

Abstract
Cellular senescence, the stable cell cycle arrest elicited by various forms of stress, is an important facet of tumor suppression. Although much is known about the key players in the implementation of senescence, including the pRb and p53 axes and the cyclin dependent kinase inhibitors p16INK4a and p21CIP1, many details remain unresolved. In studying conditional senescence in human fibroblasts that express a temperature sensitive SV40 large T-antigen (T-Ag), we uncovered an unexpected role for CDK4. At the permissive temperature, where pRb and p53 are functionally compromised by T-Ag, cyclin D-CDK4 complexes are disrupted by the high p16INK4a levels and reduced expression of p21CIP1. In cells arrested at the non-permissive temperature, p21CIP1 promotes reassembly of cyclin D-CDK4 yet pRb is in a hypo-phosphorylated state, consistent with cell cycle arrest. In exploring whether the reassembled cyclin D-CDK4-p21 complexes are functional, we found that shRNA-mediated knockdown or chemical inhibition of CDK4 prevented the increase in cell size associated with the senescent phenotype by allowing the cells to arrest in G1 rather than G2/M. The data point to a role for CDK4 kinase activity in a G2 checkpoint that contributes to senescence.
Keywords: CDK4, human fibroblasts, p16INK4a, p21CIP1, p53, retinoblastoma protein, senescence, SV40 T-antigen
Abbreviations
- HFs
human fibroblasts
- CDK
cyclin dependent kinase
- pRb
retinoblastoma protein
- SV40
simian virus 40
- TERT
telomerase reverse transcriptase
- SA-βgal
senescence-associated β-galactosidase activity
- ts
temperature sensitive
- shRNA
short-hairpin RNA
- FACS
fluorescence actvated cell sorting
- PI
propidium iodide
- BrdU
bromodeoxyuridine
Introduction
Cellular senescence is a state of stable cell cycle arrest that is induced by various types of stress, including telomere attrition, oxidative damage and aberrant proliferative signaling.1-4 In physiological contexts, senescence prevents the outgrowth of oncogenically compromised cells, promotes a number of age-related debilities and has a role in normal embryonic development.1-6
Many of the classical studies on senescence have been conducted in primary human fibroblasts (HFs). In standard tissue culture conditions, these cells undergo replicative senescence, primarily as a consequence of telomere erosion, and display oncogene-induced senescence upon expression of a mutant oncogene.7–9 A common factor is the triggering of a persistent DNA damage response (DDR) that, unlike a classical cell cycle checkpoint, appears to be effectively irreversible.1,2 The cells remain metabolically viable and adopt a number of distinctive features, the most obvious being an enlarged, irregular shape and increased expression of lysosomal β-galactosidase, generally referred to as senescence-associated β-galactosidase activity,10 although these phenotypic changes are not essential for cell cycle arrest.11,12
Senescent cells also express elevated levels of the cyclin dependent kinase inhibitors p21CIP1 and p16INK4a (refs13–17) and ectopic expression of either of these proteins produces a senescence-like phenotype in early passage HFs.18 This is consistent with long standing evidence that senescence can be delayed or avoided by interfering with the pRb and p53 tumor suppressors.19 Loss of p53 results in a dramatic reduction in the levels of p21CIP1 (ref20) and loss of pRb renders cells insensitive to the effects of p16INK4a (refs 21–23). Whereas p21CIP1 associates with multiple cyclin-CDK complexes at different stages of the cell cycle, p16INK4a interacts specifically with CDK4 and CDK6 and prevents their association with D-type cyclins.24,25 The binding of p16INK4a to CDK4/6 also causes redistribution of p21CIP1 (and p27KIP1) from the cyclin D-CDK complexes onto cyclin E-CDK2 complexes.17,26-31 As these kinases are responsible for the sequential phosphorylation of pRb required for entry into S-phase and progression through the cell cycle,32–36 their inhibition causes cell cycle arrest that is dependent on pRb.
While pRb is in its active hypo-phosphorylated state in senescent cells, and cyclin E-CDK2 activity is extinguished,37 we and others have noted that senescent HFs retain a substantial amount of the ternary complexes formed by D-type cyclins, CDK4 and p21CIP1 (refs 17 and 38). It is not clear whether these complexes are functional as the CIP/KIP family act as assembly factors as well as inhibitors for cyclin D-CDK complexes39,40 (reviewed in refs 24 and 25). There are differences of opinion whether the ternary complexes are catalytically active in vivo or simply serve as a buffering system that controls the availability of the CIP/KIP proteins to inhibit CDK2.40-44
To try to gain insight into this question, we made use of 2 independently generated strains of HFs that undergo conditional senescence following inactivation of a temperature sensitive (ts) allele of simian virus 40 large tumor antigen (SV40 T-Ag). At the permissive temperature (34°C), with both pRb and p53 inactivated by T-Ag, the cells proliferate rapidly despite lacking detectable cyclin D-CDK complexes, recapitulating the original observations of subunit rearrangement.26,45,46 When shifted to the non-permissive temperature (39°C), at which T-Ag is inactivated, the cells undergo a senescence-like arrest accompanied by the reassembly of cyclin D1-CDK4-p21CIP1 complexes and inhibition of CDK2-associated kinase activity. Knockdown of p21CIP1 with shRNA allowed the cells to avoid arrest by restoring CDK2 activity. Remarkably, shRNA-mediated knockdown or chemical inhibition of CDK4 prevented the increase in cell size associated with the senescent phenotype by allowing the cells to evade a G2 checkpoint and arrest in the subsequent G1 phase. The data imply that the reassembled cyclin D1-CDK4-p21CIP1 complexes are functionally contributing to the senescent phenotype, presumably by acting on a substrate(s) other than pRb.
Results
Conditional senescence following inactivation of SV40 T-Ag
SVts8 cells were originally derived by introducing a temperature-sensitive (ts) allele of SV40 T-Ag into the TIG3 strain of human fibroblasts.47,48 They have endogenous telomerase activity that is not affected by the switch from permissive to non-permissive temperature (Supplementary Fig. S1). BJ-TERT-tsLT cells were generated more recently by infecting BJ fibroblasts with retroviral vectors encoding hTERT and ts T-Ag.49 In principle, the presence of telomerase should have enabled both of these strains to avoid replicative senescence.8,9 However, when switched to the restrictive temperature, 39°C, the cells arrested with characteristics of senescent human fibroblasts.48-50 They appeared considerably larger and more irregular than cells maintained at the permissive temperature, changes that could be readily observed by microscopic comparisons of stained cells (Fig. 1A) and documented by cell size measurements (Fig. 1B). Although T-Ag is inactivated within 12 h of the temperature shift,15 the physical differences developed over several days, at which point the cells also stained positively for SA-βgal activity (Fig. 1A). FACS analyses, combining PI staining and BrdU incorporation, suggested that most of the cells were arrested in G2, with a substantial proportion showing a >4N DNA content (Fig. 1C and Fig. S2A).
Figure 1.
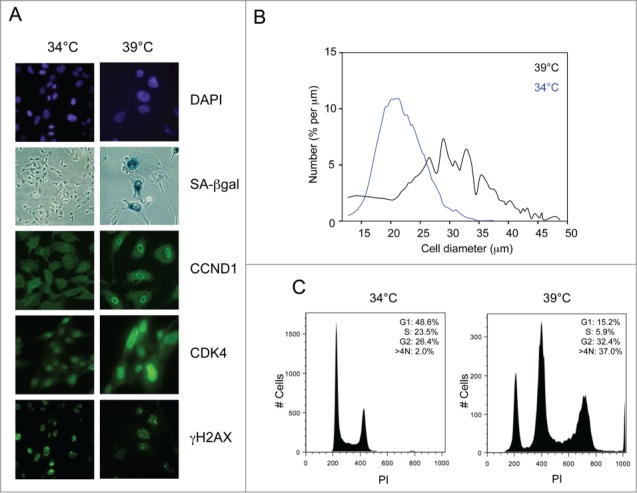
Conditional senescence in SVts8 cells. (A) Representative images of SVts8 cells at the permissive (34°C) and non-permissive (39°C) temperatures, stained with DAPI or for SA-βgal activity, CCND1, CDK4 and γH2AX as indicated. (B) Cell size measurements of SVts8 cells grown at 34°C or switched to 39°C for 5 days. (C) Cell cycle profiles of SVts8 cells grown at 34°C or switched to 39°C for 5 days. The percentage of cells in the G1, S, and G2/M phases and with a >4N DNA content are indicated in each panel.
As both cell lines have telomerase activity, we suspected that they might have one or more irreparable telomeres or some other form of persistent DNA damage.51,52 In line with this idea, the cells stained positively for γH2AX and phosphorylated ATM at both the permissive and the non-permissive temperatures (Fig. 1A and data not shown) and both p53 and Chk2 were phosphorylated on residues characteristic of a DNA damage response. At the permissive temperature, T-Ag presumably renders the cells oblivious to the DNA damage checkpoint by inactivating both pRb and p53.
Role of pRb, p53 and p21CIP1 in conditional senescence
To pursue this idea, we assessed the status of the pRb and p53 pathways by immunoblotting. In the cells proliferating at 34°C, pRb was hyper-phosphorylated on a number of the key residues (e.g Ser807, Ser 811 and Thr826) that are thought to influence its ability to repress transcription (Fig. 2A). These modifications appeared to be independent of CDK4/6 activity as they were refractory to PD033291, a specific CDK4/6 inhibitor, at concentrations that cause G1 arrest in normal HFs.53 Upon shifting to 39°C, pRb became hypo-phosphorylated and there was a marked decline in the level of cyclin A, consistent with a shutdown of E2F-dependent transcription and cell cycle arrest (Fig. 2B).
Figure 2.
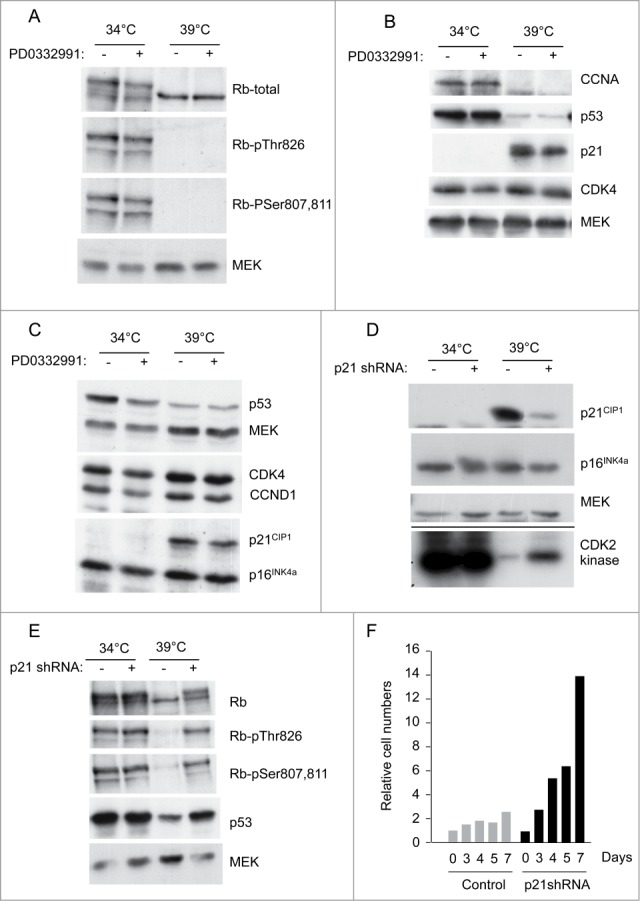
Status of cell cycle regulatory components at the permissive and non-permissive temperatures. (A) SVts8 cells cultured in the presence and absence of PD0332991 were switched from 34°C to 39°C for 6 d and cell lysates were fractionated by SDS-PAGE and immunoblotted with antibodies that detect specific phosphorylation events on pRb. MEK was used as a loading control. (B) Similar analysis of BJ-TERT-tsLT cells 6 d after temperature shift with antibodies against cyclin A (CCNA), p53, p21CIP1 and CDK4. (C) Effects of temperature shift on p21CIP1 and p16INK4a levels in SVts8 cells. (D) SVts8 cells at 34°C were infected with a retrovirus encoding shRNA against p21CIP1 (+) or an empty vector control (−). After antibiotic selection, cells were replated and grown at 34°C or 39°C for 48 h. Equivalent amounts of cell lysate were either analyzed directly by immunoblotting for p21CIP1, p16INK4a and MEK or immunoprecipitated with an antibody against CDK2 and assayed for their ability to phosphorylate GST-pRb in the presence of [γ-32P]ATP. (E) Phosphorylation status of pRb in SVts8 cells transduced with p21CIP1 or control shRNA. Equivalent amounts of cell lysate were analyzed directly by immunoblotting with the indicated antibodies. (F) SVts8 cells transduced with p21CIP1-specific shRNA continue to grow at 39°C for at least 7 d The plots record cell numbers relative to those before the temperature shift.
As binding to T-Ag results in the stabilization of p53 (ref 54), the levels of p53 were generally higher in the cells growing at 34°C, albeit to variable extents in different experiments (Figs. 2B and 2C). However, inactivation of T-Ag at 39°C resulted in a dramatic increase in p21CIP1 expression, consistent with restoration of the transcriptional activity of p53 (Figs. 2B and 2C). In contrast, p16INK4a was highly expressed at both temperatures. At 34°C, p16INK4a expression is de-repressed by the functional inactivation of pRb15 but, as an inherently stable protein,46 its levels barely changed when pRb activity was restored at 39°C (Fig. 2C).
Taken together, therefore, the data suggested that p21CIP1 is likely to play a critical role in the senescence-like arrest by impeding the phosphorylation of pRb. To substantiate this idea, we infected proliferating SVts8 cells with a retrovirus encoding a previously validated shRNA against p21CIP1 (ref 38). When the cells were shifted to 39°C, we observed a substantial reduction in the level of p21CIP1 in the shRNA-transduced cells compared to the empty vector controls (Fig. 2D). As predicted, knockdown of p21CIP1 caused a partial reactivation of CDK2 kinase activity, as measured using the C-terminal domain of pRb as a substrate (Fig. 2D). It also partially restored phosphorylation of Ser807/811 and Thr826 (Fig. 2E). Importantly, knockdown of p21CIP1 allowed some of the cells to avoid the senescence-like arrest at 39°C, as judged by increasing cell numbers, FACS analyses and cell size measurements (Fig. 2F and data not shown). The results are consistent with previous reports that the arrest of BJ-TERT-tsLT cells can be alleviated by shRNA against p53 (ref 49).
Status of Cyclin D-CDK complexes at the permissive and non-permissive temperatures
As p21CIP1 acts as both an assembly factor and inhibitor of cyclin D–CDK complexes,39,40 we compared the status of these complexes at the permissive and non-permissive temperatures. Consistent with the original reports of subunit rearrangement,26,45,46 we were unable to detect either cyclin D1 or p21CIP1 in CDK4 immunoprecipitates from cells grown at 34°C, whereas p16INK4a was clearly co-precipitated (Fig. 3A). However, when the cells were shifted to 39°C, the levels of p21CIP1 were restored and both cyclin D1 and p21CIP1 were found to co-precipitate with CDK4 (Fig. 3A). Similar results were obtained with SVts8 and BJ-TERT-tsLT cells and with the other D-cyclins (not shown). Reciprocal immunoprecipitation with cyclin D1 and p21CIP1 antibodies confirmed the restoration of the cyclin D1-CDK4-p21CIP1 complexes at 39°C (Fig. 3B), with a concomitant reduction in the amount of CDK4 that co-precipitates with p16INK4a. Gel filtration analyses confirmed that cyclin D1, CDK4 and p21CIP1 assembled in high molecular weight (150–200 kDa) complexes at 39°C but not at 34°C, whereas p16INK4a was invariably either monomeric or in binary complexes with CDK4 or CDK6 (not shown). Note that CDK6 is much less abundant than CDK4 in HFs, as judged by RNA sequencing data55 and we have previously shown that most of the CDK6 in HFs is associated with either p18INKc or p16INK4a (ref 56).
Figure 3.
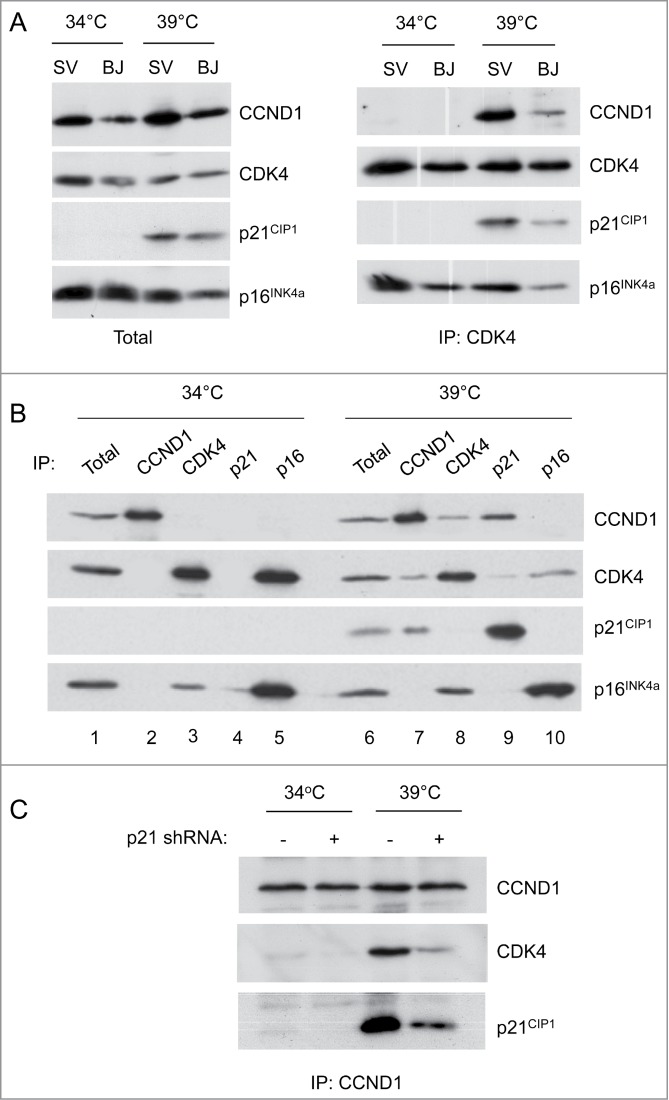
Comparison of cyclin-CDK-CKI interactions at the permissive and non-permissive temperatures. (A) Cell lysates were prepared from SVts8 (SV) or BJ-TERT-tsLT (BJ) cells grown at 34°C or 6 d after shifting to 39°C. Equal amounts of protein were either analyzed directly by SDS-PAGE and immunoblotting (left panel) or after immunoprecipitation with an antibody against CDK4 (right panel). (B) Equal amounts of cell lysate (300 μg of total protein) from SVts8 cells grown at 34°C and 39°C were immunoprecipitated with antisera against CCND1, CDK4, p21CIP1 and p16INK4a as indicated. After fractionation by SDS-PAGE, the precipitated proteins were immunoblotted for CCND1, CDK4, p21CIP1 and p16INK4a as indicated. The total amounts of CCND1, CDK4, p21CIP1 and p16INK4a were monitored by direct immunoblotting of 30 μg of each protein sample (Total). (C) Similar analysis of CCND1 immunoprecipitates from SVts8 cells transduced with an shRNA against p21CIP1 or non-specific shRNA.
We also noted a change in the localization of cyclin D1 when the cells were shifted between the different temperatures. In normal fibroblasts, cyclin D1 is predominantly nuclear40,57,58 but in SVts8 cells grown at the permissive temperature, cyclin D1 appeared to be distributed throughout the cell, with a substantial proportion in the cytoplasm (see Fig. 1A). When the cells were shifted to the non-permissive temperature, the nuclear localization of cyclin D1 was restored. This would be consistent with the idea that the nuclear localization of cyclin D1 is promoted by association with CIP/KIP proteins.39,40,59
Taken together, the results implied the rather paradoxical situation that in cells proliferating at 34°C, pRb is hyper-phosphorylated in the absence of cyclin D-CDK complexes whereas, in cells arresting at 39°C, pRb becomes hypo-phosphorylated despite the reassembly of cyclin D-CDK complexes. Moreover, in the cells treated with p21CIP1 shRNA, there was less CDK4 associated with cyclin D1 despite release from the arrest (Fig. 3C).
A role for CDK4 in conditional senescence
As the reassembly of the cyclin D1-CDK4-p21CIP1 complexes at 39°C did not lead to phosphorylation of pRb, even on residues reputed to be specific targets of D-cyclins,32-36 we were interested to know whether the complexes were contributing in any way to the observed phenotype. To address this question, we used shRNAs to specifically knock down the levels of CDK4 and CDK6. As documented in Figures S3A and S3B, we were able to achieve ≥90 % reduction in the levels of CDK4 and CDK6 and the shRNAs were specific for the respective kinases. Knockdown of either of the CDKs had little if any effect on the cells growing at 34°C, in line with the fact that the kinases are not associated with their regulatory cyclins. However, when shifted to 39°C, the cells transduced with CDK4 shRNA were visibly smaller and more uniform in size than the control cells transduced with a non-specific shRNA (Fig. 4A). Cell size measurements confirmed these impressions and suggested that, when measured 5–6 d after temperature shift, the mean cell diameter was only marginally larger than that of actively proliferating cells maintained at 34°C (Figs. 4B, S3C and S3D). However, the cells were not growing exponentially and the cell numbers only increased by approximately 2-fold (not shown). The effects of CDK4 shRNA were therefore distinct from those of p21CIP1 shRNA.
Figure 4.
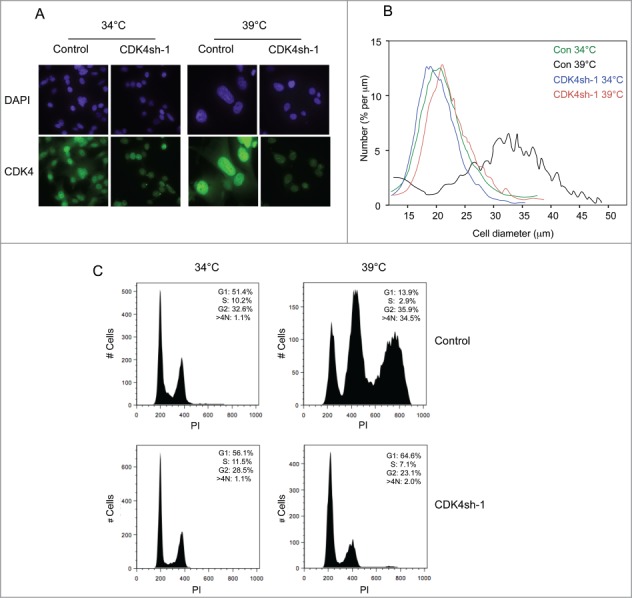
Effects of CDK4 knockdown on the senescence phenotype. (A) Representative images of SVts8 cells transduced with CDK4 or control shRNA at the permissive (34°C) and non-permissive (39°C) temperatures. Cells were stained with DAPI or for CDK4 as indicated. (B) Cell size measurements of SVts8 cells transduced with CDK4 or control shRNA at 34°C or switched to 39°C for 6 days. (C) Cell cycle profiles of SVts8 cells transduced with CDK4 shRNA or control shRNA at 34°C, or switched to 39°C for 5 days.
The FACS profiles confirmed that the cells treated with CDK4 shRNA were not cycling. In contrast to the control cells, which at 39°C arrested primarily with a G2 cell cycle profile and a substantial proportion of >4 N cells, the cells transduced with CDK4 shRNA were mostly in G1 (Figs. 4C and S2B). These findings suggested that CDK4 knockdown enabled the cells to avoid the G2 arrest and to stop at the subsequent G1 phase, hence their smaller size. In line with this idea, CDK4 knockdown did not restore the phosphorylation of pRb that was observed following p21CIP1 knockdown.
Note that the effects on cell size and FACS profiles were reproduced with at least 2 independent shRNAs against CDK4 (Figs. S3C, S3D and S3E) but not with an shRNA against CDK6, despite very effective and specific knockdown (Fig. S3F). However, as noted above, the expression of CDK6 in human fibroblasts is an order of magnitude lower than that of CDK4.
Role of CDK4 kinase activity in conditional senescence
These findings suggested that the cyclin D-CDK4 complexes that were re-assembled in cells shifted to the non-permissive temperature were contributing to the senescence phenotype. One possibility is that they could act by sequestering p21CIP and other members of the CIP/KIP family of inhibitors but, in principle, this should promote rather than retard cell cycle progression. Conversely, knocking down CDK4 would be expected to release p21CIP1 and enforce rather than undermine senescence. We therefore surmised that the assembled complexes might regain kinase activity and act by phosphorylating an as yet unidentified substrate involved in the G2 arrest.
To explore this possibility, we treated the cells with PD0332991, a specific CDK4/6 inhibitor.53 In line with expectations, concentrations of PD0332991 that caused complete arrest of normal HFs had no effect on the proliferation of SVts8 or BJ-TERT-tsLT at the permissive temperature. However, when the cells were shifted to 39°C, there was a noticeable difference between the treated and untreated cells. The characteristic size increase associated with senescence was largely attenuated in the cultures treated with PD0332991 (Figs. 5A and 5B) and their FACS profile was more indicative of G1 arrest (Fig. 5C). The effects were less striking than those observed following CDK4 knockdown, perhaps reflecting incomplete inhibition of the kinase activity, but toxicity associated with higher doses of the drug precluded further investigation.
Figure 5.
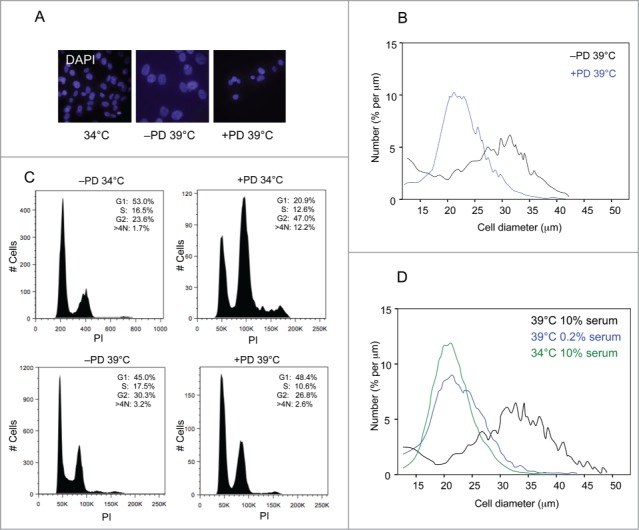
Effects of CDK4 inhibition on the senescence phenotype. (A) Representative images of SVts8 cells at the permissive temperature (34°C) or after shifting to the non-permissive temperature (39°C) in the presence or absence of the CDK4 inhibitor PD0332991 (4μM). The cells were stained with DAPI. (B) Mean cell size comparisons of SVts8 at the non-permissive temperature in the presence or absence of the CDK4 inhibitor PD0332991. (C) Cell cycle profiles of SVts8 cells at 34°C or 39°C in the presence or absence of the CDK4 inhibitor PD0332991. (D) Mean cell size comparisons of SVts8 at 34°C or after shifting to 39°C in either 0.2% or 10% serum.
In the FACS analyses, one of most obvious changes elicited by CDK4 shRNA was the loss of cells with a >4N DNA content. This suggested that CDK4 might be contributing to a block in cytokinesis, reminiscent of the effects of serum concentration reported in an earlier study.50 Interestingly, SVts8 cells held in 0.2% serum did not increase in size when shifted to 39°C (Fig. 5D) and FACS analyses suggested that they were largely arrested in G1, recapitulating the effects of CDK4 knockdown.
Discussion
Although there are many experimental contexts in which cellular senescence can be studied, both in whole animals and cultured cells, there are relatively few models in which to investigate reversibility. The tumor-suppressor effects of oncogene-induced senescence, for example, must presumably be overcome when benign lesions progress to malignancy yet experiments with cultured cells suggest that senescence represents a permanent exit from the cell cycle. Once senescence is established, disabling the pRb and p53 axes, either with virally encoded agents or RNA interference, can restore DNA synthesis but not cell proliferation.60-62 However, the arrest remains reversible if cells are deprived of serum or if signaling via the mTOR pathway is blocked.50,63,64 These observations imply that the classical senescence phenotype develops as a 2-stage process in which an initially reversible cell cycle arrest is rendered irreversible as a consequence of growth promoting stimuli.65
A potential explanation for irreversibility would be the altered state of chromatin organization that develops in senescent cells within the first few days of the initial arrest.66 However, there is a growing awareness of Rb- and p53-independent processes that reinforce the arrest in the G2/M phase of the cell cycle, in part by impeding cytokinesis.50,67,68 Here we present evidence that in cells undergoing conditional senescence, CDK4, a cyclin D-dependent kinase generally associated with pro-proliferative events in the G1 phase of the cell cycle, contributes to a G2/M blockade.
Both of the independently derived cell models that we used in the study, SVts8 and BJ-TERT-tsLT, revealed an apparently paradoxical situation in which the cyclin D-CDK complexes that are deemed to be essential for initiating the phosphorylation of pRb in G1 are undetectable in cells proliferating at 34°C but are re-established when cells arrest at 39°C. Indeed, one of the main components of these complexes, cyclin D1, is mainly cytoplasmic in the proliferating cells and moves to the nucleus in the senescent cells. The findings are entirely consistent with the original reports of subunit rearrangement26,45,46 but raise a number of questions about some widely held assumptions. For example, despite the absence of cyclin D-CDK complexes, pRb is clearly hyper-phosphorylated at 34°C, including modifications on residues that are reputed to be preferred/specific targets for the D-type cyclins.32-36 While it is conceivable that there was a minor population of active cyclin D-CDK4/6 complexes in the proliferating cells that evaded detection by immunoprecipitation or gel filtration, the phosphorylation of pRb persisted in cells treated with the CDK4/6 inhibitor PD0332991. As the pRb-protein in these cells is already compromised by T-Ag, their proliferation was unaffected by the inhibitor at concentrations that arrest the growth of normal HFs.
The converse situation applied at the non-permissive temperature where the cells arrest with pRb in its hypo-phosphorylated state despite the reassembly of cyclin D-CDK-p21CIP1 complexes. An obvious explanation would be that the reassembled complexes are catalytically inert; however, treatment with PD0332991 or shRNA-mediated knockdown of CDK4 had a marked effect on the phenotype of the arrested cells. Whereas the untreated cells had a predominantly G2/M DNA content, with a substantial proportion of >4 N cells, ablation of CDK4 resulted in a predominantly G1 population of considerably smaller cells. An obvious interpretation would be that CDK4 is functionally contributing to the G2/M arrest and that depletion of CDK4 is allowing cells to proceed to and arrest at the subsequent G1 phase. pRb remains hypo-phosphorylated in this situation, in contrast to the situation following the knock down of p21CIP1.
Our findings are consistent with the observation that the overexpression of Cdk4 in mouse astrocytes results in tetraploidy, presumably preceded by endoreduplication and an aberrant G2 arrest.69 They are also in line with previous reports that cyclin D-dependent CDKs act at different phases of the cell cycle70-72 and can target substrates other than the pRb family of pocket proteins.73-76 It is therefore tempting to speculate that in the context of cells undergoing senescence, the phosphorylation of an as yet unknown substrate by CDK4 can impede cytokinesis and promote chromosomal instability. On the one hand, the G2/M blockade could represent an additional safeguard against the propagation of oncogenically compromised cells but as the D-cyclins and their CDK partners are often over-expressed in cancer cells, it has the potential to drive the emergence of aneuploidy. Identifying the relevant substrate(s) could greatly enhance our understanding of the molecular mechanisms that underlie senescence and malignant conversion, but poses a formidable technical challenge.75
Materials and Methods
Cell culture and retroviral infection
SVts8 and BJ-TERT-tsLT cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum at either 34oC, the permissive temperature, or 39°C, the non-permissive temperature for T-antigen function. Other cell strains were cultured under the same conditions at 37°C. To permit uptake of ecotropic retroviruses, cells were infected with an amphotropic retrovirus (pWXL-Neo-Eco) encoding the mouse basic amino acid transporter as described.18
Ecotropic retroviral supernatants were produced by transfecting the BOSC-23 packaging cell line with the relevant plasmid DNAs, using calcium phosphate precipitation. Viral supernatants were filtered through a 0.45 μm filter, and infections were performed in the presence of 4 μg/ml polybrene (Sigma). Drug selection was performed with 2.5 μg/ml puromycin or 100 μg/ml hygromycin. Staining for senescence-associated β-galactosidase activity was as described.10
Immunofluorescence
Cells seeded in chamber slides (Lab Tek) were washed 3 times for 1 min in phosphate-buffered saline (PBSA) and fixed in warm 3.7% formaldehyde in PBSA for 15 min at room temperature. After three additional washes in PBSA, the cells were permeabilized in 0.1% Triton X-100 in PBSA for 15 min at room temperature and washed again. Four drops of Image-iT FX Signal Enhancer (Molecular Probes) were added to each well followed by incubation for 30 min at room temperature in a humid atmosphere and further washes. The cells were then blocked in 3% BSA in PBSA for 1 h and incubated with primary antibody (previously diluted in 3% BSA in PBSA) overnight at 4°C or for 1 h at room temperature. After washing, the relevant fluorescently conjugated secondary antibody (Alexa Fluor 488, 555 or 595 from Molecular Probes) was applied and incubated for 30–60 min at room temperature in the dark. Slides were washed and sealed with coverslips mounted using ProLong Gold Antifade reagent with DAPI (Molecular Probes).
Cell size measurements
Cell diameters were measured using a Z2 Beckman Coulter counter, following the manufacturer's recommended protocols. Briefly, cells were recovered by trypsinization and approximately 5×104 cells were analyzed in a sample volume of 0.5 ml using a 100 μm aperture and calibration constant of 60. Data were processed using AccuCom software.
Immunoprecipitation and immunoblotting
The procedures used for immunoprecipitation and immunoblotting were as described elsewhere.38
Antisera
The γH2AX antibody was from Upstate (07–164) and the following antibodies were obtained from Santa Cruz: CDK2 (sc-163 and sc-6248), CDK4 (sc-601), p21CIP1 (sc-397), CHK2 (sc-9064), cyclin E (sc-248), cyclin A (sc-596) and pRb (sc-050). The MEK1/2 antibody (#9122) and polyclonal antibodies specific for CHK2 phosphorylated at Thr68 (#2661S), p53 phosphorylated at Ser15 (#9284S), and pRb phosphorylated at Ser780 (#9307), Ser795 (#9301) and Ser807/811 (#9308) were obtained from Cell Signaling Technology. The phosphor-Thr826 pRb antibody (OPA1–0302) was from Thermo Scientific. Immunoprecipitation and immunoblotting of cyclin D1, CDK6 and p16INK4a was performed using the 287.3, LB01 and DPAR12 polyclonal antibodies, respectively, as described.46,77 We also used the monoclonal antibody JC8 to detect p16INK4a (kindly provided by J. Koh and E. Harlow) and the DCS6, DCS28 and DCS31 antibodies respectively against cyclin D1, cyclin D3 and CDK4 (from J. Bartek and Novus Biochemicals NB120–6315).
Kinase assays
Cells were lysed by freeze-thawing in Tween lysis buffer (50 mM HEPES, pH8.0, 1 mM EDTA, 2.5 mM EGTA, 150 mM NaCl, 1 mM dithiothreitol, 0.1% Tween 20, 1mM sodium fluoride, 0.1 mM sodium orthovanadate, 2 μg/ml aprotinin and 100 μg/ml phenylmethylsulfonylfluoride) and the lysates were clarified by centrifugation. Following immunoprecipitation with antiserum to CDK2 (sc-163) or normal rabbit IgG from Santa Cruz (sc-2027), immune complexes were washed 3 times in Tween lysis buffer and twice with kinase reaction buffer (50 mM HEPES, pH 8.0, 10 mM MgCl2, 2.5 mM EGTA, 1 mM dithiothreitol). The complexes were then suspended in 25 μl of kinase reaction buffer containing 2 μg of GST-Rb (C-terminal domain), 25 μM ATP and 10 μCi of [γ-32P]ATP. After incubation at 30°C for 30 min, reactions were stopped by adding 25 μl of 2x sample buffer. Samples were boiled for 5 min and resolved by SDS-PAGE in a 12% gel. The phosphorylated substrate was detected by autoradiography.
Telomerase assay
Telomerase activity was analyzed using the PCR-based TRAP assay.78 Each sample contained 103 cells. Negative control samples were inactivated by heating to 85oC for 5 min prior to the TRAP assay. Samples (5 μl) of each reaction were analyzed by electrophoresis in an 18% acrylamide gel in the presence of 7M urea.
Short hairpin RNA vectors
The shRNA vectors targeting human CDK4, CDK6 and p21CIP1 were generated using the following 19-nucleotide sequences: CDK4sh-1: 5′-GAGAATGGCTACCTCTCGA-3′
CDK4sh-2: 5′-AGGCCTAGATTTCCTTCAT-3′
CDK6sh-1: 5′-GTTCAGATGTTGATCAACT-3′
p21CIP1: 5′-CTTCGACTTTGTCACCGAG-3′.
These sequences were used as the basis for complementary 59-mer oligonucleotides capable of forming a hairpin and flanked by sites for the BglII and HindIII restriction enzymes. The annealed oligonucleotides were cloned into pRetroSuper vectors79 that confer either puromycin or hygromycin resistance. (kindly provided by R. Agami and R. Bernards).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors are grateful to Rene Bernards for providing the BJ-TERT-tsLT cells, to Jim Koh, Ed Harlow and Jiri Bartek for monoclonal antibodies and to Pfizer for providing PD0332991. They also thank Sukhveer Purewal and Derek Davies for help with the FACS analyses and the many past members of the Molecular Oncology laboratory who have made direct and indirect and contributions to this study.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
Funding
The work was supported through a core grant from Cancer Research UK to the London Research Institute.
References
- 1. Campisi J, d'Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev 2007; 8:729-40; PMID:17667954; http://dx.doi.org/ 10.1038/nrm2233. [DOI] [PubMed] [Google Scholar]
- 2. Collado M, Blasco MA, Serrano M. Cellular senescence in cancer and aging. Cell 2007; 130:223-33; PMID:17662938; http://dx.doi.org/ 10.1016/j.cell.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 3. Kuilman T, Michaloglou C, Mooi WJ, Peeper DS. The essence of senescence. Genes Dev 2010; 24:2463-79; PMID:21078816; http://dx.doi.org/ 10.1101/gad.1971610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chandler H, Peters G. Stressing the cell cycle in senescence and aging. Curr Opin Cell Biol 2013; 25:765-71; PMID:23916530; http://dx.doi.org/ 10.1016/j.ceb.2013.07.005. [DOI] [PubMed] [Google Scholar]
- 5. Munoz-Espin D, Canamero M, Maraver A, Gomez-Lopez G, Contreras J, Murillo-Cuesta S, Rodriguez-Baeza A, Varela-Nieto I, Ruberte J, Collado M, et al. . Programmed cell senescence during mammalian embryonic development. Cell 2013; 155:1104-18; PMID:24238962; http://dx.doi.org/ 10.1016/j.cell.2013.10.019. [DOI] [PubMed] [Google Scholar]
- 6. Storer M, Mas A, Robert-Moreno A, Pecoraro M, Ortells MC, Di Giacomo V, Yosef R, Pilpel N, Krizhanovsky V, Sharpe J, et al. . Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell 2013; 155:1119-30; PMID:24238961; http://dx.doi.org/ 10.1016/j.cell.2013.10.041. [DOI] [PubMed] [Google Scholar]
- 7. Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 1997; 88:593-602; PMID:9054499; http://dx.doi.org/ 10.1016/S0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- 8. Bodnar AG, Ouellette M, Frolkis M, Holt SE, Chiu CP, Morin GB, Harley CB, Shay JW, Lichtsteiner S, Wright WE. Extension of life-span by introduction of telomerase into normal human cells. Science 1998; 279:349-52; http://dx.doi.org/ 10.1126/science.279.5349.349. [DOI] [PubMed] [Google Scholar]
- 9. Vaziri H, Benchimol S. Reconstitution of telomerase activity in normal human cells leads to elongation of telomeres and extended replicative life span. Curr Biol 1998; 8:279-82; PMID:9501072; http://dx.doi.org/ 10.1016/S0960-9822(98)70109-5. [DOI] [PubMed] [Google Scholar]
- 10. Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira-Smith O, et al. . A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci U S A 1995; 92:9363-7; PMID:7568133; http://dx.doi.org/ 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lee BY, Han JA, Im JS, Morrone A, Johung K, Goodwin EC, Kleijer WJ, DiMaio D, Hwang ES. Senescence-associated β-galactosidase is lysosomal β-galactosidase. Aging cell 2006; 5:187-95; PMID:16626397; http://dx.doi.org/ 10.1111/j.1474-9726.2006.00199.x. [DOI] [PubMed] [Google Scholar]
- 12. Lin AW, Barradas M, Stone JC, van Aelst L, Serrano M, Lowe SW. Premature senescence involving p53 and p16 is activated in response to constitutive MEK/MAPK mitogenic signaling. Genes Dev 1998; 12:3008-19; PMID:9765203; http://dx.doi.org/ 10.1101/gad.12.19.3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Noda A, Ning Y, Venable SF, Pereira-Smith OM, Smith JR. Cloning of senescent cell-derived inhibitors of DNA synthesis using an expression screen. Exp Cell Res 1994; 211:90-8; PMID:8125163; http://dx.doi.org/ 10.1006/excr.1994.1063. [DOI] [PubMed] [Google Scholar]
- 14. Alcorta DA, Xiong Y, Phelps D, Hannon G, Beach D, Barrett JC. Involvement of the cyclin-dependent kinase inhibitor p16 (INK4a) in replicative senescence of normal human fibroblasts. Proc Natl Acad Sci U S A 1996; 93:13742-7; PMID:8943005; http://dx.doi.org/ 10.1073/pnas.93.24.13742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hara E, Smith R, Parry D, Tahara H, Stone S, Peters G. Regulation of p16CDKN2 expression and its implications for cell immortalization and senescence. Mol Cell Biol 1996; 16:859-67; PMID:8622687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wong H, Riabowol K. Differential CDK-inhibitor gene expression in aging human diploid fibroblasts. Exp Gerontol 1996; 31:311-25; PMID:8706801; http://dx.doi.org/ 10.1016/0531-5565(95)00025-9. [DOI] [PubMed] [Google Scholar]
- 17. Stein GH, Drullinger LF, Soulard A, Dulic V. Differential roles for cyclin-dependent kinase inhibitors p21 and p16 in the mechanisms of senescence and differentiation in human fibroblasts. Mol Cell Biol 1999; 19:2109-17; PMID:10022898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. McConnell BB, Starborg M, Brookes S, Peters G. Inhibitors of cyclin-dependent kinases induce features of replicative senescence in early passage human diploid fibroblasts. Curr Biol 1998; 8:351-4; PMID:9512419; http://dx.doi.org/ 10.1016/S0960-9822(98)70137-X. [DOI] [PubMed] [Google Scholar]
- 19. Shay JW, Pereira-Smith OM, Wright WE. A role for both RB and p53 in the regulation of human cellular senescence. Exp Cell Res 1991; 196:33-9; PMID:1652450; http://dx.doi.org/ 10.1016/0014-4827(91)90453-2. [DOI] [PubMed] [Google Scholar]
- 20. el-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, Lin D, Mercer WE, Kinzler KW, Vogelstein B. WAF1, a potential mediator of p53 tumor suppression. Cell 1993; 75:817-25; PMID:8242752; http://dx.doi.org/ 10.1016/0092-8674(93)90500-P. [DOI] [PubMed] [Google Scholar]
- 21. Koh J, Enders GH, Dynlacht BD, Harlow E. Tumour-derived p16 alleles encoding proteins defective in cell-cycle inhibition. Nature 1995; 375:506-10; PMID:7777061; http://dx.doi.org/ 10.1038/375506a0. [DOI] [PubMed] [Google Scholar]
- 22. Lukas J, Parry D, Aagaard L, Mann DJ, Bartkova J, Strauss M, Peters G, Bartek J. Retinoblastoma-protein-dependent cell-cycle inhibition by the tumour suppressor p16. Nature 1995; 375:503-6; PMID:7777060; http://dx.doi.org/ 10.1038/375503a0. [DOI] [PubMed] [Google Scholar]
- 23. Medema RH, Herrera RE, Lam F, Weinberg RA. Growth suppression by p16ink4 requires functional retinoblastoma protein. Proc Natl Acad Sci U S A 1995; 92:6289-93; PMID:7603984; http://dx.doi.org/ 10.1073/pnas.92.14.6289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev 1999; 13:1501-12; PMID:10385618; http://dx.doi.org/ 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]
- 25. Pei XH, Xiong Y. Biochemical and cellular mechanisms of mammalian CDK inhibitors: a few unresolved issues. Oncogene 2005; 24:2787-95; PMID:15838515; http://dx.doi.org/ 10.1038/sj.onc.1208611. [DOI] [PubMed] [Google Scholar]
- 26. Xiong Y, Kuppuswamy D, Li Y, Livanos EM, Hixon M, White A, Beach D, Tlsty TD. Alteration of cell cycle kinase complexes in human papillomavirus E6- and E7-expressing fibroblasts precedes neoplastic transformation. J Virol 1996; 70:999-1008; PMID:8551641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jiang H, Chou HS, Zhu L. Requirement of cyclin E-Cdk2 inhibition in p16(INK4a)-mediated growth suppression. Mol Cell Biol 1998; 18:5284-90; PMID:9710613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. McConnell BB, Gregory FJ, Stott FJ, Hara E, Peters G. Induced expression of p16(INK4a) inhibits both CDK4- and CDK2-associated kinase activity by reassortment of cyclin-CDK-inhibitor complexes. Mol Cell Biol 1999; 19:1981-9; PMID:10022885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mitra J, Dai CY, Somasundaram K, El-Deiry WS, Satyamoorthy K, Herlyn M, Enders GH. Induction of p21(WAF1/CIP1) and inhibition of Cdk2 mediated by the tumor suppressor p16(INK4a). Mol Cell Biol 1999; 19:3916-28; PMID:10207115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Morisaki H, Ando A, Nagata Y, Pereira-Smith O, Smith JR, Ikeda K, Nakanishi M. Complex mechanisms underlying impaired activation of Cdk4 and Cdk2 in replicative senescence: roles of p16, p21, and cyclin D1. Exp Cell Res 1999; 253:503-10; PMID:10585273; http://dx.doi.org/ 10.1006/excr.1999.4698. [DOI] [PubMed] [Google Scholar]
- 31. Parry D, Mahony D, Wills K, Lees E. Cyclin D-CDK subunit arrangement is dependent on the availability of competing INK4 and p21 class inhibitors. Mol Cell Biol 1999; 19:1775-83; PMID:10022865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kitagawa M, Higashi H, Jung HK, Suzuki-Takahashi I, Ikeda M, Tamai K, Kato J, Segawa K, Yoshida E, Nishimura S, et al. . The consensus motif for phosphorylation by cyclin D1-Cdk4 is different from that for phosphorylation by cyclin A/E-Cdk2. Embo J 1996; 15:7060-9; PMID:9003781. [PMC free article] [PubMed] [Google Scholar]
- 33. Connell-Crowley L, Harper JW, Goodrich DW. Cyclin D1/Cdk4 regulates retinoblastoma protein-mediated cell cycle arrest by site-specific phosphorylation. Mol Biol Cell 1997; 8:287-301; PMID:9190208; http://dx.doi.org/ 10.1091/mbc.8.2.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zarkowska T, Mittnacht S. Differential phosphorylation of the retinoblastoma protein by G1/S cyclin-dependent kinases. J Biol Chem 1997; 272:12738-46; PMID:9139732; http://dx.doi.org/ 10.1074/jbc.272.19.12738. [DOI] [PubMed] [Google Scholar]
- 35. Lundberg AS, Weinberg RA. Functional inactivation of the retinoblastoma protein requires sequential modification by at least two distinct cyclin-cdk complexes. Mol Cell Biol 1998; 18:753-61; PMID:9447971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Harbour JW, Luo RX, Dei Santi A, Postigo AA, Dean DC. Cdk phosphorylation triggers sequential intramolecular interactions that progressively block Rb functions as cells move through G1. Cell 1999; 98:859-69; PMID:10499802; http://dx.doi.org/ 10.1016/S0092-8674(00)81519-6. [DOI] [PubMed] [Google Scholar]
- 37. Dulic V, Drullinger LF, Lees E, Reed SI, Stein GH. Altered regulation of G1 cyclins in senescent human diploid fibroblasts: accumulation of inactive cyclin E-Cdk2 and cyclin D1-Cdk2 complexes. Proc Natl Acad Sci U S A 1993; 90:11034-8; PMID:8248208; http://dx.doi.org/ 10.1073/pnas.90.23.11034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ruas M, Gregory F, Jones R, Poolman R, Starborg M, Rowe J, Brookes S, Peters G. CDK4 and CDK6 delay senescence by kinase-dependent and p16INK4a-independent mechanisms. Mol Cell Biol 2007; 27:4273-82; PMID:17420273; http://dx.doi.org/ 10.1128/MCB.02286-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. LaBaer J, Garrett MD, Stevenson LF, Slingerland JM, Sandhu C, Chou HS, Fattaey A, Harlow E. New functional activities for the p21 family of CDK inhibitors. Genes Dev 1997; 11:847-62; PMID:9106657; http://dx.doi.org/ 10.1101/gad.11.7.847. [DOI] [PubMed] [Google Scholar]
- 40. Cheng M, Olivier P, Diehl JA, Fero M, Roussel MF, Roberts JM, Sherr CJ. The p21(Cip1) and p27(Kip1) CDK 'inhibitors' are essential activators of cyclin D-dependent kinases in murine fibroblasts. Embo J 1999; 18:1571-83; PMID:10075928; http://dx.doi.org/ 10.1093/emboj/18.6.1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bagui TK, Jackson RJ, Agrawal D, Pledger WJ. Analysis of cyclin D3-cdk4 complexes in fibroblasts expressing and lacking p27(kip1) and p21(cip1). Mol Cell Biol 2000; 20:8748-57; PMID:11073976; http://dx.doi.org/ 10.1128/MCB.20.23.8748-8757.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bagui TK, Mohapatra S, Haura E, Pledger WJ. P27Kip1 and p21Cip1 are not required for the formation of active D cyclin-cdk4 complexes. Mol Cell Biol 2003; 23:7285-90; PMID:14517297; http://dx.doi.org/ 10.1128/MCB.23.20.7285-7290.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Olashaw N, Bagui TK, Pledger WJ. Cell cycle control: a complex issue. Cell Cycle 2004; 3:263-4; PMID:14726706; http://dx.doi.org/ 10.4161/cc.3.3.720. [DOI] [PubMed] [Google Scholar]
- 44. Sugimoto M, Martin N, Wilks DP, Tamai K, Huot TJ, Pantoja C, Okumura K, Serrano M, Hara E. Activation of cyclin D1-kinase in murine fibroblasts lacking both p21(Cip1) and p27(Kip1). Oncogene 2002; 21:8067-74; PMID:12444543; http://dx.doi.org/ 10.1038/sj.onc.1206019. [DOI] [PubMed] [Google Scholar]
- 45. Xiong Y, Zhang H, Beach D. Subunit rearrangement of the cyclin-dependent kinases is associated with cellular transformation. Genes Dev 1993; 7:1572-83; PMID:8101826; http://dx.doi.org/ 10.1101/gad.7.8.1572. [DOI] [PubMed] [Google Scholar]
- 46. Parry D, Bates S, Mann DJ, Peters G. Lack of cyclin D-Cdk complexes in Rb-negative cells correlates with high levels of p16INK4/MTS1 tumour suppressor gene product. Embo J 1995; 14:503-11; PMID:7859739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ide T, Tsuji Y, Nakashima T, Ishibashi S. Progress of aging in human diploid cells transformed with a tsA mutant of simian virus 40. Exp Cell Res 1984; 150:321-8; PMID:6319164; http://dx.doi.org/ 10.1016/0014-4827(84)90575-5. [DOI] [PubMed] [Google Scholar]
- 48. Tsuyama N, Miura M, Kitahira M, Ishibashi S, Ide T. SV40 T-antigen is required for maintenance of immortal growth in SV40-transformed human fibroblasts. Cell Struct Funct 1991; 16:55-62; PMID:1851674; http://dx.doi.org/ 10.1247/csf.16.55. [DOI] [PubMed] [Google Scholar]
- 49. Berns K, Hijmans EM, Mullenders J, Brummelkamp TR, Velds A, Heimerikx M, Kerkhoven RM, Madiredjo M, Nijkamp W, Weigelt B, et al. . A large-scale RNAi screen in human cells identifies new components of the p53 pathway. Nature 2004; 428:431-7; PMID:15042092; http://dx.doi.org/ 10.1038/nature02371. [DOI] [PubMed] [Google Scholar]
- 50. Takahashi A, Ohtani N, Yamakoshi K, Iida S, Tahara H, Nakayama K, Nakayama KI, Ide T, Saya H, Hara E. Mitogenic signalling and the p16INK4a-Rb pathway cooperate to enforce irreversible cellular senescence. Nat Cell Biol 2006; 8:1291-7; PMID:17028578; http://dx.doi.org/ 10.1038/ncb1491. [DOI] [PubMed] [Google Scholar]
- 51. Fumagalli M, Rossiello F, Clerici M, Barozzi S, Cittaro D, Kaplunov JM, Bucci G, Dobreva M, Matti V, Beausejour CM, et al. . Telomeric DNA damage is irreparable and causes persistent DNA-damage-response activation. Nat Cell Biol 2012; 14:355-65; PMID:22426077; http://dx.doi.org/ 10.1038/ncb2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hewitt G, Jurk D, Marques FD, Correia-Melo C, Hardy T, Gackowska A, Anderson R, Taschuk M, Mann J, Passos JF. Telomeres are favoured targets of a persistent DNA damage response in ageing and stress-induced senescence. Nat Commun 2012; 3:708; PMID:22426229; http://dx.doi.org/ 10.1038/ncomms1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Fry DW, Harvey PJ, Keller PR, Elliott WL, Meade M, Trachet E, Albassam M, Zheng X, Leopold WR, Pryer NK, et al. . Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol Cancer Ther 2004; 3:1427-38; PMID:15542782. [PubMed] [Google Scholar]
- 54. Lane DP, Simanis V, Bartsch R, Yewdell J, Gannon J, Mole S. Cellular targets for SV40 large T-antigen. Proc R Soc Lond B Biol Sci 1985; 226:25-42; http://dx.doi.org/ 10.1098/rspb.1985.0077. [DOI] [PubMed] [Google Scholar]
- 55. Pemberton H, Anderton E, Patel H, Brookes S, Chandler H, Palermo R, Stock J, Rodriguez-Niedenfuhr M, Racek T, de Breed L, et al. . Genome-wide co-localization of Polycomb orthologs and their effects on gene expression in human fibroblasts. Genome Biol 2014; 15:R23; PMID:24485159; http://dx.doi.org/ 10.1186/gb-2014-15-2-r23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Gagrica S, Brookes S, Anderton E, Rowe J, Peters G. Contrasting behavior of the p18INK4c and p16INK4a tumor suppressors in both replicative and oncogene-induced senescence. Cancer Res 2012; 72:165-75; PMID:22080569; http://dx.doi.org/ 10.1158/0008-5472.CAN-11-2552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Baldin V, Lukas J, Marcote MJ, Pagano M, Draetta G. Cyclin D1 is a nuclear protein required for cell cycle progression in G1. Genes Dev 1993; 7:812-21; PMID:8491378; http://dx.doi.org/ 10.1101/gad.7.5.812. [DOI] [PubMed] [Google Scholar]
- 58. Diehl JA, Cheng M, Roussel MF, Sherr CJ. Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis and subcellular localization. Genes Dev 1998; 12:3499-511; PMID:9832503; http://dx.doi.org/ 10.1101/gad.12.22.3499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Alt JR, Gladden AB, Diehl JA. p21(Cip1) Promotes cyclin D1 nuclear accumulation via direct inhibition of nuclear export. J Biol Chem 2002; 277:8517-23; PMID:11751903; http://dx.doi.org/ 10.1074/jbc.M108867200. [DOI] [PubMed] [Google Scholar]
- 60. Ide T, Tsuji Y, Ishibashi S, Mitsui Y. Reinitiation of host DNA synthesis in senescent human diploid cells by infection with Simian virus 40. Exp Cell Res 1983; 143:343-9; PMID:6299766; http://dx.doi.org/ 10.1016/0014-4827(83)90060-5. [DOI] [PubMed] [Google Scholar]
- 61. Gorman SD, Cristofalo VJ. Reinitiation of cellular DNA synthesis in BrdU-selected nondividing senescent WI-38 cells by simian virus 40 infection. J Cell Physiol 1985; 125:122-6; PMID:2995423; http://dx.doi.org/ 10.1002/jcp.1041250116. [DOI] [PubMed] [Google Scholar]
- 62. Beausejour CM, Krtolica A, Galimi F, Narita M, Lowe SW, Yaswen P, Campisi J. Reversal of human cellular senescence: roles of the p53 and p16 pathways. Embo J 2003; 22:4212-22; PMID:12912919; http://dx.doi.org/ 10.1093/emboj/cdg417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Leontieva OV, Blagosklonny MV. CDK4/6-inhibiting drug substitutes for p21 and p16 in senescence: duration of cell cycle arrest and MTOR activity determine geroconversion. Cell Cycle 2013; 12:3063-9; PMID:23974099; http://dx.doi.org/ 10.4161/cc.26130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Imai Y, Takahashi A, Hanyu A, Hori S, Sato S, Naka K, Hirao A, Ohtani N, Hara E. Crosstalk between the Rb pathway and AKT signaling forms a quiescence-senescence switch. Cell Rep 2014; 7:194-207; PMID:24703840; http://dx.doi.org/ 10.1016/j.celrep.2014.03.006. [DOI] [PubMed] [Google Scholar]
- 65. Blagosklonny MV. Geroconversion: irreversible step to cellular senescence. Cell Cycle 2014; 13:3628-35; PMID:25483060; http://dx.doi.org/ 10.4161/15384101.2014.985507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Narita M, Nunez S, Heard E, Narita M, Lin AW, Hearn SA, Spector DL, Hannon GJ, Lowe SW. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell 2003; 113:703-16; PMID:12809602; http://dx.doi.org/ 10.1016/S0092-8674(03)00401-X. [DOI] [PubMed] [Google Scholar]
- 67. Chicas A, Wang X, Zhang C, McCurrach M, Zhao Z, Mert O, Dickins RA, Narita M, Zhang M, Lowe SW. Dissecting the unique role of the retinoblastoma tumor suppressor during cellular senescence. Cancer cell 2010; 17:376-87; PMID:20385362; http://dx.doi.org/ 10.1016/j.ccr.2010.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Humbert N, Navaratnam N, Augert A, Da Costa M, Martien S, Wang J, Martinez D, Abbadie C, Carling D, de Launoit Y, et al. . Regulation of ploidy and senescence by the AMPK-related kinase NUAK1. EMBO J 2010; 29:376-86; PMID:19927127; http://dx.doi.org/ 10.1038/emboj.2009.342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Holland EC, Hively WP, Gallo V, Varmus HE. Modeling mutations in the G1 arrest pathway in human gliomas: overexpression of CDK4 but not loss of INK4a-ARF induces hyperploidy in cultured mouse astrocytes. Genes Dev 1998; 12:3644-9; PMID:9851971; http://dx.doi.org/ 10.1101/gad.12.23.3644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Hitomi M, Stacey DW. Cyclin D1 production in cycling cells depends on ras in a cell-cycle-specific manner. Curr Biol 1999; 9:1075-84; PMID:10531005; http://dx.doi.org/ 10.1016/S0960-9822(99)80476-X. [DOI] [PubMed] [Google Scholar]
- 71. Gabrielli BG, Sarcevic B, Sinnamon J, Walker G, Castellano M, Wang XQ, Ellem KA. A cyclin D-Cdk4 activity required for G2 phase cell cycle progression is inhibited in ultraviolet radiation-induced G2 phase delay. J Biol Chem 1999; 274:13961-9; PMID:10318807; http://dx.doi.org/ 10.1074/jbc.274.20.13961. [DOI] [PubMed] [Google Scholar]
- 72. Burgess A, Wigan M, Giles N, Depinto W, Gillespie P, Stevens F, Gabrielli B. Inhibition of S/G2 phase CDK4 reduces mitotic fidelity. J Biol Chem 2006; 281:9987-95; PMID:16476733; http://dx.doi.org/ 10.1074/jbc.M512714200. [DOI] [PubMed] [Google Scholar]
- 73. Prall OW, Sarcevic B, Musgrove EA, Watts CK, Sutherland RL. Estrogen-induced activation of Cdk4 and Cdk2 during G1-S phase progression is accompanied by increased cyclin D1 expression and decreased cyclin-dependent kinase inhibitor association with cyclin E-Cdk2. J Biol Chem 1997; 272:10882-94; PMID:9099745; http://dx.doi.org/ 10.1074/jbc.272.16.10882. [DOI] [PubMed] [Google Scholar]
- 74. Matsuura I, Denissova NG, Wang G, He D, Long J, Liu F. Cyclin-dependent kinases regulate the antiproliferative function of Smads. Nature 2004; 430:226-31; PMID:15241418; http://dx.doi.org/ 10.1038/nature02650. [DOI] [PubMed] [Google Scholar]
- 75. Anders L, Ke N, Hydbring P, Choi YJ, Widlund HR, Chick JM, Zhai H, Vidal M, Gygi SP, Braun P, et al. . A systematic screen for CDK4/6 substrates links FOXM1 phosphorylation to senescence suppression in cancer cells. Cancer cell 2011; 20:620-34; PMID:22094256; http://dx.doi.org/ 10.1016/j.ccr.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Wierstra I, Alves J. Transcription factor FOXM1c is repressed by RB and activated by cyclin D1/Cdk4. Biol Chem 2006; 387:949-62; PMID:16913845. [DOI] [PubMed] [Google Scholar]
- 77. Bates S, Parry D, Bonetta L, Vousden K, Dickson C, Peters G. Absence of cyclin D/cdk complexes in cells lacking functional retinoblastoma protein. Oncogene 1994; 9:1633-40; PMID:8183557. [PubMed] [Google Scholar]
- 78. Kim NW, Piatyszek MA, Prowse KR, Harley CB, West MD, Ho PL, Coviello GM, Wright WE, Weinrich SL, Shay JW. Specific association of human telomerase activity with immortal cells and cancer. Science 1994; 266:2011-5; http://dx.doi.org/ 10.1126/science.7605428. [DOI] [PubMed] [Google Scholar]
- 79. Brummelkamp TR, Bernards R, Agami R. A system for stable expression of short interfering RNAs in mammalian cells. Science 2002; 296:550-3; http://dx.doi.org/ 10.1126/science.1068999. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.