

Abstract
Pregnancy in rodents is associated with a two- to threefold increase in β-cell mass, which is attributable to large increases in β-cell proliferation, complimented by increases in β-cell size, survival, and function and mediated mainly by the lactogenic hormones prolactin (PRL) and placental lactogens. In humans, however, β-cell mass does not increase as dramatically during pregnancy, and PRL fails to activate proliferation in human islets in vitro. To determine why, we explored the human PRL–prolactin receptor (hPRLR)–Janus kinase 2 (JAK2)–signal transducer and activator of transcription 5 (STAT5)–cyclin–cdk signaling cascade in human β-cells. Surprisingly, adult human β-cells express little or no PRLR. As expected, restoration of the hPRLR in human β-cells rescued JAK2-STAT5 signaling in response to PRL. However, rescuing hPRLR-STAT5 signaling nevertheless failed to confer proliferative ability on adult human β-cells in response to PRL. Surprisingly, mouse (but not human) Stat5a overexpression led to upregulation of cyclins D1–3 and cdk4, as well as their nuclear translocation, all of which are associated with β-cell cycle entry. Collectively, the findings show that human β-cells fail to proliferate in response to PRL for multiple reasons, one of which is a paucity of functional PRL receptors, and that murine Stat5 overexpression is able to bypass these impediments.
Introduction
Types 1 and 2 diabetes and gestational diabetes mellitus (GDM) result partially or completely from a lack of requisite numbers of functional human β-cells. Adult human β-cells are remarkably resistant to the induction of proliferation, likely for many reasons (1–10). One contributing factor may be the sequestration of cyclins A, E, D1, and D3, as well as their cdk partners (cdks 1,2, 4 and 6), in the cytoplasm in quiescent adult human β-cells (9–12). Forced overexpression of cyclins/cdks permits induction of cell cycle entry associated with nuclear translocation of cyclins and cdks, suggesting that trafficking and proliferative events are linked (9–12). Interestingly, cyclin D2—in contrast to its abundance and essential presence for rodent β-cell proliferation (13–15)—is either absent or present at very low levels in human β-cells (16–19). Although the reasons for this difference are unknown, overexpression of cyclin D2 can induce human β-cell cycle entry (17). Therefore, identification of any factor or signal in human β-cells to increase cyclins/cdks and their nuclear trafficking may provide a useful hint to promote human β-cell proliferation and expansion for diabetes therapy.
GDM in humans and rodents is attributable to insulin resistance resulting from pregnancy-associated hormonal changes, as well as an inadequate β-cell response to this resistance (20–36). During normal rodent pregnancy, β-cell proliferation together with an increase in individual β-cell size result in a 200–300% increase in β-cell mass (27–31). Further, increases in glucokinase activity result in a shift in the glucose-stimulated insulin secretion curve, such that more insulin is secreted per β-cell at any given glucose concentration (21–23), changes attributed to production of placental lactogens (PLs) as well as pituitary-derived prolactin (PRL) (21–36). PRL and PLs signal through multiple pathways, including Janus kinase 2 (JAK2)–signal transducer and activator of transcription 5 (STAT5) signaling (10,24–26), to activate pathways farther downstream, such as a Bcl6-menin-p18INK4/p27CIP 34, Tph1/2-serotonin-5HTR (32,35), FoxM1 (30), and HGF-cMet (33,37) pathways, as well as cross-talk with phosphoinositide 3-kinase (PI3K)–Akt–mammalian target of rapamycin and mitogen-activated protein kinase (MAPK) signaling (38). In rodent models, these changes require the interaction of PL/PRL with PRL receptors (PRLRs), the reduction of which in vivo models leads to β-cell failure and GDM (31,32).
In contrast to rodents, in the single large series of human β-cell adaptation to pregnancy, there was only a minor (40%) increase in β-cell mass. This was attributable not to β-cell proliferation but, rather, to neogenesis of small islet clusters (8). Remarkably, there was no measurable increase in β-cell proliferation or size. This neogenesis-driven increase in β-cell mass is presumably sufficient to overcome the insulin resistance of pregnancy. The reasons for this discrepancy between gravid rodents and humans are uncertain, but they may reflect differences in age or interspecies differences. Human genome-wide association studies suggest that polymorphisms in the hPRLR gene increase the risk for GDM (39).
Here, we explored the regulation of d-cyclins and cdks by upstream signaling pathways in human β-cells, hoping to define a complete pathway from a cell surface receptor, through a signaling cascade, to activation of cell cycle machinery. This led us to the lactogenic signaling pathway and to the surprising observation that adult human β-cells contain few, if any, PRLRs. This paucity seems to explain, albeit only partially, the failure of human β-cells to proliferate in response to PRL/PL.
Research Design and Methods
Adenoviruses
Recombinant adenoviruses expressing cytomegalovirus (CMV)-driven human constitutively activated protein kinase B (PKB), mouse Stat5a, human c-MYC, green fluorescent protein (GFP), or β-galactosidase (LacZ) have been described (11,12,16,17,28,40–42). The adenovirus expressing full-length hPRLR was purchased from Signagen Laboratories (Gaithersburg, MD). Ad.STAT5A was generated using the pAd/CMV/V5-DEST GATEWAY recombination system (Life Technologies) and a pDONR221 plasmid containing the human full-length STAT5A cDNA obtained from PlasmID at Dana Farber Cancer Center, Boston, MA.
Human and Rat Islets, Human Pancreas, Mammary Gland Epithelia, and Cancer Cell Lines
Fifty-nine human islet preparations from nondiabetic human donors (mean age ± SEM of 45 ± 7 years; range 19–68 years; mean BMI ± SEM if 27 ± 4 kg/m2; 32 males, 27 females) were provided by the Integrated Islet Distribution Program (http://iidp.coh.org). Upon arrival, islets were transferred to RPMI-1640 complete medium supplemented with 5.5 mmol/L glucose, 0.5% fetal bovine serum, 0.2% bovine serum albumin, and 2% penicillin/streptomycin for overnight recovery at 37°C (5% CO2). Rat islets were isolated from 1- to 2-month-old male Sprague-Dawley rats (Charles River Laboratories) and cultured overnight before experiments as described elsewhere (40) and as approved by the Institutional Animal Care and Use Committee at Mount Sinai. Normal human mammary gland epithelia were provided by the Komen Foundation. Normal adult human pancreas sections from four de-identified surgical specimens were obtained from the Department of Pathology at Mount Sinai Hospital. Human tumor cell lines (MCF-7, T47D, HCT-116, HepG2, or HEK-293) were from American Type Culture Collection.
Adenovirus Transduction, Culture Conditions, and Cell Lines
Islets were dispersed into single cells with trypsin/EDTA and distributed into 96-well plates (for immunoblot and real-time RT-PCR assays) or poly-d-lysine/laminin-coated chamber slides (BD Biosciences; for immunostaining assays). Cells were transduced with adenoviruses at the multiplicity of infection (MOI) described in the text or figure legends for 2 h in serum-free RMPI-1640 medium (11,12,16,17,40). RPMI-1640 complete medium with 10% FBS was then added, and the cells were cultured and treated further, as described below. For human PRL (hPRL) treatment, islet cells were incubated in complete culture medium containing 5.5 mmol/L glucose, 0.5% FBS, 0.2% bovine serum albumin, and 300 or 400 ng/mL recombinant hPRL (Sigma or R&D Systems) as indicated. Medium was refreshed every 2 or 3 days. For BrdU studies, islet cells were treated with BrdU (50 μmol/L) (Sigma) for 24 h before fixation; MCF7 and T47D cells were stimulated with 300 ng/mL PRL for 16 h, followed by 10 μmol/L BrdU for 30 min.
Immunoblots
Cell lysates were resolved on 10% SDS-PAGE (Fisher Scientific), transferred to polyvinylidene fluoride membranes (Bio-Rad), blocked for 1 h in 5% milk/Tris-buffered saline with Tween-20, probed overnight with primary antisera (Supplementary Table 1), and then probed with horseradish peroxidase–conjugated secondary antibodies and visualized using enhanced chemiluminescence (GE Healthcare or Advansta) on X-ray film. Densitometric quantification was performed using ImageJ software.
Histology, Immunocytochemistry, and Morphological Analysis
Dispersed islet cells on chamber slides were fixed in 2% paraformaldehyde/PBS for 15 min and immunolabeled (11,12,16,17,41,42). Briefly, after antigen retrieval with NaCitrate (Dako), cells were incubated overnight with primary antisera (Supplementary Table 2). For BrdU or Ki-67 immunostaining, a second antigen retrieval was performed using 2 N HCl at 25°C for 10 min before primary antibody incubation. Antigen–antibody complexes were incubated further with secondary antibodies conjugated with either Cy3, fluorescein isothiocyanate (Jackson ImmunoResearch), or AlexaFluor (Life Technologies). Cells were counterstained with DAPI. Images were captured on a Leica SP5 DM confocal microscope and analyzed using ImagePro software. In each BrdU incorporation experiment, a minimum of 1,000 insulin-positive cells were counted.
TaqMan Real-Time RT-PCR
Total RNA was purified using the Absolutely RNA Miniprep purification kit (Agilent Technologies) according to the manufacturer’s instructions. cDNA was prepared using the Superscript double-stranded cDNA synthesis kit (Life Technologies). Quantitative real-time RT-PCR was performed using an ABI Prism 7500 detection system (Applied Biosystems) with TaqMan or SYBR green RT-PCR Master Mix reagents, primer/probe sets, and 10–100 ng of cDNA as the template. The ratio of mRNA for the gene of interest to the amount of internal control mRNA of cyclophilin A (PPIA) was calculated, and the ratio for each gene was normalized to its median. Primers and probes are shown in Supplementary Tables 3 and 4.
Statistics
Analysis was performed using unpaired, two-tailed Student t test or by one-way ANOVA with Bonferroni post hoc test for multiple group comparisons. Data are presented as means ± SEMs. Significance was set at P ≤ 0.05.
Results
Expression, Abundance, and Stability of d-cyclins and Cdks in Human Islets
We first validated d-cyclin and cdk4/6 antisera directed against each of the three d-cyclins and cdk4/6 in human islets; antisera were confirmed as being specific for cyclins D1, D2, and D3 and cdk4/6, failing to cross-react with other d-cyclins or cdks (Supplementary Fig. 1A). As reported previously (16–19), human islets contain the easily detectable cyclins D1 and D3 and cdk4/6, but little if any cyclin D2 (Supplementary Fig. 1A and F–J). We next assessed steady-state cyclin D1, D2, and D3 mRNA abundance in five human islet preparations (H1–H5) and in three human cell lines (HepG2, HK2, and Calu-6) (Supplementary Fig. 1B). Cyclin D2 mRNA abundance was variable but readily detected in all five human islet preparations, and it seemed to be comparable to the three human cell lines. Within the constraints of different primer affinities, cyclin D2 mRNA abundance in human islets was not notably different than the abundance of cyclins D1 and D3. Studies of cycloheximide half-life (T1/2) of human islets revealed that cyclins D1 and D3 had T1/2 values of 40–60 min (Supplementary Fig. 1C and E). Since cyclin D2 is not easily detectable in human islets, cyclin D2 was expressed adenovirally at levels approximating those of endogenous cyclins D1 and D3 (Supplementary Fig. 1D). The T1/2 of cyclin D2 (35 min) was slightly briefer than that of the other d-cyclins, suggesting that slightly more rapid degradation may contribute to the small amounts of cyclin D2 in human islets. As expected, cdk4 and cdk6 were very stable, with T1/2 values of >24 h (Supplementary Fig. 1F and G). The proteasome inhibitor MG132 was used to further assess d-cyclin stability (Supplementary Fig. 1H–J). Abundance of each of the three d-cyclins increased with MG132 treatment, with cyclin D1 and D3 increasing by four- to sixfold by 2 h, and cyclin D2 increasing by twofold. Collectively, these studies document that all three d-cyclins are rapidly degraded by the proteasome in human islets, with the possibility of cyclin D2 being degraded slightly more rapidly. Since cyclin D2 mRNA is present in human β-cells and cyclin D2 protein is increased with proteasome inhibition, it seems that at least some cyclin D2 mRNA is translated into protein, albeit perhaps not as efficiently as are cyclins D1 and D3. Cdks 4 and 6 are very stable (T1/2 >24 h) in human islets.
Comparison of Effects of Akt/PKB, c-MYC, and Stat5a Induction on d-cyclin and Cdk Abundance and Nuclear Translocation in the Human β-Cell
We next queried whether the d-cyclins and cdks 4/6 could be upregulated by adenoviral activation of Akt/PKB, c-MYC, and Stat5a (10,24–26,41,42). Adenoviral delivery of Akt/PKB, c-MYC, and Stat5 was confirmed by immunoblot and immunocytochemistry (Fig. 1A and B), which also demonstrated increased phosphorylation of Akt/PKB and Stat5a and the nuclear translocation of c-MYC and Stat5a. Also, all three signaling pathways induced all three of the d-cyclins; Stat5a also induced cdk4 (Fig. 2A and B). Cdk6 was unaffected. In addition to changes at the protein level, Akt/PKB, c-MYC, and Stat5a variably increased mRNA expression of cyclin D3, with nonsignificant increases in cyclins D1 and D2 (Fig. 2C). Cdk4 mRNA expression was also increased by Stat5a. Collectively, these studies reveal that d-cyclins and cdk4 can be induced in human islets by the activation of upstream signaling pathways.
Figure 1.
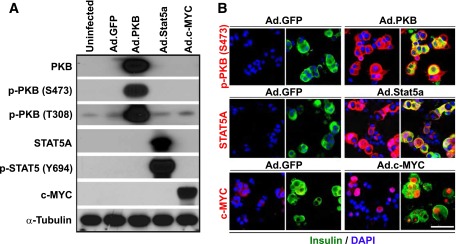
Validation of adenovirus-mediated overexpression of PKB, Stat5a, or c-MYC in dispersed adult human islets. A: Immunoblots of indicated proteins in islets not transduced (Uninfected) or transduced with a control adenovirus expressing GFP (Ad.GFP) or adenoviruses expressing constitutively active Akt/PKB, Stat5a, or c-MYC (all at 300 MOI for 96 h). Note that each of the three proteins is expressed efficiently and that Akt/PKB and Stat5 undergo appropriate phosphorylation. B: Immunofluorescent labeling of indicated proteins in islets after viral transduction. Compared with islets transduced with Ad.GFP (left), islets transduced with Akt/PKB, Stat5a, or c-MYC adenoviruses (right) display marked increases in the three molecules in several islet cell types, notably insulin-positive (β) cells. Also note that some β-cells display nuclear labeling of Stat5a or c-MYC. Scale bar = 25 μm. Both A and B are representative of five separate experiments from different islet donors.
Figure 2.

Expression of PKB, Stat5a, or c-MYC induces d-cyclin and CDK4 expression and nuclear trafficking associated with cell cycle entry in adult human β-cells. Dispersed islets were transduced with the adenoviruses shown (300 MOI each) for 96 h. A: Immunoblots of the indicated cyclins and cdks. B: Densitometric quantitation of immunoblots from multiple experiments (as in A). C: Real-time RT-PCR assays of the corresponding mRNAs. These three panels (A–C) reveal that expression of Ad.Akt/PKB, Stat5, or c-MYC leads to protein and mRNA induction of each of the three d-cyclins and CDK4, but not CDK6. Data are depicted as means ± SEs and include six or seven independent islet donors for each group. *P < 0.05, **P < 0.01 vs. Ad-GFP. D: Immunofluorescent labeling for the same proteins in β-cells. The first two pairs of columns show unmerged and merged images, and the final two columns show only merged images. Note that d-cyclins D1 and D3 and cdks 4 and 6 can be observed at low levels in β-cells under control (Ad.GFP) conditions, but each of the three d-cyclins (in particular D2 and D3) and CDK4 increase with Akt/PKB, Stat5, or c-MYC exposure. More important, each of the d-cyclins, and to a lesser extent CDK4, also appear in the nuclear compartment of insulin-positive cells. Similar results were obtained from multiple experiments using three or four independent islet donors. Scale bar = 25 μm. E: Representative micrographs showing BrdU incorporation into adenovirus-transduced human insulin-positive β-cells. F: Quantitation of the percentage of BrdU-positive/insulin-positive cells from multiple experiments (as in E) using six independent islet donors. Note that Ad-Akt/PKB, Stat5, and c-MYC all induce BrdU incorporation, with Stat5 yielding the largest increment. Bars show means ± SEs. *P < 0.01 vs. Ad.GFP.
In quiescent human β-cells, d-cyclins and cdks are largely cytoplasmic but can be induced to traffic to the nucleus by d-cyclin and cdk overexpression (9,11,12). We used immunocytochemistry to assess whether activation of upstream signaling pathways might also induce their cytoplasmic nuclear trafficking and to determine whether the increases in d-cyclins and cdks observed on immunoblots (Fig. 2A and B) occur within β-cells. Most d-cyclins and cdk4/6 appeared at low levels in the cytoplasmic compartment of β-cells under control (Ad.GFP) conditions (Fig. 2D). By contrast, adenoviral expression of Akt/PKB, c-MYC, and Stat5a led to obvious increases in the abundance of all three d-cyclins and cdk4/6 in β-cells. Further, all three d-cyclins were now observed in the nuclear compartment. These observations indicate that it is not necessary to artificially overexpress d-cyclins to observe their nuclear translocation in human β-cells; they also illustrate that the nuclear trafficking of d-cyclins in human β-cells can be influenced by upstream signaling.
Comparison of Effects of Akt/PBK, c-MYC, and Stat5a Induction on Human β-Cell Proliferation
Adenoviral expression of Akt/PKB and c-MYC have been reported to activate cell cycle entry in human β-cells (41,42), but Stat5a has not been assessed. While we observed that all three agents induce BrdU incorporation in human β-cells (Fig. 2E and F), Stat5a yielded the largest increase. Because of the particularly robust BrdU labeling index induced by Stat5a, and because STAT5 proliferation in human β-cells has not previously been studied, we focused on the PRL-PRLR-JAK2-STAT5 pathway for the remainder of the study.
Effects of Prolactin on Human β-Cell Proliferation
Recombinant hPRL induced MCF7 and T47D human breast cancer cells and rat β-cells to enter the cell cycle within 3 days but had no mitogenic effect on human β-cells at 3 days (Supplementary Fig. 2). The mitogenic effect in rat islets was enhanced further by 6 days, but again, no proliferation was observed in human β-cells at 6 days, although BrdU incorporation was readily observed in the same islet preparations with Ad.Stat5a (Fig. 3A–C). Since the same hPRL preparation was able to drive rat β-cell proliferation (Fig. 3B and C; Supplementary Fig. 2), the failure of human β-cells to replicate cannot be ascribed to biologic inactivity of the hPRL preparation used. This confirms previous reports that hPRL can drive rodent (21–37), but not human (5,8), β-cell proliferation.
Figure 3.

Failure of replication in response to PRL, and inventory of PRLR-JAK-STAT components in adult human β-cells. A and B: Immunofluorescent labeling of insulin and BrdU in dispersed human (A) and rat (B) islets following PRL treatment for 6 days or Ad.Stat5a transduction for 4 days. C: Quantitation of the percentage of BrdU-positive/insulin-positive cells from separate experiments using three or four independent donors for each group. The graph also includes the experiments shown in Supplementary Fig. 2B, in which islets were treated with PRL for 3 days. *P < 0.05, **P < 0.01 vs. vehicle. Unlike rat β-cells, adult human β-cells fail to replicate in response to PRL, despite the ample BrdU incorporation by Ad.Stat5 in the same islet preparations. D: Real-time RT-PCR analysis of PRLR, GHR, and JAK-STAT signaling pathway components in human islets, purified human mammary epithelia (M. Epithelia), two human breast cancer cell lines (T47D and MCF-7), and three other human tumor cell lines (HCT-116, HepG2, and HEK-293). ICA512 (also known as IA2 or PTPRN) is abundant in human β-cells and confirms the quality of the human islet mRNA. Note in particular that human PRLR is barely detectable (ΔCT values 29–35) in human islets (using primers that detect all isoforms of PRLR) and is substantially lower than expression in normal human mammary epithelium, the two breast cancer cell lines, and HEK-293 cells. By contrast, GHR expression in adult human islets is comparable to the levels in MCF-7 and T47D cells (inset), even though it is much lower than that in normal mammary epithelium. Meanwhile, JAK2, STAT5A, and STAT5B are abundant in adult human islets as compared with the various controls. See Supplementary Fig. 3 for SOCS family members. Each dot represents a measurement from one individual specimen, with 3–10 specimens per group. The red bars represent the mean values of all measurements of each group. E and F: Immunofluorescent labeling of PRLR, JAK2, and STAT5A in MCF-7 cells and dispersed adult human islets (E), and intact adult human pancreas sections (F). Two different hPRLR antibodies (Abs), including a mouse monoclonal Ab (E and F) and a rabbit antiserum (F), were used. Both Abs specifically detected hPRLR protein, as evidenced by dramatically reduced PRLR labeling in MCF-7 cells (E, inset) or pancreas sections (F) with pretreatment of a PRLR competing polypeptide, or in pancreas sections treated with control nonimmune rabbit serum (F). Note that in human islets, cytoplasmic JAK2 and STAT5A are detectable in insulin-positive (green) β-cells; however, PRLR is undetectable in adult human β-cells, either in dispersed islets or in intact normal pancreas. By contrast, PRLR is present in other insulin-negative, non-β-cells. Similar results were obtained from multiple experiments using four to six independent human islet or pancreas donors. See Supplementary Fig. 4 for additional details. Scale bar = 25 μm.
Components of the hPRLR-JAK2-STAT5 Signaling Pathway in Human Islets
We used quantitative PCR (qPCR) to further explore the hPRLR-JAK2-STAT5 pathway, comparing human islets with multiple controls: two human breast cancer cell lines (MCF7 and T47D), primary human mammary epithelia, and three additional human cell lines (HCT116, HepG2, and HEK293 cells). In contrast to the other cell types, the human islets contained abundant quantities of the β-cell-enriched mRNA ICA-512 (also known as IA-2 and PTPRN), confirming the quality of the mRNA (Fig. 3D). By contrast, human islets contained essentially undetectable quantities of hPRLR mRNA, although hPRLR mRNA was abundant in both human breast cancer cell lines, normal mammary epithelium, and HEK293 cells. JAK2 and STAT5A/B mRNA were present in human islets in amounts comparable to or exceeding the other cell types, exceeded only by normal mammary epithelium. Human growth hormone receptor (GHR) mRNA also was present. Exploration of potential inhibitory pathways revealed no striking differences between human islets and most other tissues, although SOCS1, SOCS3, and SOCS5 seemed to be increased in human islets as compared with the other cell types (Supplementary Fig. 3).
Confirmation That Most Human β-Cells Contain Little or No hPRLR
Immunocytochemistry confirmed that the large majority of dispersed human β-cells lack the hPRLR (Fig. 3E, Supplementary Fig. 4A), whereas control MCF7 cells contain easily detectable hPRLR. The specificity of the immunolabeling was confirmed by coincubation with an hPRLR-blocking peptide. Immunohistochemistry of normal adult islets in intact pancreas surgical specimens using two different antisera (Supplementary Table 2) and appropriate positive and negative controls also confirmed the absence of hPRLR in adult human β-cells (Fig. 3F). By contrast, JAK2 and STAT5A were readily detectable in human β-cells (Fig. 3E), corroborating the qPCR data in Fig. 3D. Closer inspection of Fig. 3E reveals that some islet cells do express hPRLR, but most are not β-cells. Co-labeling experiments with hPRLR and glucagon, insulin, pancreatic polypeptide (PP), or somatostatin revealed that hPRLR can be found in α-cells and PP cells, but not δ-cells (Supplementary Fig. 4A). This pattern was also confirmed in intact human pancreatic sections, which revealed co-labeling of α-cells and PP cells with hPRLR (Supplementary Fig. 4B). In contrast to the lack of hPRLR on most human β-cells, immunofluorescent labeling of human GHR (hGHR) on β-cells using anti-hGHR antiserum AL47 (Supplementary Table 2) was readily detectable, both in dispersed islets and in situ in the intact pancreas (Supplementary Fig. 5), as well as in T47D breast cancer cells—observations that are consistent with qPCR analysis (Fig. 3D). hGHR was also visible on some non-β islet cells (Supplementary Fig. 5).
Adenoviral Reconstitution With hPRLR Restores JAK2-STAT5 Responsiveness to PRL
Transduction of human islets with a CMV-driven adenovirus expressing native hPRLR resulted in strong expression of the hPRLR, as assessed by immunoblot, with identical size and at levels exceeding those of the native hPRLR in T47D cells (Fig. 4A). Further, strong cell surface expression was observed in β-cells (Fig. 4B). Treatment of hPRLR-transduced human islets with PRL induced phosphorylation of both JAK2 as well as STAT5; induction of phospho-JAK2 was comparable to that observed in PRL-treated T47D cells, and induction of phospho-STAT5 (p-STAT5) exceeded that observed in T47D cells (Fig. 4C). A time-course study of human islets with Ad.hPRLR + PRL revealed rapid (<5 min) induction of JAK2 phosphorylation, followed by immediate and sustained increases in STAT5 phosphorylation, results not observed in control human islets (Fig. 4D). PRL treatment of hPRLR-expressing human β-cells also led to translocation of 694Y-p-STAT5 into the nuclear compartment (Fig. 4E), as well as a brisk increase in STAT5A mRNA expression; STAT5B was not affected (Supplementary Fig. 6A). Thus, adenoviral transduction led to expression of a functional hPRLR of appropriate size on the β-cell surface; this hPRLR had no apparent basal activity but was capable of activating downstream JAK2-STAT5 signaling, including nuclear translocation of 694Y-p-STAT5, all with efficacy comparable to or greater than the native receptor in T47D cells.
Figure 4.
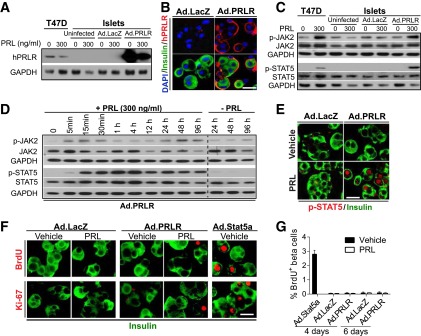
Reconstituting hPRLR restores PRL signaling but fails to induce adult human β-cell replication in response to PRL. Dispersed adult human islets were transduced with 200 MOI Ad.PRLR or control adenovirus (Ad.LacZ) for 48 h, followed by treatment without or with 300 ng/mL PRL for the indicated times. A: Immunoblot of PRLR revealing that Ad.PRLR yields more abundant PRLR protein in human islets than in T47D human breast cancer cells and has the expected Mr of native PRLR. B Immunofluorescent labeling showing that overexpressed PRLR (red) by Ad.PRLR is expressed on the surface of insulin-positive (green) β-cells. C: Immunoblot of the indicated total and phosphorylated proteins. After 2 h of PRL treatment, adult human islets with reconstituted hPRLR, like T47D cells, are able to respond appropriately to PRL, with increased JAK2 and STAT5 phosphorylation. D: The time course of JAK2 and STAT5 phosphorylation in Ad.PRLR-transduced human islets with PRL treatment for the times indicated. Note that in human islets with reconstituted PRLR expression, phosphorylation of endogenous JAK2 and STAT5 was activated rapidly, and p-STAT5 was sustained up to 96 h after PRL treatment, results not observed without PRL treatment. E: Immunofluorescent labeling showing that abundant nuclear 694Y-p-STAT5 is observed in the Ad.PRLR-transduced β-cells, whereas no nuclear p-STAT5 is observed in the Ad.PRLR-transduced β-cells 96 h after PRL treatment. The experiments in A–E were repeated using at least three different donor islets. F: Immunofluorescent labeling for BrdU and Ki-67 in transduced human islets with or without PRL treatment for 96 h. As a control, the same islet preparations were also transduced with Ad.Stat5a for 96 h. G: Quantitation of the percentage of BrdU-positive β-cells from multiple experiments (as in F) using six to eight human donor islets. Bars indicate mean ± SE. Collectively, A–E demonstrate that functional hPRLR can be restored to human β-cells, yet F and G show that this does not lead to restoration of PRL-inducible human β-cell proliferation.
Rescuing Functional hPRLR on β-Cells Fails to Restore Mitogenic Responses to PRL
Surprisingly, reconstitution of the functional hPRLR on human β-cells followed by PRL treatment failed to permit cell cycle entry, as assessed using either BrdU or Ki-67 (Fig. 4F and G). By contrast, Ad.Stat5 treatment induced robust induction of both BrdU and Ki-67 (Figs. 2F and 4F and G). Dose–response studies revealed that treatment with Ad.Stat5 generated substantially higher levels of Stat5 and p-Stat5 in human islets than did the Ad.hPRLR + PRL combination (Fig. 5A and B); in addition, Ad.Stat5 also generated more intense and more frequent nuclear appearance of p-Stat5 (Fig. 5C). Further, at low Ad.Stat5 MOI (e.g., 3–15 MOI), which yielded degrees of p-Stat5 expression in human islets comparable to those obtained with Ad.hPRLR + PRL (Fig. 5A and B), no β-cell proliferation was observed in response to either Ad.Stat5 or Ad.hPRLR + PRL (Fig. 5D and E). These findings suggest that replacement of functional hPRLR on human β-cells rescues their ability to signal via the JAK2-STAT5 pathway in response to PRL, but fails to rescue their ability to replicate in response to PRL. By contrast, Ad.Stat5 overexpression leads to appropriate downstream Stat5 phosphorylation and nuclear translocation, as well as proliferation in human β-cells.
Figure 5.

Differential STAT5 phosphorylation and β-cell replication from reconstituted hPRLR signaling vs. Ad.Stat5a expression. A: Immunoblots of the PRLR, STAT5, and 694Y-p-Stat5 in dispersed human islet cells transduced with indicated adenoviruses with increasing MOI for 48 h, followed by treatment with or without 300 ng/mL PRL for 2 h. B: Densitometry of p-STAT5 compared with GAPDH (as shown in A) from three independent experiments. The ratio of p-STAT5 vs. GAPDH in islets with 5 MOI Ad.PRLR + PRL treatment was arbitrarily set as 1. Note that Ad.Stat5 generates substantially larger quantities of Stat5 and p-Stat5 than does the Ad.PRLR + PRL combination. C: Immunofluorescent labeling of 694Y-p-STAT5 (red) in insulin-positive (green) β-cells after transduction with the indicated adenoviruses for 48 h, followed by treatment with or without PRL (300 ng/mL) for 2 h. Note that there is weak nuclear p-STAT5 labeling in the PRL + Ad.PRLR–treated human β-cells, but it is far less abundant than that in the Ad-Stat5–treated β-cells. This is representative of experiments with three human islet donor preparations. D: A dose response of BrdU incorporation in adult human islet cells with transduction of Ad.Stat5 or Ad.PRLR for 48 h, followed by treatment without or with PRL (300 ng/mL) for 96 h. E: Quantitation of percentage of BrdU-positive, insulin-positive β-cells from D. Bars indicate mean ± SEM, using three independent islet donors for each group. Note that the minimum Ad.Stat5 MOI required for proliferation is 25, and this is associated with substantially more p-Stat5a protein (A) than is generated by the highest MOI of Ad.PRLR in combination with PRL. Thus, reconstituted hPRLR signaling never achieves the robust p-Stat5 induction observed with Ad-Stat5.
Effects of PRL + Ad.hPRLR on Suppressors of Cytokine Signaling, Protein Inhibitors of Activated STATS, and Cyclin-Dependent Kinase Inhibitors
We hypothesized that the failure of Ad.PRLR in combination with PRL treatment may reflect induction of inhibitory suppressors of cytokine signaling (SOCSs), protein inhibitors of activated STATs (PIASs), or cyclin-dependent kinase inhibitors (CDKIs) in human islets. To explore these possibilities, we examined expression of all of the SOCS, PIAS, and CDKI family members in response to Ad.PRLR alone or in combination with hPRL (Fig. 6A and B, Supplementary Fig. 6B). Of these 20 potential inhibitors, only 2 were induced: the SOCS family member cytokine-inducible SH2-containing protein (CISH), and the CDKI family member CDKN2C, encoding p18INK4C.
Figure 6.

Real-time RT-PCR analyses of SOCS genes and cell cycle inhibitors in adult human islets with reconstituted hPRLR. Dispersed human islets were transduced with 200 MOI Ad.PRLR or control adenovirus (Ad.LacZ) for 48 h, followed by treatment without or with 300 ng/mL PRL for 72 h, and then RT-PCR assays for SOCS genes (A) and CDKI genes (B). See additional SOCS family members in Supplementary Fig. 6B. Among the 12 SOCSs and 8 CDKIs, only two—CISH and CDKN2C—were induced by PRL signaling. Data are depicted as means ± SEs and include four independent islet donors for each group. *P < 0.05, **P < 0.01 vs. Ad-LacZ without PRL treatment.
Human STAT5A Overexpression Fails to Activate D-Cyclins, Cdk4, or Proliferation in Human β-Cells
In studies to this point, we had adenovirally delivered murine Stat5a. We next explored the effects of human STAT5A expression on human β-cell proliferation. Surprisingly, while human Ad.STAT5A was overexpressed in human islets and β-cells at levels comparable to those of mouse Ad.Stat5a, and although STAT5A was phosphorylated comparably (Fig. 7A and B), STAT5A overexpression failed to induce increments in any of the three d-cyclins or cdk4, and it failed to activate proliferation as assessed by either Ki-67 or BrdU labeling (Fig. 7C and D). By contrast, Ad.Stat5a expressed at comparable levels induced all of these events.
Figure 7.

Comparison of mouse Stat5a and human STAT5A in d-cyclin and CDK4 regulation, and β-cell replication, in dispersed adult human islets. Immunoblots (A) and immunofluorescent staining (B) of the indicated proteins in human islets transduced with the indicated adenoviruses (200 MOI) for 96 h. Note that the abundance (A, upper panels) and pattern (B, upper row) of total and phosphorylated STAT5 protein achieved by mouse Ad.Stat5a or human Ad.STAT5A were comparable in transduced human islets. Unlike mouse Stat5a, however, human STAT5A fails to induce d-cyclin and CDK4 expression (A, five lower panels) as well as nuclear trafficking of cyclin D2 and CDK4 (B, lower two rows) in adult human β-cells. Representative of experiments using three or four independent islet donors. C: BrdU and Ki-67 labeling in Ad.Stat5a- or Ad.STAT5A-transduced human insulin-positive β-cells. D: Quantitation of the percentage of BrdU-positive/insulin-positive cells from independent experiments (as in C) using four different islet donors. Note that human STAT5A is unable to drive human β-cell replication, but mouse Stat5a can. Bars show means ± SEs. Scale bars = 25 μm.
Discussion
These studies describe at least nine novel and unexpected observations relevant to the failure of human β-cell proliferation during gestation. First, they provide a detailed analysis of the lactogen-hPRLR-JAK2/STAT5-d-cyclin/cdk pathway to proliferation in the human β-cell. Second, they reveal that there are few or no hPRLRs on most adult human β-cells, but the remaining downstream components of the canonical JAK2-STAT5 signaling cascade are present. Third, they show that the hPRLR is present on α-cells and PP cells. Fourth, they demonstrate that although replacement or restoration of functional hPRLR on human β-cells restores the ability of PRL to signal, it is insufficient for induction of proliferation. Fifth, they provide insight into the failure of human β-cells to robustly express the mitogenic cyclin D2 protein. Sixth, despite the failure of the reconstituted hPRLR signaling cascade to initiate cell cycle progression, they document that induction of murine Stat5 signaling at higher levels can restore human β-cell proliferation. Seventh, they reveal that human STAT5A is a poor substitute for its murine equivalent. These observations may help to explain the failure of human β-cell proliferation during pregnancy (8). Eighth, and perhaps most important, they identify Stat5a as a potential potent and tractable target for induction of human β-cell proliferation. Finally, as with our previous report linking the loss of platelet-derived growth factor receptor-α (PDGFRα) in adult β-cells (43), they may provide evidence for a multifactorial programmatic change in adult human β-cells, which conspires to prevent replication.
Cyclin D2 is essential to normal β-cell expansion and function in rodent islets (13–15). By contrast, cyclin D1 and D3 are dispensable in rodent islets (14,15). In human islets, mRNA encoding cyclin D2 is present in amounts comparable to those in human tumor cell lines (Supplementary Fig. 1B). Most authors, however, have detected little or no cyclin D2 protein in human β-cells (16–19). Under basal conditions we also found little or no cyclin D2 protein in human islets (16–19) but did find readily detectable cyclin D2 mRNA. This relative lack of cyclin D2 protein may reflect slightly greater instability compared with cyclins D1 and D3, since its T1/2 may be slightly briefer and/or may have resulted from particularly inefficient translation of cyclin D2 mRNA in the human β-cell. Whatever the cause, we observed that the low basal abundance of cyclin D2 is readily overridden by activation of at least three different upstream signaling pathways, including c-MYC, Stat5a, and Akt/PKB signaling. The three pathways also drive upregulation of cyclins D1 and D3 as well as cdk4, and in several cases they also lead to increases in their nuclear abundance. This latter point is of some interest because in previous studies of nuclear translocation of cyclins and cdks in β-cells, this was observed only in settings of their overexpression. The current findings suggest that nuclear translocation of cyclins and cdks does not require forced overexpression, but instead can be driven by upstream signaling events. With regard to the fundamental importance of cyclin D2 in human β-cell proliferation, one might interpret its low abundance/absence to mean that it is irrelevant in human β-cells or, alternately, that cyclin D2 is essential for human β-cell proliferation, an interpretation supported by the findings that cyclin D2 can be induced in human β-cells in association with activation of mitogenic signaling, and that cyclin D2 overexpression can induce human β-cells to enter the cell cycle (16).
Among the c-MYC, Akt/PKB, and Stat5 pathways, Stat5 generated the most robust β-cell proliferation. Attention was, therefore, focused on PRL-PRLR-JAK2/STAT5 signaling. It is noteworthy that Akt/PKB and Stat5a/STAT5A are phosphorylated on 473S, 308T(AKT), and 694Y(STAT5) when expressed in human islets (Figs. 1 and 7). Which kinases may be responsible is uncertain, but several candidates are present in β-cells, including JAK2 and PI3K pathway kinases.
Notably, we confirmed previous reports suggesting that, in contrast to rodent β-cells (21–37), PRL fails to activate proliferation in human β-cells (5,8). This does not seem to result from a lack of downstream mediators of lactogenic signaling because STAT5A/B mRNA and protein are present in apparently normal amounts and because forced Stat5a expression can lead to induction of cyclins/cdks and proliferation. By contrast, the large majority of human β-cells seem to lack meaningful expression of hPRLR. While this may be a surprise based on the many examples of PRLR expression in rodent β-cells (21–37), Benner et al. (44) reported that while PRLR mRNA is abundant in rodent islets, it is far less abundant in human islets. More important, available human RNA sequencing data from FACS-purified human β-cells reported by Nica et al. (45) and Blodgett et al. (46) support the conclusion that human β-cells lack hPRLR expression. Moreover, the observation that hPRLR is readily detectable in dispersed and intact islets in α- and PP cells, but not β-cells, supports the concept that the absence of hPRLR on β-cells is a feature of native adult β-cells in situ, and not an artifact of islet isolation, dispersion, or FACS purification.
Replacement of functional hPRLR on β-cells rescued several key aspects of the PRLR signaling cascade. For example, in the presence of reconstituted hPRLR, treatment with PRL leads to rapid JAK2 phosphorylation followed by rapid and sustained 694Y-STAT5 phosphorylation, accompanied by nuclear 694Y-STAT5 translocation in β-cells. We interpret these observations to mean that the paucity of functional PRLRs on human β-cells plausibly contributes to their failure to proliferate during human pregnancy. We must qualify this conjecture because it remains possible that PRLR expression is returned to human β-cells during pregnancy; this question has not been addressed here nor elsewhere. Similarly, our data do not address the possibility that hPRLR may be present on human β-cells during development and childhood but is lost during adulthood.
Surprisingly, replacement of hPRLR did not rescue the mitogenic response to PRL, for reasons hypothesized in Fig. 8. For example, it may be that additional signaling pathways must be recruited to the PRLR-JAK2-STAT5 system in β-cells since, in some cell types, JAK2-STAT5 signaling can be coupled to PI3K, MAPK, and other signaling pathways (38). This seems unlikely since forced expression of Stat5 alone is able to activate proliferation. It is also possible that mStat5a, but not endogenous human STAT5A, is able to engage menin or serotoninergic signaling, and that this is required for mitogenic signaling. This possibility may be strengthened by the human STAT5A findings discussed below. Alternatively, it may be that the proliferation induced by forced mStat5 expression reflects a greater abundance of Stat5 and p-Stat5 than that achieved with PRL treatment in the presence of hPRLR. While this seems possible, it also seems unlikely to be the only explanation since T47D breast cancer cells proliferate in response to PRL (Supplementary Fig. 2), despite lower phospho-JAK2, and STAT5 and pSTAT5, abundance comparable to human islets with PRL + Ad.hPRLR (Fig. 4A–C), and since marked overexpression of STAT5A fails to engage the cell cycle (Fig. 7). It is also possible that that “normal” basal levels of SOCS family members such as CISH (Fig. 6A) might rapidly increase in response to PRL-hPRLR-JAK2-STAT5 activation, or that abrupt increases in key cell cycle inhibitors, such as p18INK4C (Fig. 6B), might constrain proliferation. While little is known regarding CISH in human islets, it seems to be nonessential for murine β-cell development and function (47). The only CDKI member induced was CDKN2C, encoding p18INK4C; inactivating mutations in CDKN2C have been postulated to underlie some cases of multiple endocrine neoplasia type 1a (48). Thus, further studies of human islets on CISH and CDKN2C/p18INK4C are warranted. Finally, we believe that it is possible that adult human β-cells downregulate a broad repertoire of intracellular targets essential for proliferation, including not only the hPRLR, as shown here, but also the PDGFRα described previously (43), perhaps via broad epigenetic silencing of key chromatin regions not investigated here (9). Clarifying these questions will require additional study.
Figure 8.
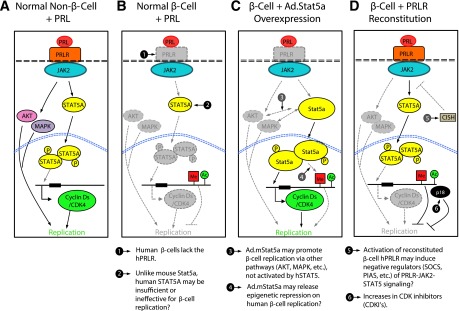
Working models for the failure of PRL-PRLR-JAK2-STAT5 signaling to activate proliferation in adult human β-cells. A: In non-β-cells, such as mammary epithelium or lymphocytes, PRL binds to the hPRLR, activating JAK2, which recruits and phosphorylates (P) STAT5, which then dimerizes and transits to the nucleus, where it binds to promoters of genes associated with proliferation, such as cyclins and cdks. In some cell types JAK2-STAT5 activation may also recruit signaling molecules that activate signaling in the PI3K/Akt/PKB pathway or the Ras/Raf/MAPK pathway, which may contribute to proliferation. B: In the normal adult β-cell, the hPRLR is absent. Therefore, PRL fails to activate JAK2-STAT5 signaling, nuclear trafficking, or proliferation. C: Adenoviral expression of high levels of mouse Stat5a bypasses the absent hPRLR, leads to Stat5 phosphorylation by undefined kinases, activates all three d-cyclins and cdk4, permits their nuclear translocation, and results in proliferation. It is possible, but unknown, whether overexpression of Stat5 may influence PI3K/Akt/PKB or MAPK signaling in human islets or whether it alters restrictive DNA or histone modifications such as methylation (Me) or acetylation (Ac) that may prevent replication. D: Reconstitution of functional hPRLR on human β-cells restores JAK2-STAT5 phosphorylation and STAT5A nuclear translocation but still fails to activate proliferation. This could result from unknown irreversible epigenetic changes, increases in suppressors of cytokine signaling (e.g., CISH), increases in CDKIs (e.g., p18/CDKN2C), or other unknown mechanisms in adult human β-cells. See text for details.
It is surprising that human STAT5A and mouse Stat5a differ so remarkably in their relative abilities to induce human β-cells to replicate, given the 96% homology between these two peptides. Examination of Supplementary Fig. 7 provides no obvious clues as to why this might occur. Future studies with adenoviral STAT5A/Stat5a chimeric variants may shed light on this issue.
Finally, while the hPRLR is absent on most human β- or δ-cells, it is present to some degree on α-cells, PP cells, and potentially other islet cells. This may explain the observations from several laboratories that low levels of hPRLR mRNA are present in whole human islets. The meaning of these intriguing results is uncertain but may suggest novel paracrine signaling pathways within the islet, another attractive target for future investigation. For example, the existence of hPRLRs on non-β-cells may explain the protective effects of lactogens on human islets against lipotoxic insults (28). Alternatively, it is also possible that putative lactogenic hormone effects on human β-cells may be mediated via other cytokine family receptors present on β-cells. It also remains possible that a small subpopulation of human β-cells exists, as described by Schraenen et al. (29), some of which may express PRLRs. Exploring these possibilities also will require additional studies. Of course, species specificity is important here, since, for example, while human growth hormone is an agonist for the hGHR, it is a poor agonist for the murine GHR (49).
In summary, adult human β-cells contain few or no functional hPRLRs, in part explaining the failure of human β-cells to proliferate in vitro or in vivo in response to lactogenic hormones. Additional poorly defined barriers to proliferation must also exist, however, since reconstitution of functional hPRLR and overexpressing STAT5A in human β-cells fail to permit proliferation in response to lactogenic hormones. Perhaps most important in therapeutic terms is that murine Stat5a may be a potentially tractable target for human β-cell proliferation. Approaches might include the development of small-molecule mimics of Stat5 signaling or otherwise leveraging the unique features of mouse Stat5a versus human STAT5A. Of course, such a strategy would need to contend with the current lack of effective tools to target small molecules to β-cells and would need to clarify the biology underlying the discrepant β-cell responses to Stat5a versus STAT5A.
Supplementary Material
Article Information
Acknowledgments. The authors thank Drs. Rupangi C. Vasavada, Adolfo Garcia-Ocaña, and Donald K. Scott (all at Icahn School of Medicine at Mount Sinai, New York, NY), Dr. Alvin Powers (Division of Endocrinology, Vanderbilt University, Nashville, TN), and Dr. Seung Kim (Department of Developmental Biology, Stanford University, Stanford, CA) for invaluable and continuous insight. The authors thank the National Institutes of Health/National Institute of Diabetes and Digestive and Kidney Diseases Integrated Islet Distribution Program, Dr. Tatsuya Kin (Alberta Diabetes Institute, Edmonton, Alberta, Canada), and Dr. Pyotr Witkowski (Department of Surgery, University of Chicago School of Medicine, Chicago, IL) for providing human islets.
Funding. This work was supported by JDRF grant 1-2011-603; National Institutes of Health grants R-01 DK55023, R-01 DK58259, R-01 DK 46395, U-01 DK089538, and UC4 DK104211; and a James A. Haley Veterans’ Hospital Merit Review (to S.J.F.).
Duality of Interest. No potential conflicts of interest relevant to this article were reported.
Author Contributions. H.C., J.W.K., K.K.T., F.S., N.F.-T., K.P., H.L., P.W., and A.S.B. performed experiments. R.P., J.J., Y.Z., and S.J.F. contributed reagents, designed the study, and interpreted data. H.C. and A.F.S. wrote the manuscript. H.C. and A.F.S. are the guarantors of this work and, as such, had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
Prior Presentation. Parts of this study were presented in abstract form at the 74th Scientific Sessions of the American Diabetes Association, San Francisco, CA, 13–17 June 2014.
Footnotes
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db15-0083/-/DC1.
References
- 1.Gregg BE, Moore PC, Demozay D, et al. Formation of a human β-cell population within pancreatic islets is set early in life. J Clin Endocrinol Metab 2012;97:3197–3206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes 2003;52:102–110 [DOI] [PubMed] [Google Scholar]
- 3.Rahier J, Guiot Y, Goebbels RM, Sempoux C, Henquin JC. Pancreatic beta-cell mass in European subjects with type 2 diabetes. Diabetes Obes Metab 2008;10(Suppl 4):32–42 [DOI] [PubMed] [Google Scholar]
- 4.Menge BA, Tannapfel A, Belyaev O, et al. Partial pancreatectomy in adult humans does not provoke beta-cell regeneration. Diabetes 2008;57:142–149 [DOI] [PubMed] [Google Scholar]
- 5.Parnaud G, Bosco D, Berney T, et al. Proliferation of sorted human and rat beta cells. Diabetologia 2008;51:91–100 [DOI] [PubMed] [Google Scholar]
- 6.Levitt HE, Cyphert TJ, Pascoe JL, et al. Glucose stimulates human beta cell replication in vivo in human islets transplanted into NOD-severe combined immunodeficiency (SCID) mice. Diabetologia 2011;54:572–582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maedler K, Schumann DM, Schulthess F, et al. Aging correlates with decreased beta-cell proliferative capacity and enhanced sensitivity to apoptosis: a potential role for Fas and pancreatic duodenal homeobox-1. Diabetes 2006;55:2455–2462 [DOI] [PubMed] [Google Scholar]
- 8.Butler AE, Cao-Minh L, Galasso R, et al. Adaptive changes in pancreatic beta cell fractional area and beta cell turnover in human pregnancy. Diabetologia 2010;53:2167–2176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang P, Fiaschi-Taesch NM, Vasavada RC, Scott DK, Garcia-Ocaña A, Stewart AF. Diabetes mellitus–advances and challenges in human β-cell proliferation. Nat Rev Endocrinol 2015;11:201–212 [DOI] [PubMed] [Google Scholar]
- 10.Stewart AF, Hussain MA, Garcia-Ocaña A, et al. Human β-cell proliferation and intracellular signaling: part 3. Diabetes 2015;64:1872–1885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fiaschi-Taesch NM, Kleinberger JW, Salim FG, et al. Human pancreatic β-cell G1/S molecule cell cycle atlas. Diabetes 2013;62:2450–2459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fiaschi-Taesch NM, Kleinberger JW, Salim FG, et al. Cytoplasmic-nuclear trafficking of G1/S cell cycle molecules and adult human β-cell replication: a revised model of human β-cell G1/S control. Diabetes 2013;62:2460–2470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alonso LC, Yokoe T, Zhang P, et al. Glucose infusion in mice: a new model to induce beta-cell replication. Diabetes 2007;56:1792–1801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kushner JA, Ciemerych MA, Sicinska E, et al. Cyclins D2 and D1 are essential for postnatal pancreatic beta-cell growth. Mol Cell Biol 2005;25:3752–3762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Georgia S, Hinault C, Kawamori D, et al. Cyclin D2 is essential for the compensatory β-cell hyperplastic response to insulin resistance in rodents. Diabetes 2010;59:987–996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fiaschi-Taesch N, Bigatel TA, Sicari B, et al. Survey of the human pancreatic beta-cell G1/S proteome reveals a potential therapeutic role for cdk-6 and cyclin D1 in enhancing human beta-cell replication and function in vivo. Diabetes 2009;58:882–893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fiaschi-Taesch NM, Salim F, Kleinberger J, et al. Induction of human beta-cell proliferation and engraftment using a single G1/S regulatory molecule, cdk6. Diabetes 2010;59:1926–1936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lavine JA, Raess PW, Davis DB, et al. Overexpression of pre-pro-cholecystokinin stimulates beta-cell proliferation in mouse and human islets with retention of islet function [retracted in: Mol Endocrinol 2010;24:472]. Mol Endocrinol 2008;22:2716–2728 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 19.Davis DB, Lavine JA, Suhonen JI, et al. FoxM1 is up-regulated by obesity and stimulates beta-cell proliferation. Mol Endocrinol 2010;24:1822–1834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Metzger BE, Phelps RL. Diabetes mellitus and pregnancy. In Endocrinology. 6th ed. DeGroot L, Jameson L, Eds. Philadelphia, Saunders Elsevier, 2010, p. 2644–2661 [Google Scholar]
- 21.Sorenson RL, Brelje TC. Adaptation of islets of Langerhans to pregnancy: beta-cell growth, enhanced insulin secretion and the role of lactogenic hormones. Horm Metab Res 1997;29:301–307 [DOI] [PubMed] [Google Scholar]
- 22.Brelje TC, Scharp DW, Lacy PE, et al. Effect of homologous placental lactogens, prolactins, and growth hormones on islet B-cell division and insulin secretion in rat, mouse, and human islets: implication for placental lactogen regulation of islet function during pregnancy. Endocrinology 1993;132:879–887 [DOI] [PubMed] [Google Scholar]
- 23.Sorenson RL, Stout LE. Prolactin receptors and JAK2 in islets of Langerhans: an immunohistochemical analysis. Endocrinology 1995;136:4092–4098 [DOI] [PubMed] [Google Scholar]
- 24.Stout LE, Svensson AM, Sorenson RL. Prolactin regulation of islet-derived INS-1 cells: characteristics and immunocytochemical analysis of STAT5 translocation. Endocrinology 1997;138:1592–1603 [DOI] [PubMed] [Google Scholar]
- 25.Jackerott M, Møldrup A, Thams P, et al. STAT5 activity in pancreatic beta-cells influences the severity of diabetes in animal models of type 1 and 2 diabetes. Diabetes 2006;55:2705–2712 [DOI] [PubMed] [Google Scholar]
- 26.Friedrichsen BN, Richter HE, Hansen JA, et al. Signal transducer and activator of transcription 5 activation is sufficient to drive transcriptional induction of cyclin D2 gene and proliferation of rat pancreatic beta-cells. Mol Endocrinol 2003;17:945–958 [DOI] [PubMed] [Google Scholar]
- 27.Vasavada RC, Garcia-Ocaña A, Zawalich WS, et al. Targeted expression of placental lactogen in the beta cells of transgenic mice results in beta cell proliferation, islet mass augmentation, and hypoglycemia. J Biol Chem 2000;275:15399–15406 [DOI] [PubMed] [Google Scholar]
- 28.Kondegowda GN, Mozar A, Otero A, Chin C, Garcia-Ocaña A, Vasavada RC. Lactogens protect rodent and human beta cells against glucolipotoxicity-induced cell death through Janus kinase-2 (JAK2)/signal transducer and activator of transcription-5 (STAT5) signalling. Diabetologia 2012;55:1721–1732 [DOI] [PubMed] [Google Scholar]
- 29.Schraenen A, Lemaire K, de Faudeur G, et al. Placental lactogens induce serotonin biosynthesis in a subset of mouse beta cells during pregnancy. Diabetologia 2010;53:2589–2599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang H, Zhang J, Pope CF, et al. Gestational diabetes mellitus resulting from impaired β-cell compensation in the absence of FoxM1, a novel downstream effector of placental lactogen. Diabetes 2010;59:143–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Freemark M, Avril I, Fleenor D, et al. Targeted deletion of the PRL receptor: effects on islet development, insulin production, and glucose tolerance. Endocrinology 2002;143:1378–1385 [DOI] [PubMed] [Google Scholar]
- 32.Arumugam R, Fleenor D, Freemark M. Knockdown of prolactin receptors in a pancreatic cell line: effects on DNA synthesis, apoptosis, and gene expression. Endocrine 2014;46:568–576,. 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Demirci C, Ernst S, Alvarez-Perez JC, et al. Loss of HGF/c-Met signaling in pancreatic β-cells leads to incomplete maternal β-cell adaptation and gestational diabetes. Diabetes 2012;61:1143–1152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Karnik SK, Chen H, McLean GW, et al. Menin controls growth of pancreatic beta-cells in pregnant mice and promotes gestational diabetes mellitus. Science 2007;318:806–809 [DOI] [PubMed] [Google Scholar]
- 35.Kim H, Toyofuku Y, Lynn FC, et al. Serotonin regulates pancreatic beta cell mass during pregnancy. Nat Med 2010;16:804–808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rieck S, Kaestner KH. Expansion of beta-cell mass in response to pregnancy. Trends Endocrinol Metab 2010;21:151–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Johansson M, Olerud J, Jansson L, Carlsson PO. Prolactin treatment improves engraftment and function of transplanted pancreatic islets. Endocrinology 2009;150:1646–1653 [DOI] [PubMed] [Google Scholar]
- 38.Nyga R, Pecquet C, Harir N, et al. Activated STAT5 proteins induce activation of the PI 3-kinase/Akt and Ras/MAPK pathways via the Gab2 scaffolding adapter. Biochem J 2005;390:359–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Le TN, Elsea SH, Romero R, Chaiworapongsa T, Francis GL. Prolactin receptor gene polymorphisms are associated with gestational diabetes. Genet Test Mol Biomarkers 2013;17:567–571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cozar-Castellano I, Takane KK, Bottino R, Balamurugan AN, Stewart AF. Induction of beta-cell proliferation and retinoblastoma protein phosphorylation in rat and human islets using adenovirus-mediated transfer of cyclin-dependent kinase-4 and cyclin D1. Diabetes 2004;53:149–159 [DOI] [PubMed] [Google Scholar]
- 41.Rao P, Roccisana J, Takane KK, et al. Gene transfer of constitutively active Akt markedly improves human islet transplant outcomes in diabetic severe combined immunodeficient mice. Diabetes 2005;54:1664–1675 [DOI] [PubMed] [Google Scholar]
- 42.Karslioglu E, Kleinberger JW, Salim FG, et al. cMyc is a principal upstream driver of beta-cell proliferation in rat insulinoma cell lines and is an effective mediator of human beta-cell replication. Mol Endocrinol 2011;25:1760–1772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen H, Gu X, Liu Y, et al. PDGF signalling controls age-dependent proliferation in pancreatic β-cells. Nature 2011;478:349–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Benner C, van der Meulen T, Cacéres E, Tigyi K, Donaldson CJ, Huising MO. The transcriptional landscape of mouse beta cells compared to human beta cells reveals notable species differences in long non-coding RNA and protein-coding gene expression. BMC Genomics 2014;15:620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nica AC, Ongen H, Irminger J-C, et al. Cell-type, allelic, and genetic signatures in the human pancreatic beta cell transcriptome. Genome Res 2013;23:1554–1562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Blodgett DM, Nowosielska A, Afik S, et al. Novel observations from next generation RNA sequencing of highly purified human adult and fetal islet cell subsets. Diabetes 30 April 2015 [Epub ahead of print] [DOI] [PMC free article] [PubMed]
- 47.Jiao Y, Rieck S, Le Lay J, Kaestner KH. CISH has no non-redundant functions in glucose homeostasis or beta cell proliferation during pregnancy in mice. Diabetologia 2013;56:2435–2445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Agarwal SK, Mateo CM, Marx SJ. Rare germline mutations in cyclin-dependent kinase inhibitor genes in multiple endocrine neoplasia type 1 and related states. J Clin Endocrinol Metab 2009;94:1826–1834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Brouwers B, de Faudeur G, Osipovich AB, et al. Impaired islet function in commonly used transgenic mouse lines due to human growth hormone minigene expression. Cell Metab 2014;20:979–990 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.