), JNK inhibitors SP600125 (1 µM) or JNK-IN-8 (10 nM), or vehicle were added to cells 15 minutes before the desensitizing stimulus. (C) Cells were pretreated with U50488 (1 µM), Sal-A (1 µM), or vehicle (Veh) for 15 minutes (desensitizing stimulus), followed by either U50488 (100 nM), Sal-A (100 nM), or DAMGO (100 nM) (test stimulus) for 15 minutes. Data are expressed as the percentage of PGE2-stimulated cAMP levels and represent the mean ± S.E.M. of four experiments. Pretreatment with either U50488 or Sal-A had no effect on basal or PGE2-stimulated cAMP levels, which were 0.23 ± 0.04 pmol per well and 284 ± 67% above basal levels, respectively, mean ± S.E.M., n = 8. **P < 0.01 compared with corresponding vehicle pretreatment.
Figure 6A shows that an ethoxymethyl derivative of Sal-A (EOM-Sal-B) produced a monotonic dose-response curve for antinociception, unlike the inverted U-shaped curve of Sal-A (Fig. 1; Supplemental Fig. 2). nor-BNI (0.3 ng) antagonized the response to 30 µg of EOM-Sal-B. Neither the ERK inhibitor U0126 nor the JNK inhibitor L-JNKi-1 altered the maximal response to EOM-Sal-B. In peripheral sensory neurons in culture, EOM-Sal-B inhibited PGE2-stimulated cAMP accumulation, with a higher potency than Sal-A (pEC50 = 10.07 ± 0.07 [0.09 nM] versus 8.29 ± 0.24 [5 nM] for EOM-Sal-B versus Sal-A, respectively) but similar efficacy (Emax = 59 ± 2% versus 55 ± 2% for EOM-Sal-B versus Sal-A, respectively) (Fig. 6B). The higher potency of EOM-Sal-B compared with Sal-A is consistent with previous reports (Cunningham et al., 2011). However, unlike Sal-A, EOM-Sal-B did not activate JNK in peripheral sensory neurons but did activate ERK (Fig. 6, C and D). Given that activation of ERK was responsible for the downward phase of the dose-response curve for U50488, we were surprised to find that activation of ERK by EOM-Sal-B did not reduce the antinociceptive effect of EOM-Sal-B (Fig. 6A). It has been shown that the mechanism by which ERK is activated can dictate the cellular response to activated ERK (Azzi et al., 2003; Tohgo et al., 2003; Ahn et al., 2004; Zheng et al., 2008). Figure 6D shows that whereas the activation of ERK by U50488 was sensitive to PTx, indicating mediation by Gαi G proteins, activation of ERK by EOM-Sal-B was insensitive to PTx, suggesting a mechanism of ERK activation different from that of U50488. Figure 6E shows that pretreatment with EOM-Sal-B did not reduce the response to a second application of EOM-Sal-B as Sal-A and U50488 did (Fig. 5)
Fig. 6.

Effect of EOM-Sal-B on peripheral sensory neurons in vivo and ex vivo. (A) Rats received i.pl. injections of nor-BNI (0.3 ng), L-JNKi-1 (30 µg), U0126 (10 µg), or vehicle (Veh), followed by coinjection (i.pl.) of PGE2 (0.3ug) with vehicle or EOM-Sal-B (doses indicated) 15 minutes later. PWLs in response to radiant heat applied to the ventral surface of the hindpaw were measured in duplicate before injection and at 5-minute intervals for 20 minutes after the last injection. Data are expressed as the mean ± S.E.M. of the area under the time-course curves for individual rats. Each data point represents mean ± S.E.M. of five to six animals. (B) Peripheral sensory neurons in culture were treated with PGE2 (1 µM), with various concentrations of EOM-Sal-B for 15 minutes. Data are expressed as the percentage of PGE2-stimulated cAMP levels and represent the mean ± S.E.M. of five to seven experiments. The concentration response curve for Sal-A from Fig. 3 is shown in gray for comparison. (C) Peripheral sensory neurons in culture were treated with vehicle, EOM-Sal-B (100 nM), or Sal-A (100 nM) for 5 minutes, and the level of phospho-JNK (pJNK) was measured using Western analysis. Immunoblots were analyzed using Li-COR Odyssey Imaging software. Data points represent pJNK levels normalized to actin as a percentage of basal levels and represent the mean ± S.E.M. of four experiments. *P < 0.05 and **P < 0.01 compared with corresponding vehicle treatment. Inset: representative immunoblot. (D) Peripheral sensory neurons in culture were pretreated with vehicle or PTx (400 ng/ml, 24 hours) followed by U50488 (100 nM) or EOM-Sal-B (100 nM) for the times indicated. The level of phosphorylated ERK (pERK) was measured using the pERK Surefire assay kit from PerkinElmer Life and Analytical Sciences according to the manufacturer’s protocol. Data are expressed as the percentage increase in pERK over basal (no ligand) activity and represent the mean ± S.E.M. of four experiments. Data for U50488 with vehicle pretreatment are from Fig. 4 for comparison. (E) Peripheral sensory neurons in culture were pretreated with EOM-Sal-B (1.0 µM) or vehicle for 15 minutes (desensitizing stimulus), washed, and then incubated with PGE2 with EOM-Sal-B (100 nM) for an additional 15 minutes (test stimulus). Data are expressed as the percentage of PGE2-stimulated cAMP levels and represent the mean ± S.E.M. of eight experiments. Pretreatment with EOM-Sal-B did not alter basal or PGE2-stimulated cAMP levels, which were 0.75 ± 0.07 pmol per well and 240 ± 28% above basal levels, mean ± S.E.M., n = 8, respectively. **P < 0.01 compared with vehicle/vehicle treatment.
Discussion
Previously, we reported that peripherally restricted doses of the KOR agonist U50488 can produce robust antinociceptive responses in rats (Berg et al., 2011), suggesting that peripheral KOR may be a favorable target for the development of peripherally restricted opioid analgesics. Here, we further characterized the effect of KOR activation on the function of peripheral sensory neurons in vivo (inhibition of thermal allodynia in the rat hindpaw) and ex vivo (signaling in primary cultures) in response to U50488 and another highly selective KOR agonist, Sal-A (Roth et al., 2002; Cunningham et al., 2011).
Both U50488 and Sal-A reduced PGE2-elicited thermal allodynia when injected directly in the rat hindpaw. Consistent with previous results (Berg et al., 2011), the dose-response curve for U50488 had an inverted U shape, with the peak antinociceptive response occurring at a dose of 0.1 µg. As the dose increased beyond 0.1 µg, the antinociceptive effect of U50488 decreased. Similarly, the dose-response curve for Sal-A was also an inverted U shape, with a peak antinociceptive response at 0.1 µg that returned to baseline at higher doses. Both phases of the dose-response curves to U50488 and Sal-A were antagonized equivalently by the selective KOR antagonist nor-BNI, suggesting that both effects were mediated by the activation of KOR.
Interestingly, the downward phases of the dose-response curves for the two agonists were differentially sensitive to kinase inhibitors. The downward phase of the dose-response curve to U50488, but not that of Sal-A, was blocked by the inhibitor of ERK activation U0126, suggesting that the decline in antinociceptive efficacy of U50488 was due to ERK. The downward phase of the dose-response curve to Sal-A was partially blocked by inhibition of JNK activity with SP600125 or L-JNKi-1, suggesting that activation of JNK played a role in the decline in antinociceptive efficacy of Sal-A. Although U0126 and SP600125 are highly selective for their respective targets (English and Cobb, 2002), caution is required for the interpretation of results since all pharmacological inhibitors can have off-target effects. Moreover, ERK and JNK, along with other signaling molecules (e.g., p38 mitogen activated protein kinase), each can regulate several cellular functions that could influence KOR-mediated responses in vivo (Cargnello and Roux, 2011). To better understand the roles of ERK and JNK in regulating KOR function in peripheral sensory neurons, it would be important to evaluate the effects of inhibitors on the complete dose-response curves of U50488 and Sal-A to help delineate cellular mechanisms that underlie KOR regulation.
Although the efficacy of U50488 and Sal-A to inhibit PGE2-stimulated cAMP accumulation in peripheral sensory neuron cultures was similar, their ability to regulate ERK and JNK activity differed. U50488, but not Sal-A, activated ERK. Conversely, Sal-A, but not U50488, activated JNK. These results agree with the behavior experiments, where inhibition of ERK activity blocked the downward phase of the U50488 dose-response curve, but not that of Sal-A, whereas inhibition of JNK reduced the effect of Sal-A, but not that of U50488. In heterologous expression systems and in brain tissues, both U50488 and Sal-A have been reported to activate both ERK and JNK (Bruchas and Chavkin, 2010). The selective regulation of ERK and JNK by U50488 and Sal-A here illustrates the uniqueness of opioid receptor function in peripheral sensory neurons (Patwardhan et al., 2005; Berg et al., 2007, 2011; Rowan et al., 2010; Sullivan et al., 2015).
The different pattern of KOR signaling elicited by U50488 versus that of Sal-A is consistent with the concept of ligand functional selectivity or biased agonism (Urban et al., 2007; Kenakin and Christopoulos, 2013), where different ligands promote the formation of ligand-specific receptor conformations that differentially regulate the activity of various signal transduction systems coupled to the receptor. Several reports have demonstrated KOR ligand functional selectivity in HEK cells (Bruchas and Chavkin, 2010; Rives et al., 2012; Schattauer et al., 2012; White et al., 2014, 2015) and immortalized astrocytes (McLennan et al., 2008), and it has been suggested that ligands biased for G protein activation may be more effective analgesics (Bruchas et al., 2007a; White et al., 2015). Although well established in heterologous expression systems, there are relatively few studies that have established the functional selectivity of ligands toward cellular signaling pathways in physiologically relevant systems (Bailey et al., 2009; Melief et al., 2010; Schmid and Bohn, 2010; Bosier et al., 2012; Morgan et al., 2014; White et al., 2015). The differential, ligand-dependent signaling of KOR to ERK and JNK presented here is also evidence of the functional selectivity of KOR ligands in peripheral sensory neurons, both in primary culture and in vivo.
The mechanism underlying the downward phase of the dose-response curves to U50488 and Sal-A may be due to homologous desensitization. In sensory neuron cultures, pretreatment with U50488 abolished inhibition of adenylyl cyclase activity by a second application of U50488. This desensitization was blocked by inhibition of ERK activation. Since inhibition of ERK also abolished the downward phase of the U50488 dose-response curve for antinociception, this suggests that the downward phase is due to desensitization. Similarly, inhibition of JNK activity blocked desensitization of Sal-A–mediated inhibition of adenylyl cyclase activity and reduced the downward phase of the Sal-A antinociception dose-response curve. Although several cellular mechanisms underlie GPCR desensitization (Gainetdinov et al., 2004; Kelly et al., 2008), the lack of U50488- or Sal-A–induced desensitization of DAMGO signaling suggests that signaling components in common with MOR and KOR signaling (e.g., Gαi proteins and adenylyl cyclase) are not the targets of desensitization mechanisms activated by U50488 or Sal-A. Moreover, our finding that U50488 pretreatment did not desensitize the response to Sal-A and vice versa suggests that ligand-occupied KOR itself may be the target of desensitization mechanisms. Thus, the conformation of KOR occupied by Sal-A may not be a target for desensitization mechanisms activated by pretreatment with U50488, as is the conformation of KOR occupied by U50488 and vice versa.
Further evidence that desensitization may underlie the downward phase of the dose-response curves of U50488 and Sal-A comes from experiments with the ethoxymethyl derivative of Sal-A EOM-Sal-B. EOM-Sal-B had a similar efficacy to inhibit adenylyl cyclase activity as U50488 and Sal-A did in cultured neurons, but did not activate JNK as Sal-A did. Importantly, EOM-Sal-B pretreatment did not desensitize signaling by a second application of EOM-Sal-B as Sal-A and U50488 did. Accordingly, the dose-response curve for antinociception for EOM-Sal-B did not have a downward phase but instead was monotonic. Taken together, these results suggest that the downward phase of the antinociception dose-response curves for U50488 and Sal-A is due to ERK- and JNK-mediated desensitization of KOR signaling, respectively.
Interestingly, although Sal-A did not activate ERK in peripheral sensory neurons, EOM–Sal-B activated ERK to an extent similar to that of U50488. Thus, the substitution of the C2 position acetyl group of Sal-A with an ethoxymethyl ether changed the signaling specificity of Sal-A. Given that the downward phase of the antinociception dose-response curve to U50488 was mediated by ERK, we were surprised that EOM–Sal-B did not have a dose-response curve with a downward phase and the ERK inhibitor U0126 did not change the maximal antinociceptive response to EOM–Sal-B. Since the magnitude of ERK activation by EOM–Sal-B was somewhat less than that of U50488, it is possible that the lower level of ERK activity is insufficient to produce desensitization. Alternatively, perhaps the EOM–Sal-B KOR conformation is not a target for desensitization mechanisms activated by EOM–Sal-B. However, the mechanism by which EOM–Sal-B activated ERK (PTx insensitive) differed from that of U50488 (PTx sensitive). It has been shown that the consequences of the activation of ERK can differ markedly, depending upon the mechanism by which ERK is activated (Azzi et al., 2003; Tohgo et al., 2003; Ahn et al., 2004; Kohout et al., 2004; Shenoy et al., 2006; Zheng et al., 2008). For MOR, when ERK activation occurs via a non–G protein, β-arrestin–dependent mechanism, activated ERK translocates to the nucleus (Zheng et al., 2008, 2010a,b). However, when ERK activation is G protein dependent, activated ERK remains in the cytoplasm and phosphorylates various cytosolic targets (Yoon and Seger, 2006). Thus, the differential cellular mechanisms of ERK activation via U50488 versus that via EOM–Sal-B may explain why EOM–Sal-B did not produce ERK-mediated desensitization of KOR signaling.
Our results suggest that the KOR ligands U50488, Sal-A, and EOM-Sal-B elicit distinct patterns of signaling in peripheral sensory neurons and that as a result, antinociceptive behavioral responses are differentially regulated. The ability of ligands acting at the same receptor subtype to differentially regulate specific cellular signaling pathways holds great promise for new drug development. Just as selectivity for different receptor subtypes has played a crucial role in targeting drugs for specific therapeutic indications, the development of ligands with selectivity for specific signaling pathways (or specific patterns of signaling pathways) that mediate beneficial effects and with minimal activity at pathways that lead to adverse effects would be expected to further improve therapeutic efficacy. Further, this work highlights the importance of measuring ligand activity at multiple cellular signaling pathways to properly characterize ligand efficacy and improve predictions of therapeutic efficacy in vivo.
Supplementary Material
Acknowledgments
The authors thank Peter LoCoco for his helpful comments.
Abbreviations
- ANOVA
analysis of variance
- BK
bradykinin
- CNS
central nervous system
- DAMGO
(D-Ala2, N-MePhe4, Gly-ol)-enkephalin
- EOM-Sal-B
ethoxymethyl derivative of salvinorin A
- ERK
extracellular signal-regulated kinase
- i.pl.
intraplantar
- JNK
c-Jun N-terminal kinase
- KOR
kappa opioid receptor
- MOR
µ-opioid receptor
- nor-BNI
nor-binaltorphimine dihydrochloride
- PBS
phosphate buffered saline
- PGE2
prostaglandin E2
- PTx
pertussis toxin
- PWL
paw withdrawal latency
- Sal-A
salvinorin A
- U50488
(–)-(trans)-3,4-dichloro-N-methyl-N-[2-(1-pyrrolidinyl)cyclohexyl]benzeneacetamide
Authorship Contributions:
Participated in research design: Berg, Clarke, Jamshidi, Prisinzano
Conducted experiments: Jamshidi, Jacobs, Sullivan, Chavera, Saylor
Performed data analysis: Berg, Clarke, Jamshidi
Wrote or contributed to the writing of the manuscript: Berg, Clarke, Jamshidi, Prisinzano
Footnotes
This work was supported by U.S. Public Health Service grants from the National Institutes of Health [Grants RO1 DA 024865, R01 GM 106035, and R01 DA 018151]; the William and Ella Owens Medical Research Foundation; and U.S. Public Health Service training grants from the National Institute of Dental and Craniofacial Research [COSTAR Training Grant T32DE14318 to B.A.J.], the National Institute of General Medicine [Grant T32 GM 008545] to R.M.S., and the National Institute on Drug Abuse [Grant T32 DA 031115] to L.C.S.
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.
References
- Ahn S, Shenoy SK, Wei H, Lefkowitz RJ. (2004) Differential kinetic and spatial patterns of beta-arrestin and G protein-mediated ERK activation by the angiotensin II receptor. J Biol Chem 279:35518–35525. [DOI] [PubMed] [Google Scholar]
- Azzi M, Charest PG, Angers S, Rousseau G, Kohout T, Bouvier M, Piñeyro G. (2003) Beta-arrestin-mediated activation of MAPK by inverse agonists reveals distinct active conformations for G protein-coupled receptors. Proc Natl Acad Sci USA 100:11406–11411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey CP, Oldfield S, Llorente J, Caunt CJ, Teschemacher AG, Roberts L, McArdle CA, Smith FL, Dewey WL, Kelly E, et al. (2009) Involvement of PKC alpha and G-protein-coupled receptor kinase 2 in agonist-selective desensitization of mu-opioid receptors in mature brain neurons. Br J Pharmacol 158:157–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg KA, Patwardhan AM, Sanchez TA, Silva YM, Hargreaves KM, Clarke WP. (2007) Rapid modulation of micro-opioid receptor signaling in primary sensory neurons. J Pharmacol Exp Ther 321:839–847. [DOI] [PubMed] [Google Scholar]
- Berg KA, Rowan MP, Gupta A, Sanchez TA, Silva M, Gomes I, McGuire BA, Portoghese PS, Hargreaves KM, Devi LA, et al. (2012) Allosteric interactions between δ and κ opioid receptors in peripheral sensory neurons. Mol Pharmacol 81:264–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg KA, Rowan MP, Sanchez TA, Silva M, Patwardhan AM, Milam SB, Hargreaves KM, Clarke WP. (2011) Regulation of κ-opioid receptor signaling in peripheral sensory neurons in vitro and in vivo. J Pharmacol Exp Ther 338:92–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosier B, Muccioli GG, Mertens B, Sarre S, Michotte Y, Lambert DM, Hermans E. (2012) Differential modulations of striatal tyrosine hydroxylase and dopamine metabolism by cannabinoid agonists as evidence for functional selectivity in vivo. Neuropharmacology 62:2328–2336. [DOI] [PubMed] [Google Scholar]
- Bruchas MR, Chavkin C. (2010) Kinase cascades and ligand-directed signaling at the kappa opioid receptor. Psychopharmacology (Berl) 210:137–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruchas MR, Land BB, Aita M, Xu M, Barot SK, Li S, Chavkin C. (2007a) Stress-induced p38 mitogen-activated protein kinase activation mediates kappa-opioid-dependent dysphoria. J Neurosci 27:11614–11623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruchas MR, Yang T, Schreiber S, Defino M, Kwan SC, Li S, Chavkin C. (2007b) Long-acting kappa opioid antagonists disrupt receptor signaling and produce noncompetitive effects by activating c-Jun N-terminal kinase. J Biol Chem 282:29803–29811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cargnello M, Roux PP. (2011) Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol Mol Biol Rev 75:50–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JJ, Dymshitz J, Vasko MR. (1997) Regulation of opioid receptors in rat sensory neurons in culture. Mol Pharmacol 51:666–673. [DOI] [PubMed] [Google Scholar]
- Cunningham CW, Rothman RB, Prisinzano TE. (2011) Neuropharmacology of the naturally occurring kappa-opioid hallucinogen salvinorin A. Pharmacol Rev 63:316–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai Y, Iwata K, Fukuoka T, Kondo E, Tokunaga A, Yamanaka H, Tachibana T, Liu Y, Noguchi K. (2002) Phosphorylation of extracellular signal-regulated kinase in primary afferent neurons by noxious stimuli and its involvement in peripheral sensitization. J Neurosci 22:7737–7745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- English JM, Cobb MH. (2002) Pharmacological inhibitors of MAPK pathways. Trends Pharmacol Sci 23:40–45. [DOI] [PubMed] [Google Scholar]
- Fields HL, Emson PC, Leigh BK, Gilbert RF, Iversen LL. (1980) Multiple opiate receptor sites on primary afferent fibres. Nature 284:351–353. [DOI] [PubMed] [Google Scholar]
- Gainetdinov RR, Premont RT, Bohn LM, Lefkowitz RJ, Caron MG. (2004) Desensitization of G protein-coupled receptors and neuronal functions. Annu Rev Neurosci 27:107–144. [DOI] [PubMed] [Google Scholar]
- Gupta A, Bodin L, Holmström B, Berggren L. (2001) A systematic review of the peripheral analgesic effects of intraarticular morphine. Anesth Analg 93:761–770. [DOI] [PubMed] [Google Scholar]
- Jagla C, Martus P, Stein C. (2014) Peripheral opioid receptor blockade increases postoperative morphine demands--a randomized, double-blind, placebo-controlled trial. Pain 155:2056–2062. [DOI] [PubMed] [Google Scholar]
- Ji RR, Gereau RW, 4th, Malcangio M, Strichartz GR. (2009) MAP kinase and pain. Brain Res Brain Res Rev 60:135–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalso E, Smith L, McQuay HJ, Andrew Moore R. (2002) No pain, no gain: clinical excellence and scientific rigour--lessons learned from IA morphine. Pain 98:269–275. [DOI] [PubMed] [Google Scholar]
- Kelly E, Bailey CP, Henderson G. (2008) Agonist-selective mechanisms of GPCR desensitization. Br J Pharmacol 153 (Suppl 1):S379–S388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenakin T, Christopoulos A. (2013) Signalling bias in new drug discovery: detection, quantification and therapeutic impact. Nat Rev Drug Discov 12:205–216. [DOI] [PubMed] [Google Scholar]
- Kivell B, Prisinzano TE. (2010) Kappa opioids and the modulation of pain. Psychopharmacology (Berl) 210:109–119. [DOI] [PubMed] [Google Scholar]
- Kohout TA, Nicholas SL, Perry SJ, Reinhart G, Junger S, Struthers RS. (2004) Differential desensitization, receptor phosphorylation, beta-arrestin recruitment, and ERK1/2 activation by the two endogenous ligands for the CC chemokine receptor 7. J Biol Chem 279:23214–23222. [DOI] [PubMed] [Google Scholar]
- Labuz D, Mousa SA, Schäfer M, Stein C, Machelska H. (2007) Relative contribution of peripheral versus central opioid receptors to antinociception. Brain Res 1160:30–38. [DOI] [PubMed] [Google Scholar]
- McLennan GP, Kiss A, Miyatake M, Belcheva MM, Chambers KT, Pozek JJ, Mohabbat Y, Moyer RA, Bohn LM, Coscia CJ. (2008) Kappa opioids promote the proliferation of astrocytes via Gbetagamma and beta-arrestin 2-dependent MAPK-mediated pathways. J Neurochem 107:1753–1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melief EJ, Miyatake M, Bruchas MR, Chavkin C. (2010) Ligand-directed c-Jun N-terminal kinase activation disrupts opioid receptor signaling. Proc Natl Acad Sci USA 107:11608–11613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melief EJ, Miyatake M, Carroll FI, Béguin C, Carlezon WA, Jr, Cohen BM, Grimwood S, Mitch CH, Rorick-Kehn L, Chavkin C. (2011) Duration of action of a broad range of selective κ-opioid receptor antagonists is positively correlated with c-Jun N-terminal kinase-1 activation. Mol Pharmacol 80:920–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan MM, Reid RA, Saville KA. (2014) Functionally selective signaling for morphine and fentanyl antinociception and tolerance mediated by the rat periaqueductal gray. PLoS One 9:e114269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obara I, Parkitna JR, Korostynski M, Makuch W, Kaminska D, Przewlocka B, Przewlocki R. (2009) Local peripheral opioid effects and expression of opioid genes in the spinal cord and dorsal root ganglia in neuropathic and inflammatory pain. Pain 141:283–291. [DOI] [PubMed] [Google Scholar]
- Patwardhan AM, Berg KA, Akopain AN, Jeske NA, Gamper N, Clarke WP, Hargreaves KM. (2005) Bradykinin-induced functional competence and trafficking of the delta-opioid receptor in trigeminal nociceptors. J Neurosci 25:8825–8832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rives ML, Rossillo M, Liu-Chen LY, Javitch JA. (2012) 6′-Guanidinonaltrindole (6′-GNTI) is a G protein-biased κ-opioid receptor agonist that inhibits arrestin recruitment. J Biol Chem 287:27050–27054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivière PJ. (2004) Peripheral kappa-opioid agonists for visceral pain. Br J Pharmacol 141:1331–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth BL, Baner K, Westkaemper R, Siebert D, Rice KC, Steinberg S, Ernsberger P, Rothman RB. (2002) Salvinorin A: a potent naturally occurring nonnitrogenous kappa opioid selective agonist. Proc Natl Acad Sci USA 99:11934–11939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowan MP, Berg KA, Milam SB, Jeske NA, Roberts JL, Hargreaves KM, Clarke WP. (2010) 17beta-estradiol rapidly enhances bradykinin signaling in primary sensory neurons in vitro and in vivo. J Pharmacol Exp Ther 335:190–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowan MP, Ruparel NB, Patwardhan AM, Berg KA, Clarke WP, Hargreaves KM. (2009) Peripheral delta opioid receptors require priming for functional competence in vivo. Eur J Pharmacol 602:283–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schattauer SS, Miyatake M, Shankar H, Zietz C, Levin JR, Liu-Chen LY, Gurevich VV, Rieder MJ, Chavkin C. (2012) Ligand directed signaling differences between rodent and human κ-opioid receptors. J Biol Chem 287:41595–41607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid CL, Bohn LM. (2010) Serotonin, but not N-methyltryptamines, activates the serotonin 2A receptor via a ß-arrestin2/Src/Akt signaling complex in vivo. J Neurosci 30:13513–13524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenoy SK, Drake MT, Nelson CD, Houtz DA, Xiao K, Madabushi S, Reiter E, Premont RT, Lichtarge O, Lefkowitz RJ. (2006) Beta-arrestin-dependent, G protein-independent ERK1/2 activation by the beta2 adrenergic receptor. J Biol Chem 281:1261–1273. [DOI] [PubMed] [Google Scholar]
- Stein C, Lang LJ. (2009) Peripheral mechanisms of opioid analgesia. Curr Opin Pharmacol 9:3–8. [DOI] [PubMed] [Google Scholar]
- Stein C, Schäfer M, Machelska H. (2003) Attacking pain at its source: new perspectives on opioids. Nat Med 9:1003–1008. [DOI] [PubMed] [Google Scholar]
- Stein C, Zollner C. (2009) Opioids and sensory nerves. Handb Exp Pharmacol 495–518. [DOI] [PubMed]
- Sullivan LC, Berg KA, Clarke WP. (2015) Dual regulation of δ-opioid receptor function by arachidonic acid metabolites in rat peripheral sensory neurons. J Pharmacol Exp Ther 353:44–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tohgo A, Choy EW, Gesty-Palmer D, Pierce KL, Laporte S, Oakley RH, Caron MG, Lefkowitz RJ, Luttrell LM. (2003) The stability of the G protein-coupled receptor-beta-arrestin interaction determines the mechanism and functional consequence of ERK activation. J Biol Chem 278:6258–6267. [DOI] [PubMed] [Google Scholar]
- Urban JD, Clarke WP, von Zastrow M, Nichols DE, Kobilka B, Weinstein H, Javitch JA, Roth BL, Christopoulos A, Sexton PM, et al. (2007) Functional selectivity and classical concepts of quantitative pharmacology. J Pharmacol Exp Ther 320:1–13. [DOI] [PubMed] [Google Scholar]
- Vadivelu N, Mitra S, Hines RL. (2011) Peripheral opioid receptor agonists for analgesia: a comprehensive review. J Opioid Manag 7:55–68. [DOI] [PubMed] [Google Scholar]
- Vanderah TW. (2010) Delta and kappa opioid receptors as suitable drug targets for pain. Clin J Pain 26 (Suppl 10):S10–S15. [DOI] [PubMed] [Google Scholar]
- Weibel R, Reiss D, Karchewski L, Gardon O, Matifas A, Filliol D, Becker JA, Wood JN, Kieffer BL, Gaveriaux-Ruff C. (2013) Mu opioid receptors on primary afferent nav1.8 neurons contribute to opiate-induced analgesia: insight from conditional knockout mice. PLoS One 8:e74706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White KL, Robinson JE, Zhu H, DiBerto JF, Polepally PR, Zjawiony JK, Nichols DE, Malanga CJ, Roth BL. (2015) The G protein-biased κ-opioid receptor agonist RB-64 is analgesic with a unique spectrum of activities in vivo. J Pharmacol Exp Ther 352:98–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White KL, Scopton AP, Rives ML, Bikbulatov RV, Polepally PR, Brown PJ, Kenakin T, Javitch JA, Zjawiony JK, Roth BL. (2014) Identification of novel functionally selective κ-opioid receptor scaffolds. Mol Pharmacol 85:83–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon S, Seger R. (2006) The extracellular signal-regulated kinase: multiple substrates regulate diverse cellular functions. Growth Factors 24:21–44. [DOI] [PubMed] [Google Scholar]
- Zheng H, Loh HH, Law PY. (2008) Beta-arrestin-dependent mu-opioid receptor-activated extracellular signal-regulated kinases (ERKs) translocate to nucleus in contrast to G protein-dependent ERK activation. Mol Pharmacol 73:178–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H, Zeng Y, Chu J, Kam AY, Loh HH, Law PY. (2010a) Modulations of neuroD activity contribute to the differential effects of morphine and fentanyl on dendritic spine stability. J Neurosci 30:8102–8110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H, Zeng Y, Zhang X, Chu J, Loh HH, Law PY. (2010b) mu-Opioid receptor agonists differentially regulate the expression of miR-190 and NeuroD. Mol Pharmacol 77:102–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang ZY, Wen YR, Zhang DR, Borsello T, Bonny C, Strichartz GR, Decosterd I, Ji RR. (2006) A peptide c-Jun N-terminal kinase (JNK) inhibitor blocks mechanical allodynia after spinal nerve ligation: respective roles of JNK activation in primary sensory neurons and spinal astrocytes for neuropathic pain development and maintenance. J Neurosci 26:3551–3560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zöllner C, Mousa SA, Fischer O, Rittner HL, Shaqura M, Brack A, Shakibaei M, Binder W, Urban F, Stein C, et al. (2008) Chronic morphine use does not induce peripheral tolerance in a rat model of inflammatory pain. J Clin Invest 118:1065–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.