

Abstract
Unrestrained p53 activity during development, as occurs upon loss of the p53 negative regulators Mdm2 or Mdmx, causes early embryonic lethality. Surprisingly, co-expression of wild-type p53 and a transcriptionally-dead variant of p53, with mutations in both transactivation domains (p53L25Q,W26S,F53Q,F54S), also causes lethality, but later in gestation and in association with a host of very specific phenotypes reminiscent of a syndrome known as CHARGE. Molecular analyses revealed that wild-type p53 is inappropriately activated in p535,26,53,54/+ embryos, triggering cell-cycle arrest or apoptosis during development to cause CHARGE phenotypes. In addition, CHARGE syndrome is typically caused by mutations in the CHD7 chromatin remodeler, and we have shown that activated p53 contributes to phenotypes caused by CHD7-deficiency. Together, these studies provide new insight into CHARGE syndrome and expand our understanding of the role of p53 in diseases other than cancer.
Keywords: p53, transcriptional activation, CHARGE syndrome, CHD7, embryonic development, Ribosomopathy, Mdm2, neural Crest
Introduction
The p53 protein plays a fundamental role in suppressing cancer, as evidenced by the findings that p53 is mutated in over half of human tumors and that p53 null mice display a completely penetrant cancer predisposition.1-4 The ability of p53 to restrain malignancy is thought to relate to its ability to respond to a wide spectrum of cellular stresses by inducing cell-cycle arrest or apoptosis to limit expansion of neoplastic cells.5,6 p53 is a transcriptional activator, which binds to specific DNA elements throughout the genome, leading to the induction of p53 target genes involved in the apoptosis or cell-cycle arrest programs, although p53 can also act through transactivation-independent mechanisms such as direct induction of apoptosis through cytoplasmic interactions with Bcl-2 family members.7,8 In addition to an unequivocal role in cancer suppression, the ability of p53 to respond to cellular stresses has broader significance, as p53 participates in additional physiological and pathological processes. For example, p53 activation drives some of the deleterious phenotypic effects of stroke, neurodegenerative disease, and genotoxic cancer therapies.9,10 Thus, p53 activity can be beneficial or detrimental, depending on the context, underscoring the importance of keeping it tightly controlled.
Keeping the Brakes on p53
The critical importance of restraining p53 in a physiological context was originally appreciated through analyses of knockout mice deficient for the p53 negative regulators Mdm2 and Mdmx.11-16 Mdm2 binds and inhibits p53's 2 transcriptional activation domains (TADs) as well as acting as an E3 ubiquitin ligase to promote p53 degradation,17,18 while Mdmx binds and inhibits the p53 TADs but only affects stability through interactions with Mdm2 rather than ubiquitylating p53 itself.19-21 Loss of Mdm2 in mice causes early embryonic lethality at ∼E3.5, attributable to inappropriate p53 activity, as evidenced by the complete viability of Mdm2−/−;p53−/− mice. These observations provided the first demonstration that unrestrained p53 activity during development can drive embryonic lethality.13,15,16 Similarly, Mdmx−/− mice are also embryonic lethal, although at a later timepoint (between E7.5–11.5), and this lethality is again fully rescued by p53 deficiency.11,12,14 Subsequent studies using Mdm2 and Mdmx conditional mouse strains further revealed the necessity of regulating p53 in various tissues, including the heart, haematopoietic system, skin, bone, and neuroepithelium, where p53 activation causes increased cell-cycle arrest, senescence or apoptosis and perturbs proper tissue development.19,22-29 Thus, the appropriate restraint of p53 is fundamental for proper embryo and tissue development.
Elucidating the Role of Transactivation in p53-driven Developmental Phenotypes
We sought to follow-up on these studies by investigating the mechanisms through which p53 activation during embryogenesis promotes developmental phenotypes. In particular, given the various biochemical activities of p53, we set out to address the requirement for p53 transcriptional activation potential in driving developmental phenotypes. Toward this end, we leveraged p53 mutant knock-in mouse strains that we generated previously as tools to assess the contribution of p53 transactivation activity to different biological functions.30,31 In these strains, mutations were introduced into the first (L25Q,W26S) or both (L25Q,W26S,F53Q,F54S) of p53's 2 amino-terminal TADs (Fig. 1A) to create the p5325,26 and p5325,26,53,54 mutants. These mutations were originally shown to compromise p53's transactivation activity in reporter assays in vitro, and we previously utilized these knock-in mice to study p53 transcriptional programs underlying responses to genotoxic cancer therapy and tumor suppression.30-34 Using genome-wide expression profiling, we found that p5325,26 displays selective transactivation function, being severely compromised for the ability to activate most p53 target genes, but fully active on a subset of p53 target genes, while p5325,26,53,54 is completely transactivation-deficient (Fig. 1B).30,35 Moreover, our studies revealed that the p5325,26,53,54 mutant is unable to promote apoptosis in vivo in response to gamma-irradiation or to suppress tumorigenesis in various mouse cancer models, indicating the transactivation function is critical for these biological processes.30,36 Interestingly, the p5325,26 mutant showed separation-of-function activity, where it could not trigger cell-cycle arrest or apoptosis in response to genotoxic damage but could suppress growth of a variety of cancers,30,31,36 suggesting that the selective transcriptional activity manifested by this mutant accounts for its function in tumor suppression. Hence, we set out to similarly address the importance of transcriptional activation for p53-driven developmental phenotypes, using the same mutants. Importantly, the mutations at residues 25 and 26 disrupt the Mdm2-p53 interaction, resulting in stabilized p53 (Fig. 1B).37 Thus, it is possible to examine the mechanism of p53-induced embryonic lethality using these mice, as Mdm2 binding and transactivation are both perturbed (although transactivation is compromised to different extents in the 2 strains). The p53 TAD mutant knock-in mice conditionally express p53 upon deletion of an upstream lox-stop-lox cassette by the Cre recombinase, and therefore they were crossed to CMV-Cre mice, which ubiquitously express Cre.38 Widespread expression of a single copy of p5325,26, in p5325,26/− embryos, resulted in mid-gestational embryonic lethality, which we hypothesized reflected the ability of p5325,26 to retain efficient transactivation of a subset of p53 target genes, including Bax, which had been shown to be partially responsible for the lethality in Mdm2-null embryos.15 The importance of residual transactivation activity in triggering embryonic lethality in p5325,26/− embryos was confirmed upon evaluation of p5325,26,53,54/− embryos, where we found that widespread expression of a single copy of the transactivation-dead p5325,26,53,54 mutant was compatible with the generation of viable adults. These data therefore reveal that p53 transactivation potential is essential for the embryonic lethality resulting from stabilized p53.
Figure 1.
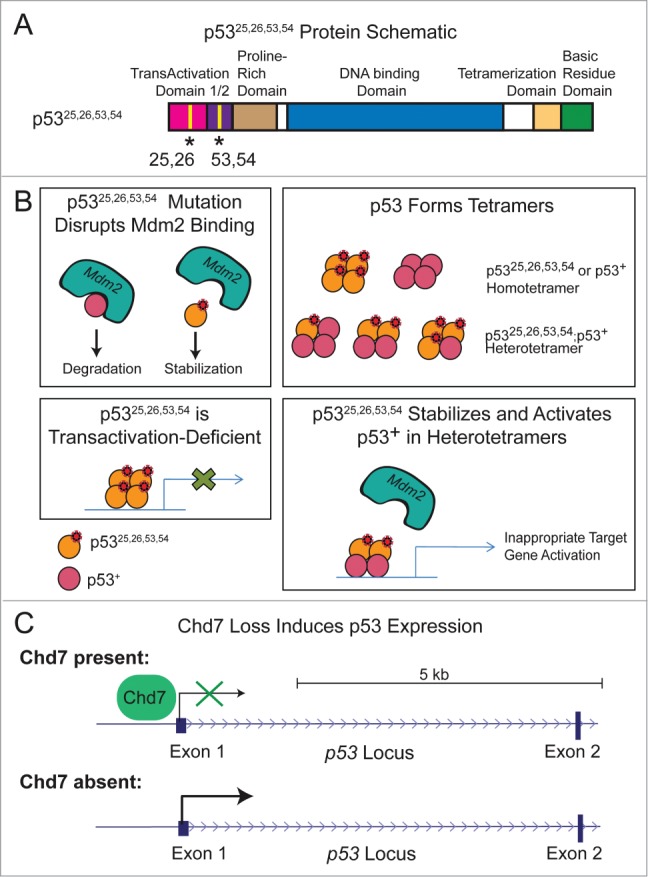
p53 is induced in p5325,26,53,54/+ embryos and Chd7-null cells. (A) Schematic of the p5325,26,53,54 protein with L25Q, W26S, F53Q, and F54S mutations and structural domains indicated. (B) Model for how p5325,26,53,54 may stabilize and activate wild-type p53 (denoted by p53+). Top Left: p5325,26,53,54 mutations disrupt the p53-interaction with Mdm2, resulting in stabilized p53 protein. Top Right: p53 can form tetramers comprised of any combination of mutant and/or wild-type p53 proteins, which can result in homotetramers or heterotetramers. Bottom Left: p5325,26,53,54 can bind to DNA but is transactivation-deficient. Bottom Right: p5325,26,53,54 can interact with wild-type p53 in heterotetramers, disrupt the p53-interaction with Mdm2, and promote wild-type p53 to transactivate target genes. Shown is an example of the type of heterotetramer that can form, although other subunit compositions are possible. C) Chd7 loss induces p53 expression. One mechanism by which Chd7 loss could induce p53 expression is through loss of Chd7 binding to the p53 promoter, resulting in derepression of p53 expression.
A Model for CHARGE Syndrome
In conducting these studies, we discovered, much to our surprise, that co-expression of the transactivation-dead p5325,26,53,54 mutant and wild-type p53 triggers mid-late gestational embryonic lethality at ∼E13.5–15.5.35 This finding suggested a genetic interaction between wild-type and mutant p53, whose intricate nature we subsequently revealed by in vitro analyses.35 We found that due to the disruption of the Mdm2-p53 interaction caused by mutation of the 25,26 residues, p5325,26,53,54 is stabilized, which in turn allows it to interact with and stabilize wild-type p53 to adequately high levels for wild-type p53 to bind and transactivate select target genes and to induce apoptosis and cell-cycle arrest (Fig. 1B). However, the level of p53 activity in p5325,26,5,354/+ embryos arising from the combination of a stable transactivation-dead mutant p53 protein with wild-type p53 is clearly not equivalent to the activity levels of wild-type p53 due to Mdm2 loss, as demonstrated by the more moderate phenotypes in p5325,26,53,54/+ embryos than in Mdm2−/− embryos.13,15,16 Indeed, Mdm2-nullizygosity induces stabilized, fully active wild-type p53, while combined expression of wild-type p53 and p5325,26,53,54 in p5325,26,53,54/+ embryos merely causes modest p53 activation, as only wild-type p53 subunits are active while p5325,26,53,54 subunits are transactivation-dead (Fig. 1B). Collectively, these findings were extremely surprising and completely unexpected, as transcriptionally-dead p53 molecules typically display dominant-negative activities. This study therefore serves as a novel paradigm for how other p53 mutant proteins, as well as potentially other mutant proteins, might drive disease phenotypes. Of note, the activation of wild-type p53 by the tumor suppression-dead p5325,26,53,54 mutant suggests one reason for the lack of such transactivation-deficient mutants in human cancers.
Interestingly, the p5325,26,53,54/+ embryos exhibited an exquisitely specific spectrum of phenotypes including coloboma of the retina (failure of optic closure), heart outflow tract and atrio-ventricular cushion defects, and inner and outer ear defects – a collection of phenotypes very reminiscent of a syndrome known as CHARGE.39 CHARGE is an autosomal dominant congenital syndrome named for its characteristic features: ocular coloboma, heart defects, choanal atresia (narrowing of the nasal passage), retarded growth and development, genitourinary hypoplasia, and ear abnormalities (Fig. 2).39,40 In 70–90% of cases, CHARGE is caused by mutations in CHD7, encoding an ATP-dependent chromatin remodeler.41,42 However, while Chd7+/- mice exhibit some features of CHARGE, they have not been described to display major hallmarks, such as coloboma and heart outflow tract defects, and Chd7−/− embryos display embryonic lethality at E10.5.43-45 In contrast, our p5325,26,53,54/+ embryos survive until later in gestation and exhibit a full spectrum of CHARGE symptoms.35 In fact, the manifestation of extensive ear malformations, coloboma, and heart defects in p5325,26, 53,54/+ embryos emphasizes the value of our model for recapitulating a comprehensive form of CHARGE. In addition, while the embryonic lethality in mouse contrasts with human CHARGE, where individuals are mostly viable, it remains possible that fetuses with more severe phenotypes may die in utero, but this has not yet been demonstrated.46
Figure 2.
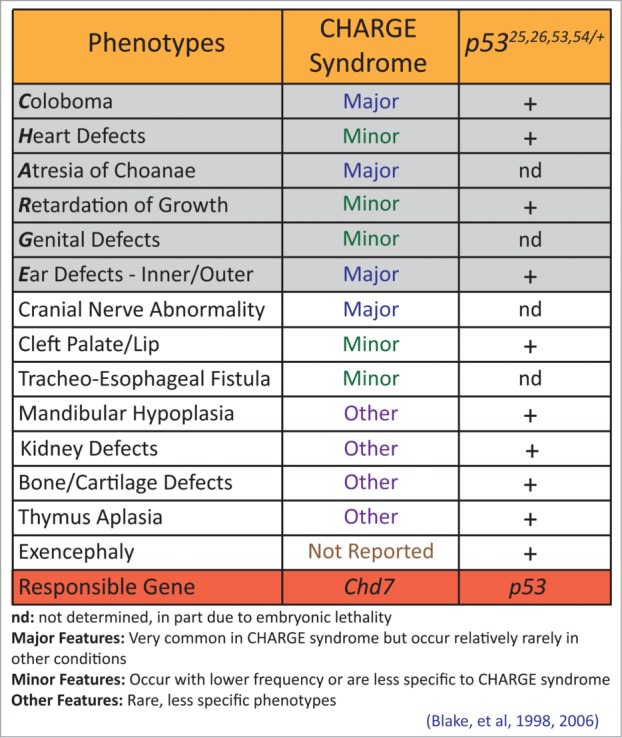
p5325,26,53,54/+ embryos display phenotypes characteristic of CHARGE syndrome. Characteristics of CHARGE syndrome are considered major features, minor features, or other features, depending on the specificity to CHARGE and the utility of the feature for diagnosing CHARGE. The phenotypes contributing to the CHARGE acronym are shaded in gray. Phenotypes observed in p5325,26,53,54/+ embryos are indicated with a +. nd: not determined in p5325,26,53,54/+ embryos, due in part to mid-late gestational embryonic lethality.
A Role for p53 Downstream of Chd7
While these mouse studies show that p53 activation can cause features of CHARGE, the role of p53 downstream of Chd7 deficiency and in human CHARGE remained unclear. We therefore investigated whether there was any crosstalk between Chd7 and p53. Although we found that p53 activation did not affect Chd7 levels, we showed that the converse was true, that in several settings Chd7 loss triggers p53 activation.35 First, as neural crest cells (NCCs) are thought to at least partially underlie CHARGE phenotypes,39,47,49 we examined mouse NCCs and found that Chd7 inactivation resulted in increased expression of p53 and select p53 target genes (Fig. 1C). Second, to extend these data to human CHARGE patient samples, we examined p53 activity in fibroblasts from control and CHD7-mutation positive CHARGE patients. We found both elevated p53 protein and heightened p53 transcriptional activity on select p53 target genes in CHARGE patient fibroblasts relative to controls. Thus, in this human cell setting as well, CHD7 deficiency results in increased p53 levels and enhanced p53 target gene expression. Finally, using thymus samples from CHD7-mutation positive CHARGE syndrome patients, we observed increased p53 stabilization by immunohistochemistry in the thymi of fetuses from CHARGE patients relative to control fetuses. Collectively, these data suggest that CHD7 loss triggers p53 activation. However, to unequivocally implicate p53 in CHARGE, an examination of whether phenotypes manifesting with Chd7 deficiency are rescued by p53 deficiency was required.44 Our analysis of embryonic phenotypes associated with Chd7-nullizygosity showed that they are partially rescued by p53-heterozygosity.35 Specifically, the developmental delay, as assessed by somite number, was completely rescued, but limb and craniofacial formation were only partially rescued. Collectively, these findings indicate that p53 does indeed contribute to phenotypes downstream of Chd7 loss and strongly implicate p53 in promoting phenotypes in CHD7 mutation-positive human CHARGE patients.
It is also possible that p53 could function independently of CHD7 to promote CHARGE phenotypes in the ∼10–30% of cases where CHD7 mutations have not been identified. For example, mutations in genes other than CHD7 could trigger p53 activation. Alternatively, p53 or components of the p53 pathway could be mutated in some CHARGE cases. Our mutational analysis of the p53 coding region in a subset of CHD7-intact CHARGE patients did not reveal any p53 mutations, although we analyzed a limited sample size.35 Moreover, the modest levels of p53 activation that promote the CHARGE-like phenotypes in our mouse studies may be difficult to achieve by point mutation of the p53 coding sequence. Instead, mutations in p53 regulatory elements or in genes encoding key p53 regulators could have the effect of slightly activating p53 to promote CHARGE phenotypes and thus may be more likely to occur.50 Collectively, our findings demonstrate a direct connection between Chd7 loss and p53 activation in CHARGE, highlighting the importance of our p5325,26,53,54/+ mouse model for studying CHARGE.
Signaling from Chd Proteins to p53
The observation that p53 is activated upon Chd7 loss raises the question of the signal transduction cascade involved. Using chromatin immunoprecipitation (ChIP), we were able to show that Chd7 binds to the p53 promoter in mouse NCCs and that Chd7 deficiency results in increased p53 transcripts in both mouse NCCs and CHARGE patient fibroblasts, together suggesting that Chd7 directly represses p53 transcription (Fig. 1C).35 However, Chd7 loss could also activate p53 indirectly, through more canonical pathways that impinge upon p53. It is well-established that p53 is activated post-translationally by stress signals, such as DNA damage or oncogene expression, via disruption of the p53-interaction with Mdm2 and consequent p53 stabilization.6 Furthermore, defects in ribosomal biogenesis, such as those triggered by ribosomal subunit mutation, also activate p53 by perturbing the Mdm2-p53 interaction.51 Interestingly, Chd7 has been shown to play a role in ribosome biogenesis, as Chd7 binds to rDNA and enhances rRNA expression.52 Chd7 inactivation results in both reduced pre-rRNA and global protein synthesis, suggestive of the existence of ribosome defects. In addition, Chd7 interacts with the nucleolar protein Treacle (encoded by Tcof1), which when mutated causes defects in ribosome biogenesis and p53 activation,52,53 and it is thus quite plausible that Chd7 loss similarly triggers p53 activation at the post-translational level through this pathway.
Chd7 is a member of a larger CHD (chromodomain helicase DNA-binding) protein family, comprising 9 ATP-dependent chromatin remodelers (Chd1-9), some of which directly bind DNA.54 Interestingly, several of these have previously been reported to impinge upon the p53 pathway.55-57 Notably, loss of Chd8, which interacts with Chd7, causes early embryonic lethality that is partially rescued by p53 deficiency.55,58 Chd8 was found to bind to p53 along with H1 to suppress p53 transactivation activity, thereby preventing p53-mediated apoptosis during embryogenesis.55 In addition, Chd4 regulates p53 at the post-translational level by deacetylating p53, such that Chd4 loss enhances p53 acetylation and activity.56 In addition, embryos with duplication of 1p36, which contains the tumor suppressor gene Chd5, exhibit enhanced senescence and apoptosis as well as perinatal lethality, which are rescued by p53 deficiency.57 Activation of p53 in this context is attributable to Chd5 positively regulating p19Arf expression, which liberates p53 from Mdm2.57 Therefore, the ability of Chd proteins to regulate p53 at the post-translational level has a strong precedent. Additional studies in the future will help to fully elaborate the molecular interactions between Chd7 and p53.
The Role of p53 in Other Diseases
Our findings that p53 plays a causative role in CHARGE syndrome phenotypes supports the emerging idea that p53 can have deleterious effects, contributing to diseases beyond cancer (Fig. 3). Support for this notion came originally from studies showing that p53-induced apoptosis in neurons contributes to the associated pathologies in stroke and neurodegenerative diseases, including Alzheimer, Parkinson and Huntington's diseases.9 Subsequently, p53 activation was implicated in several genetic diseases, including Diamond Blackfan Anemia (DBA), 5q-syndrome, and Treacher Collins syndrome (Fig. 3).53,59,61 Interestingly, these diseases are all considered ribosomopathies in which defects in ribosomal biogenesis are thought to underlie the observed symptoms. In DBA, mutation of genes encoding ribosomal proteins, such as RPS19, results in red blood cell anemia associated with developmental abnormalities, including orofacial, hand, limb, urogenital, and heart defects.61 Mutation or deletion of Rps19 in mouse models results in red blood cell anemia and skin hyperpigmentation, which are rescued by p53 deletion.60-62 In 5q-syndrome, patients carry a deletion of chromosome 5q, which includes the Rps14 gene, and these patients exhibit macrocytic anemia, megakaryocyte dysplasia, and haematopoietic progenitor cell defects, phenotypes which can all be ameliorated by loss of p53 in mouse models.59,61,63 Treacher Collins syndrome is caused by mutations in the TCOF1 gene and is characterized by craniofacial defects such as mandibular hypoplasia, cleft palate, ear defects, and coloboma of the eye in humans, and p53 deficiency can rescue the neonatal lethality and craniofacial defects observed in Tcof1 mutant mice.53,64 In these ribosomopathies, deficiency in the implicated gene products is thought to compromise ribosome biogenesis, hence triggering p53 activation. An active contribution of p53 to these syndromes has been proposed based both on observations that p53 is stabilized in mouse and human patient tissues and rescue experiments in mouse models (Fig. 3).53,59,60,65,66
Figure 3.

p53 hyperactivity is associated with multiple syndromes. Syndromes associated with p53 hyperactivity identified using mouse models and human patient samples. The phenotypes observed in human patients and the most commonly mutated genes associated with each syndrome are noted. The genotypes of the mouse models are noted. Our work identifies p53 hyperactivity in CHARGE syndrome using 3 independent criteria: p53 mutation being sufficient to cause phenotypes of the syndrome in a mouse model, p53 accumulation and/or activation in CHARGE patient samples, and p53-deficiency being able to rescue phenotypes in Chd7-deficient mice.
Beyond activation of p53 by the aforementioned stress cues, p53 mutation may also lead to alterations in p53 activity, with pathological consequences (Fig. 3). This notion is exemplified by our mouse model, where coordinate expression of p5325,26,53,54 and wild-type p53 is sufficient to drive CHARGE phenotypes.35 In addition, mice homozygously expressing a p53 truncation mutant, p53Δ31 – which lacks the C-terminal negative regulatory domain – develop dyskeratosis congenita, a syndrome characterized by nail dystrophy, oral leukoplakia, skin hyperpigmentation, aplastic anemia, and pulmonary fibrosis and attributable to telomerase dysfunction and consequent telomere attrition.67,68 A direct role for p53 in triggering phenotypes upon telomere erosion is indicated by studies in telomerase-deficient mice, where germ cell depletion is partially rescued by p53 loss.69 While telomere attrition clearly induces DNA damage, thereby activating p53, some effects may also be through ribosome biogenesis defects, as telomere maintenance proteins are also required for processing of rRNAs.70 Collectively, these data suggest that p53 contributes to developmental defects in ribosomal biogenesis syndromes. As mentioned above, it could be that CHD7 deficiency promotes CHARGE phenotypes by similarly triggering p53 activity through effects on ribosome biogenesis.
p53 is also implicated in diseases associated with defective NCC function (Fig. 3). Indeed, CHARGE is thought to arise at least in part from defects in NCC survival, proliferation, or migration.47-49 Additionally, the 22q11 deletion syndromes, including Velocardiofacial syndrome and DiGeorge syndrome, in which patients present with heart defects, hypoplastic thymus, and mild craniofacial defects, are also thought to be partially attributable to defects in NCCs.71,72 p53 suppression by genetic ablation or pharmacological inhibition can partially rescue phenotypes of Tbx1 heterozygous mice, which develop features of DiGeorge syndrome, including cardiac outflow tract defects and thymic aplasia.73 Furthermore, p53 has been reported to contribute to NCC defects in Treacher Collins syndrome, where p53 activation in Tcof1+/− embryos triggers apoptosis of neuroepithelial cells and deficiencies in the formation of migrating cranial NCCs, culminating in craniofacial malformations.53 Finally, upon inactivation of the Pax3 transcription factor, in a mouse model for Waardenburg syndrome Type I/III (in which patients exhibit pigmentation defects of the eye, hair, and skin as well as hearing loss), p53-dependent apoptosis in migrating cardiac NCCs causes outflow tract septation defects.74-77 Whether defects in neural crest cells in p5325,26,53,54/+ embryos are responsible for inducing the CHARGE-like phenotypes is unknown, but it will be interesting in future to determine the cell type(s) underlying phenotypes in p5325,26,53,54/+ embryos. Collectively, these mouse models show that p53 activation can promote an ever-broadening range of diseases – a role for p53 that until recently has been greatly underappreciated.
p53 Hyperactivity in Mice: From Embryonic Lethality to Premature Aging
Interestingly, the collection of phenotypes in p5325,26,53,54/+ embryos represents one in a range of mouse models where differing levels of p53 activity cause diverse phenotypes, ranging from embryonic lethality to premature aging (Fig. 4). Loss of Mdm2 or Mdmx results in early embryonic lethality stemming from high levels of activated wild-type p53.11-16 In contrast, our p5325,26,53,54/+ embryos display mid-late gestational embryonic lethality and CHARGE-like phenotypes, which are less severe than those observed upon Mdm2 or Mdmx loss.35 The defects in p5325,26,53,54/+ embryos are due to modest p53 activation resulting from mixed tetramers forming between wild-type p53 and transactivation-dead p5325,26,53,54.
Figure 4.

Summary of mouse strains with different levels of p53 activity and associated phenotypes. Mouse strains manifesting varying levels of p53 stabilization and/or activity exhibit diverse phenotypes ranging from embryonic lethality to premature aging to increased tumor susceptibility. The phenotypes and the p53 status in each indicated mouse strain are described.87-89
Another set of mouse strains expressing hyperactive p53 is characterized by perinatal developmental defects, commonly accompanied by early mortality within 2 months of life (Fig. 4). For example, mouse strains expressing either of 2 p53 C-terminal truncation mutants, p53Δ31 or p53ΔCTD (p53Δ24), were developed to explore the role of the C-terminal negative regulatory domain of p53.67,78 As described above, p53Δ31/ Δ31 mice exhibit lethality between 14 and 43 days along with bone marrow failure and skin hyperpigmentation as well as increased p53 activity and telomere dysfunction.67 In contrast, p53ΔCTD/ΔCTD mice display lethality by 2 weeks after birth and exhibit haematopoietic failure and impaired cerebellar development but only have activated p53 in certain tissues.78 Another study using p53TSD/− mice explored the role of phosphorylation at threonine (T) 21 and serine (S) 23 by mutating these sites to aspartic acid (D), to mimic p53 activation.79 These mice exhibit features of premature aging that is correlated with depletion of adult stem cells in various tissues due to increased activation of select p53 target genes, particularly Puma.79 Minimizing the negative regulation of p53 by Mdm2 and Mdmx has provided another approach to examine the consequences of p53 activation on mouse development (Fig. 4). Mdm2;Mdmx double heterozygous mice display partially penetrant embryonic lethality, associated with exencephaly, as well as perinatal lethality by postnatal day 14 (P14), accompanied by growth retardation, anemia, and hypoplastic tissues.80 In addition, Mdm2−/− mice homozygous for a p53 hypomorphic mutant lacking apoptotic but not cell-cycle arrest function (p53515C) displayed a failure of various progenitor cells to expand, resulting in defective haematopoietic and cerebellar development and death between P12 and P14.81 In the Mdm2Puro/Δ7–12 hypomorphic mutant mice, in which one allele is null and the second allele has reduced Mdm2 expression due to the presence of a puromycin cassette in the Mdm2 locus, the ensuing phenotypes include lymphopenia, growth retardation, and radiosensitivity, associated with enhanced p53 transcriptional activity.82,83 Interestingly, this strain displays a normal lifespan. Overall, differences in the extent of p53 activation or the spectrum of p53 target genes activated relative to strains displaying embryonic lethality may explain the reduced severity of phenotypes observed in this set of mouse strains.
Other mouse strains expressing hyperactive p53 fail to display developmental defects but ultimately succumb to premature aging (Fig. 4). In the p53 m mouse strain, a C-terminal fragment of p53 is produced, provoking premature aging in p53m/+ adults.84 However, unlike the p5325,26,53,54/+ embryos, there is no sign of increased basal p53 activity, and instead, stress signals are required to induce the p53 m protein to augment p53 stability and activity. Thus, the cumulative stress with aging may ultimately promote the aging phenotypes. Another mouse strain, overexpressing a truncated, naturally-occurring isoform of p53 that lacks the first transactivation domain (Δ40), also exhibits growth retardation and premature aging.85 However, expression of Δ40 does not affect wild-type p53 levels, and while some p53 target genes are hyperactivated, a subset of p53 target genes are actually inhibited, which could potentially blunt the effect of the activated p53.85 In contrast to these mouse strains exhibiting premature aging, the p53 super mice, carrying 1 or 2 transgenes expressing wild-type p53 on an otherwise wild-type p53 background, age normally and only exhibit enhanced p53 activity in response to DNA damage.86 The lack of premature aging in the p53 super mice is likely due to the proper negative regulation of p53 protein stability through the N-terminus. Overall, the phenotypic variability observed in these mouse strains expressing hyperactive p53 is likely due to differences in p53 stabilization, p53 transcriptional activity, and the tissues affected in the various mouse models, with the p5325,26,53,54/+ embryos exhibiting levels of p53 stability and activity at the higher end of the spectrum.
Future Perspectives and Therapeutic Implications
By unveiling p53 pathway activation as a novel mechanism contributing to the pathogenesis of CHARGE, our work provides significant new insight into the processes triggering CHARGE syndrome. Moreover, we propose that our p5325,26,53,54/+ mutant mice represent a new and more penetrant model for human CHARGE syndrome due to the highly overlapping features with human CHARGE. These mice therefore provide a great resource for better understanding the underlying cellular and molecular basis of the disease, including identifying the cell type(s) of origin. Given p53's role as a stress response protein, this new association of p53 with CHARGE syndrome could potentially enhance our understanding of the factors that underlie the great clinical heterogeneity in CHARGE, which could relate at least in part to the ability of p53 to respond to stress signals during pregnancy.41,42 The identification of a role for p53 in CHARGE syndrome invites investigation of the role of p53 in the genesis of additional developmental syndromes, even beyond those caused by defects in ribosomal biogenesis and neural crest cell function.
The discovery of a causative role for p53 in developmental diseases prompts the question of whether there could be any therapeutic benefit of inhibiting p53. While complete inhibition of p53 during development might be challenging due to the potential for promoting tumorigenesis in either the fetus or the mother, it may be effective to modestly decrease the activity of p53 through mild pharmacological inhibition of p53. Indeed a successful precedent for this strategy was seen with the amelioration of defects in Tcof1+/–, Pax3−/−, and Tbx1+/− embryos upon administration of the p53-inhibitor Pifithrin-α.53,73,74 Alternatively, p53 target gene products critical for p53's ability to promote CHARGE phenotypes could be pharmacologically inhibited, which could potentially circumvent the possibility of inadvertently promoting tumorigenesis. Overall, these studies have revealed both the importance of p53 transcriptional activity in triggering developmental phenotypes and a novel role for p53 in CHARGE syndrome, greatly expanding our understanding of the role of p53 in developmental diseases.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We would like to thank CA Brady, ME Bowen, and DM Martin for critical reading of the manuscript.
Funding
This work was supported by funding from the NSF and NCI (Grant Number 1F31CA167917-01) to JLF and by funding from the ACS, LLS, and NIH (RO1 CA140875) to LDA.
References
- 1. Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA, Jr, Butel JS, Bradley A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 1992; 356:215-21; PMID:1552940; http://dx.doi.org/ 10.1038/356215a0 [DOI] [PubMed] [Google Scholar]
- 2. Jacks T, Remington L, Williams BO, Schmitt EM, Halachmi S, Bronson RT, Weinberg RA. Tumor spectrum analysis in p53-mutant mice. Curr Biol 1994; 4:1-7; PMID:7922305 [DOI] [PubMed] [Google Scholar]
- 3. Purdie C, Harrison D, Peter A, Dobbie L, White S, Howie S, Salter D, Bird C, Wyllie A, Hooper M, et al. Tumour incidence, spectrum and ploidy in mice with a large deletion in the p53 gene. Oncogene 1994; 9:603-9; PMID:8290271 [PubMed] [Google Scholar]
- 4. Soussi T. Advances in carcinogenesis: a historical perspective from observational studies to tumor genome sequencing and TP53 mutation spectrum analysis. Biochim Biophys Acta 2011; 1816:199-208; PMID:21791238; http://dx.doi.org/ 10.1016/j.bbcan.2011.07.003 [DOI] [PubMed] [Google Scholar]
- 5. Vousden KH, Lu X. Live or let die: the cell's response to p53. Nat Rev 2002; 2:594-604; PMID:12154352 [DOI] [PubMed] [Google Scholar]
- 6. Vousden KH, Prives C. Blinded by the Light: The Growing Complexity of p53. Cell 2009; 137:413-31; PMID:19410540; http://dx.doi.org/ 10.1016/j.cell.2009.04.037 [DOI] [PubMed] [Google Scholar]
- 7. Green DR, Kroemer G. Cytoplasmic functions of the tumour suppressor p53. Nature 2009; 458:1127-30; PMID:19407794; http://dx.doi.org/ 10.1038/nature07986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Brady CA, Attardi LD. p53 at a glance. J Cell Sci 2010; 123:2527-32; PMID:20940128; http://dx.doi.org/ 10.1242/jcs.064501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Culmsee C, Mattson MP. p53 in neuronal apoptosis. Biochem Biophys Res Commun 2005; 331:761-77; PMID:15865932; http://dx.doi.org/ 10.1016/j.bbrc.2005.03.149 [DOI] [PubMed] [Google Scholar]
- 10. Gudkov AV, Komarova EA. The role of p53 in determining sensitivity to radiotherapy. Nat Rev 2003; 3:117-29; PMID:12563311 [DOI] [PubMed] [Google Scholar]
- 11. Migliorini D, Denchi EL, Danovi D, Jochemsen A, Capillo M, Gobbi A, Helin K, Pelicci PG, Marine J-C. Mdm4 (Mdmx) regulates p53-induced growth arrest and neuronal cell death during early embryonic mouse development. Mol Cell Biol 2002; 22:5527-38; PMID:12101245; http://dx.doi.org/ 10.1128/MCB.22.15.5527-5538.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Parant J, Chavez-Reyes A, Little NA, Yan W, Reinke V, Jochemsen AG, Lozano G. Rescue of embryonic lethality in Mdm4-null mice by loss of Trp53 suggests a nonoverlapping pathway with MDM2 to regulate p53. Nat Genet 2001; 29:92-5; PMID:11528400; http://dx.doi.org/ 10.1038/ng714 [DOI] [PubMed] [Google Scholar]
- 13. Montes de Oca Luna R, Wagner DS, Lozano G. Rescue of early embryonic lethality in mdm2-deficient mice by deletion of p53. Nature 1995; 378:203-6; PMID:7477326; http://dx.doi.org/ 10.1038/378203a0 [DOI] [PubMed] [Google Scholar]
- 14. Finch RA, Donoviel DB, Potter D, Shi M, Fan A, Freed DD, Wang CY, Zambrowicz BP, Ramirez-Solis R, Sands AT, et al. mdmx is a negative regulator of p53 activity in vivo. Cancer Res 2002; 62:3221-5; PMID:12036937 [PubMed] [Google Scholar]
- 15. Chavez-Reyes A, Parant JM, Amelse LL, de Oca Luna RM, Korsmeyer SJ, Lozano G. Switching mechanisms of cell death in mdm2- and mdm4-null mice by deletion of p53 downstream targets. Cancer Res 2003; 63:8664-9; PMID:14695178 [PubMed] [Google Scholar]
- 16. Jones SN, Roe AE, Donehower LA, Bradley A. Rescue of embryonic lethality in Mdm2-deficient mice by absence of p53. Nature 1995; 378:206-8; PMID:7477327; http://dx.doi.org/ 10.1038/378206a0 [DOI] [PubMed] [Google Scholar]
- 17. Chen J, Marechal V, Levine AJ. Mapping of the p53 and mdm-2 interaction domains. Mol Cell Biol 1993; 13:4107-14; PMID:7686617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Momand J, Zambetti GP, Olson DC, George D, Levine AJ. The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell 1992; 69:1237-45; PMID:1535557; http://dx.doi.org/ 10.1016/0092-8674(92)90644-R [DOI] [PubMed] [Google Scholar]
- 19. Marine JC, Francoz S, Maetens M, Wahl G, Toledo F, Lozano G. Keeping p53 in check: essential and synergistic functions of Mdm2 and Mdm4. Cell Death Differ 2006; 13:927-34; PMID:16543935; http://dx.doi.org/ 10.1038/sj.cdd.4401912 [DOI] [PubMed] [Google Scholar]
- 20. Shadfan M, Lopez-Pajares V, Yuan Z-M. MDM2 and MDMX: alone and together in regulation of p53. Transla Cancer Res 2012; 1:88-99; PMID:23002429 [PMC free article] [PubMed] [Google Scholar]
- 21. Wade M, Wang YV, Wahl GM. The p53 orchestra: Mdm2 and Mdmx set the tone. Trends Cell Biol 2010; 20:299-309; PMID:20172729; http://dx.doi.org/ 10.1016/j.tcb.2010.01.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Grier JD, Xiong S, Elizondo-Fraire AC, Parant JM, Lozano G. Tissue-Specific Differences of p53 Inhibition by Mdm2 and Mdm4. Mol Cell Biol 2006; 26:192-8; PMID:16354690; http://dx.doi.org/ 10.1128/MCB.26.1.192-198.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lengner CJ, Steinman HA, Gagnon J, Smith TW, Henderson JE, Kream BE, Stein GS, Lian JB, Jones SN. Osteoblast differentiation and skeletal development are regulated by Mdm2–p53 signaling. J Cell Biol 2006; 172:909-21; PMID:16533949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Maetens M, Doumont G, Clercq SD, Francoz S, Froment P, Bellefroid E, Klingmuller U, Lozano G, Marine J-C. Distinct roles of Mdm2 and Mdm4 in red cell production. Blood 2007; 109:2630-3; PMID:17105817; http://dx.doi.org/ 10.1182/blood-2006-03-013656 [DOI] [PubMed] [Google Scholar]
- 25. Zhang Q, He X, Chen L, Zhang C, Gao X, Yang Z, Liu G. Synergistic regulation of p53 by Mdm2 and Mdm4 is critical in cardiac endocardial cushion morphogenesis during heart development. J Pathol 2012; 228:416-28; PMID:22821713; http://dx.doi.org/ 10.1002/path.4077 [DOI] [PubMed] [Google Scholar]
- 26. Rinon A, Molchadsky A, Nathan E, Yovel G, Rotter V, Sarig R, Tzahor E. p53 coordinates cranial neural crest cell growth and epithelial-mesenchymal transition/delamination processes. Dev (Cambridge, England) 2011; 138:1827-38; PMID:21447558; http://dx.doi.org/ 10.1242/dev.053645 [DOI] [PubMed] [Google Scholar]
- 27. Hilliard SA, Yao X, El-Dahr SS. Mdm2 is required for maintenance of the nephrogenic niche. Dev Biol 2014; 387:1-14; PMID:24440154; http://dx.doi.org/ 10.1016/j.ydbio.2014.01.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gannon HS, Donehower LA, Lyle S, Jones SN. Mdm2–p53 signaling regulates epidermal stem cell senescence and premature aging phenotypes in mouse skin. Dev Biol 2011; 353:1-9; PMID:21334322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Xiong S, Van Pelt CS, Elizondo-Fraire AC, Liu G, Lozano G. Synergistic roles of Mdm2 and Mdm4 for p53 inhibition in central nervous system development. Proc Natl Acad Sci U S A 2006; 103:3226-31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Brady Colleen A, Jiang D, Mello Stephano S, Johnson Thomas M, Jarvis Lesley A, Kozak Margaret M, Broz Daniela K, Basak S, Park Eunice J, McLaughlin Margaret E, et al. Distinct p53 transcriptional programs dictate acute dna-damage responses and tumor suppression. Cell 2011; 145:571-83; PMID:21565614; http://dx.doi.org/ 10.1016/j.cell.2011.03.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Johnson TM, Hammond EM, Giaccia A, Attardi LD. The p53QS transactivation-deficient mutant shows stress-specific apoptotic activity and induces embryonic lethality. Nat Genet 2005; 37:145-52; PMID:15654339; http://dx.doi.org/ 10.1038/ng1498 [DOI] [PubMed] [Google Scholar]
- 32. Candau R, Scolnick DM, Darpino P, Ying CY, Halazonetis TD, Bergera SL. Two tandem and independent sub-activation domains in the amino terminus of p53 require the adaptor complex for activity. Oncogene 1997; 15:807-16; PMID:9266967; http://dx.doi.org/ 10.1038/sj.onc.1201244 [DOI] [PubMed] [Google Scholar]
- 33. Zhu J, Zhou W, Jiang J, Chen X. Identification of a novel p53 functional domain that is necessary for mediating apoptosis. J Biol Chem 1998; 273:13030-6; PMID:9582339; http://dx.doi.org/ 10.1074/jbc.273.21.13030 [DOI] [PubMed] [Google Scholar]
- 34. Venot C, Maratrat M, Sierra V, Conseiller E, Debussche L. Definition of a p53 transactivation function-deficient mutant and characterization of two independent p53 transactivation subdomains. Oncogene 1999; 18:2405-10; PMID:10327062; http://dx.doi.org/ 10.1038/sj.onc.1202539 [DOI] [PubMed] [Google Scholar]
- 35. Van Nostrand JL, Brady CA, Jung H, Fuentes DR, Kozak MM, Johnson TM, Lin C-Y, Lin C-J, Swiderski DL, Vogel H, et al. Inappropriate p53 activation during development induces features of CHARGE syndrome. Nature 2014; 514:228-32; PMID:25119037; http://dx.doi.org/ 10.1038/nature13585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jiang D, Brady CA, Johnson TM, Lee EY, Park EJ, Scott MP, Attardi LD. Full p53 transcriptional activation potential is dispensable for tumor suppression in diverse lineages. Proc Natl Acad Sci 2011; 108:17123-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lin J, Chen J, Elenbaas B, Levine AJ. Several hydrophobic amino acids in the p53 amino-terminal domain are required for transcriptional activation, binding to mdm-2 and the adenovirus 5 E1B 55-kD protein. Genes Dev 1994; 8:1235-46; PMID:7926727; http://dx.doi.org/ 10.1101/gad.8.10.1235 [DOI] [PubMed] [Google Scholar]
- 38. Schwenk F, Baron U, Rajewsky K. A cre-transgenic mouse strain for the ubiquitous deletion of loxP-flanked gene segments including deletion in germ cells. Nucleic Acids Res 1995; 23:5080-1; PMID:8559668; http://dx.doi.org/ 10.1093/nar/23.24.5080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Blake KD, Davenport SLH, Hall BD, Hefner MA, Pagon RA, Williams MS, Lin AE, Graham JM, Jr. CHARGE association: an update and review for the primary pediatrician. Clinical Pediatrics 1998; 37:159-73; PMID:9545604; http://dx.doi.org/ 10.1177/000992289803700302 [DOI] [PubMed] [Google Scholar]
- 40. Verloes A. Updated diagnostic criteria for CHARGE syndrome: a proposal. Am J Med Genet Part A 2005; 133A:306-8; PMID:15666308; http://dx.doi.org/ 10.1002/ajmg.a.30559 [DOI] [PubMed] [Google Scholar]
- 41. Janssen N, Bergman JEH, Swertz MA, Tranebjaerg L, Lodahl M, Schoots J, Hofstra RMW, van Ravenswaaij-Arts CMA, Hoefsloot LH. Mutation update on the CHD7 gene involved in CHARGE syndrome. Hum Mutat 2012; 33:1149-60; PMID:22461308; http://dx.doi.org/ 10.1002/humu.22086 [DOI] [PubMed] [Google Scholar]
- 42. Bergman JEH, Janssen N, Hoefsloot LH, Jongmans MCJ, Hofstra RMW, van Ravenswaaij-Arts CMA. CHD7 mutations and CHARGE syndrome: the clinical implications of an expanding phenotype. J Med Genet 2011; 48:334-42; PMID:21378379; http://dx.doi.org/ 10.1136/jmg.2010.087106 [DOI] [PubMed] [Google Scholar]
- 43. Bosman EA, Penn AC, Ambrose JC, Kettleborough R, Stemple DL, Steel KP. Multiple mutations in mouse Chd7 provide models for CHARGE syndrome. Hu Mol Genet 2005; 14:3463-76; PMID:16207732; http://dx.doi.org/ 10.1093/hmg/ddi375 [DOI] [PubMed] [Google Scholar]
- 44. Hurd E, Capers P, Blauwkamp M, Adams M, Raphael Y, Poucher H, Martin D. Loss of Chd7 function in gene-trapped reporter mice is embryonic lethal and associated with severe defects in multiple developing tissues. Mamm Genome 2007; 18:94-104; PMID:17334657; http://dx.doi.org/ 10.1007/s00335-006-0107-6 [DOI] [PubMed] [Google Scholar]
- 45. Hurd EA, Adams ME, Layman WS, Swiderski DL, Beyer LA, Halsey KE, Benson JM, Gong TW, Dolan DF, Raphael Y, et al. Mature middle and inner ears express Chd7 and exhibit distinctive pathologies in a mouse model of CHARGE syndrome. Hear Res 2011; 282:184-95; PMID:21875659; http://dx.doi.org/ 10.1016/j.heares.2011.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sanlaville D, Etchevers HC, Gonzales M, Martinovic J, Clément-Ziza M, Delezoide A-L, Aubry M-C, Pelet A, Chemouny S, Cruaud C, et al. Phenotypic spectrum of CHARGE syndrome in fetuses with CHD7 truncating mutations correlates with expression during human development. J Med Genet 2006; 43:211-317; PMID:16169932; http://dx.doi.org/ 10.1136/jmg.2005.036160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bajpai R, Chen DA, Rada-Iglesias A, Zhang J, Xiong Y, Helms J, Chang C-P, Zhao Y, Swigut T, Wysocka J. CHD7 cooperates with PBAF to control multipotent neural crest formation. Nature 2010; 463:958-62; PMID:20130577; http://dx.doi.org/ 10.1038/nature08733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Siebert JR, Graham JM, MacDonald C. Pathologic features of the CHARGE association: support for involvement of the neural crest. Teratology 1985; 31:331-6; PMID:4012643; http://dx.doi.org/ 10.1002/tera.1420310303 [DOI] [PubMed] [Google Scholar]
- 49. Sperry ED, Hurd EA, Durham MA, Reamer EN, Stein AB, Martin DM. The chromatin remodeling protein CHD7, mutated in CHARGE syndrome, is necessary for proper craniofacial and tracheal development. Dev Dyn 2014; 243:1055-66; PMID:24975120; http://dx.doi.org/ 10.1002/dvdy.24156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kang H-J, Feng Z, Sun Y, Atwal G, Murphy ME, Rebbeck TR, Rosenwaks Z, Levine AJ, Hu W. Single-nucleotide polymorphisms in the p53 pathway regulate fertility in humans. Proc Natl Acad Sci 2009; 106:9761-6; PMID:19470478; http://dx.doi.org/ 10.1073/pnas.0904280106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Golomb L, Volarevic S, Oren M. p53 and ribosome biogenesis stress: the essentials. FEBS Lett 2014; 588:2571-9; PMID:24747423; http://dx.doi.org/ 10.1016/j.febslet.2014.04.014 [DOI] [PubMed] [Google Scholar]
- 52. Zentner GE, Hurd EA, Schnetz MP, Handoko L, Wang C, Wang Z, Wei C, Tesar PJ, Hatzoglou M, Martin DM, et al. CHD7 functions in the nucleolus as a positive regulator of ribosomal RNA biogenesis. Hum Mol Genet 2010; 19:3491-501; PMID:20591827; http://dx.doi.org/ 10.1093/hmg/ddq265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Jones NC, Lynn ML, Gaudenz K, Sakai D, Aoto K, Rey J-P, Glynn EF, Ellington L, Du C, Dixon J, et al. Prevention of the neurocristopathy Treacher Collins syndrome through inhibition of p53 function. Nat Med 2008; 14:125-33; PMID:18246078; http://dx.doi.org/ 10.1038/nm1725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Layman WS, Hurd EA, Martin DM. Chromodomain proteins in development: lessons from CHARGE syndrome. Clin Genet 2010; 78:11-20; PMID:20507341; http://dx.doi.org/ 10.1111/j.1399-0004.2010.01446.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Nishiyama M, Oshikawa K, Tsukada Y-I, Nakagawa T, Iemura S-I, Natsume T, Fan Y, Kikuchi A, Skoultchi AI, Nakayama KI. CHD8 suppresses p53-mediated apoptosis through histone H1 recruitment during early embryogenesis. Nat Cell Biol 2009; 11:172-82; PMID:19151705; http://dx.doi.org/ 10.1038/ncb1831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Polo SE, Kaidi A, Baskcomb L, Galanty Y, Jackson SP. Regulation of DNA-damage responses and cell-cycle progression by the chromatin remodelling factor CHD4. EMBO J 2010; 29:3130-9; PMID:20693977; http://dx.doi.org/ 10.1038/emboj.2010.188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Bagchi A, Papazoglu C, Wu Y, Capurso D, Brodt M, Francis D, Bredel M, Vogel H, Mills AA. CHD5 Is a tumor suppressor at human 1p36. Cell 2007; 128:459-75; PMID:17289567; http://dx.doi.org/ 10.1016/j.cell.2006.11.052 [DOI] [PubMed] [Google Scholar]
- 58. Batsukh T, Pieper L, Koszucka AM, von Velsen N, Hoyer-Fender S, Elbracht M, Bergman JEH, Hoefsloot LH, Pauli S. CHD8 interacts with CHD7, a protein which is mutated in CHARGE syndrome. Hum Mol Genet 2010; 19:2858-66; PMID:20453063; http://dx.doi.org/ 10.1093/hmg/ddq189 [DOI] [PubMed] [Google Scholar]
- 59. Barlow JL, Drynan LF, Hewett DR, Holmes LR, Lorenzo-Abalde S, Lane AL, Jolin HE, Pannell R, Middleton AJ, Wong SH, et al. A p53-dependent mechanism underlies macrocytic anemia in a mouse model of human 5q- syndrome. Nat Med 2010; 16:59-66; PMID:19966810; http://dx.doi.org/ 10.1038/nm.2063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. McGowan KA, Li JZ, Park CY, Beaudry V, Tabor HK, Sabnis AJ, Zhang W, Fuchs H, de Angelis MH, Myers RM, et al. Ribosomal mutations cause p53-mediated dark skin and pleiotropic effects. Nat Genet 2008; 40:963-70; PMID:18641651; http://dx.doi.org/ 10.1038/ng.188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Boultwood J, Pellagatti A, Wainscoat JS. Haploinsufficiency of ribosomal proteins and p53 activation in anemia: diamond-blackfan anemia and the 5q- syndrome. Adv Biol Regul 2012; 52:196-203; PMID:21930148; http://dx.doi.org/ 10.1016/j.advenzreg.2011.09.008 [DOI] [PubMed] [Google Scholar]
- 62. Danilova N, Sakamoto KM, Lin S. Ribosomal protein S19 deficiency in zebrafish leads to developmental abnormalities and defective erythropoiesis through activation of p53 protein family. Blood 2008; 112(13):5228-37; PMID:18515656; http://dx.doi.org/18515656 [DOI] [PubMed] [Google Scholar]
- 63. Gaballa M, Besa E. Myelodysplastic syndromes with 5q deletion: pathophysiology and role of lenalidomide. Ann Hematol 2014; 93:723-33; PMID:24627193; http://dx.doi.org/ 10.1007/s00277-014-2022-3 [DOI] [PubMed] [Google Scholar]
- 64. Kadakia S, Helman SN, Badhey AK, Saman M, Ducic Y. Treacher collins syndrome: the genetics of a craniofacial disease. Int J Pediatr Otorhinolaryngol 2014; 78:893-8; PMID:24690222; http://dx.doi.org/ 10.1016/j.ijporl.2014.03.006 [DOI] [PubMed] [Google Scholar]
- 65. Pellagatti A, Marafioti T, Paterson JC, Barlow JL, Drynan LF, Giagounidis A, Pileri SA, Cazzola M, McKenzie ANJ, Wainscoat JS, et al. Induction of p53 and up-regulation of the p53 pathway in the human 5q− syndrome. 2010; 115(13):2721-3; http://dx.doi.org/ 10.1182/blood-2009-12-259705 [DOI] [PubMed] [Google Scholar]
- 66. Dutt S, Narla A, Lin K, Mullally A, Abayasekara N, Megerdichian C, Wilson FH, Currie T, Khanna-Gupta A, Berliner N, et al. Haploinsufficiency for ribosomal protein genes causes selective activation of p53 in human erythroid progenitor cells. Blood 2011; 117(9):2567-76; PMID:21068437; http://dx.doi.org/ 10.1182/blood-2010-07-295238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Simeonova I, Jaber S, Draskovic I, Bardot B, Fang M, Bouarich-Bourimi R, Lejour V, Charbonnier L, Soudais C, Bourdon J-C, et al. Mutant mice lacking the p53 C-terminal domain model telomere syndromes. Cell Rep 2013; 3:2046-58; PMID:23770245; http://dx.doi.org/ 10.1016/j.celrep.2013.05.028 [DOI] [PubMed] [Google Scholar]
- 68. Khincha PP, Savage SA. Genomic characterization of the inherited bone marrow failure syndromes. Semin Hematol 2013; 50:333-47; PMID:24246701; http://dx.doi.org/ 10.1053/j.seminhematol.2013.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Chin L, Artandi SE, Shen Q, Tam A, Lee S-L, Gottlieb GJ, Greider CW, DePinho RA. p53 deficiency rescues the adverse effects of telomere loss and cooperates with telomere dysfunction to accelerate carcinogenesis. Cell 1999; 97:527-38; PMID:10338216; http://dx.doi.org/ 10.1016/S0092-8674(00)80762-X [DOI] [PubMed] [Google Scholar]
- 70. Pereboom TC, van Weele LJ, Bondt A, MacInnes AW. A zebrafish model of dyskeratosis congenita reveals hematopoietic stem cell formation failure resulting from ribosomal protein-mediated p53 stabilization. Blood 2011; 118(20):5458-65; PMID:21921046; 10.1182/blood-2011-04-351460 [DOI] [PubMed] [Google Scholar]
- 71. Zhang Z, Huynh T, Baldini A. Mesodermal expression of Tbx1 is necessary and sufficient for pharyngeal arch and cardiac outflow tract development. Dev (Cambridge, England) 2006; 133:3587-95; PMID:16914493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Gao S, Li X, Amendt B. Understanding the role of Tbx1 as a candidate gene for 22q11.2 deletion syndrome. Curr Allergy Asthma Rep 2013; 13:613-21; PMID:23996541; http://dx.doi.org/ 10.1007/s11882-013-0384-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Caprio C, Baldini A. p53 suppression partially rescues the mutant phenotype in mouse models of DiGeorge syndrome. Proc Natl Acad Sci 2014; 111(37):13385-90; PMID:25197075; http://dx.doi.org/ 10.1073/pnas.1401923111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Morgan SC, Lee H-Y, Relaix F, Sandell LL, Levorse JM, Loeken MR. Cardiac outflow tract septation failure in Pax3-deficient embryos is due to p53-dependent regulation of migrating cardiac neural crest. Mech Dev 2008; 125:757-67; PMID:18672055; http://dx.doi.org/ 10.1016/j.mod.2008.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Pani L, Horal M, Loeken MR. Rescue of neural tube defects in Pax-3-deficient embryos by p53 loss of function: implications for Pax-3- dependent development and tumorigenesis. Genes Dev 2002; 16:676-80; PMID:11914272; http://dx.doi.org/ 10.1101/gad.969302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Pingault V, Ente D, Dastot-Le Moal F, Goossens M, Marlin S, Bondurand N. Review and update of mutations causing Waardenburg syndrome. Hum Mutat 2010; 31:391-406; PMID:20127975; http://dx.doi.org/ 10.1002/humu.21211 [DOI] [PubMed] [Google Scholar]
- 77. Wang XD, Morgan SC, Loeken MR. Pax3 Stimulates p53 ubiquitination and degradation independent of transcription. PLoS One 2011; 6:e29379; PMID:22216266; http://dx.doi.org/ 10.1371/journal.pone.0029379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Hamard P-J, Barthelery N, Hogstad B, Mungamuri SK, Tonnessen CA, Carvajal LA, Senturk E, Gillespie V, Aaronson SA, Merad M, et al. The C terminus of p53 regulates gene expression by multiple mechanisms in a target- and tissue-specific manner in vivo. Genes Dev 2013; 27:1868-85; PMID:24013501; http://dx.doi.org/ 10.1101/gad.224386.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Liu D, Ou L, Clemenson GD, Chao C, Lutske ME, Zambetti GP, Gage FH, Xu Y. Puma is required for p53-induced depletion of adult stem cells. Nat Cell Biol 2010; 12:993-8; PMID:20818388; http://dx.doi.org/ 10.1038/ncb2100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Terzian T, Wang Y, Van Pelt CS, Box NF, Travis EL, Lozano G. Haploinsufficiency of Mdm2 and Mdm4 in tumorigenesis and development. Mol Cell Biol 2007; 27:5479-85; PMID:17526734; http://dx.doi.org/ 10.1128/MCB.00555-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Liu G, Terzian T, Xiong S, Van Pelt CS, Audiffred A, Box NF, Lozano G. The p53-Mdm2 network in progenitor cell expansion during mouse postnatal development. J Pathol 2007; 213:360-8; PMID:17893884; http://dx.doi.org/ 10.1002/path.2238 [DOI] [PubMed] [Google Scholar]
- 82. Mendrysa SM, McElwee MK, Michalowski J, O'Leary KA, Young KM, Perry ME. mdm2 is critical for inhibition of p53 during lymphopoiesis and the response to ionizing irradiation. Mol Cell Biol 2003; 23:462-72; PMID:12509446; http://dx.doi.org/ 10.1128/MCB.23.2.462-473.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Mendrysa SM, O'Leary KA, McElwee MK, Michalowski J, Eisenman RN, Powell DA, Perry ME. Tumor suppression and normal aging in mice with constitutively high p53 activity. Genes Dev 2006; 20:16-21; PMID:16391230; http://dx.doi.org/ 10.1101/gad.1378506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Tyner SD, Venkatachalam S, Choi J, Jones S, Ghebranious N, Igelmann H, Lu X, Soron G, Cooper B, Brayton C, et al. p53 mutant mice that display early ageing-associated phenotypes. Nature 2002; 415:45-53; PMID:11780111; http://dx.doi.org/ 10.1038/415045a [DOI] [PubMed] [Google Scholar]
- 85. Maier B, Gluba W, Bernier B, Turner T, Mohammad K, Guise T, Sutherland A, Thorner M, Scrable H. Modulation of mammalian life span by the short isoform of p53. Genes Dev 2004; 18:306-19; PMID:14871929; http://dx.doi.org/ 10.1101/gad.1162404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Garcia-Cao I, Garcia-Cao M, Martin-Caballero J, Criado LM, Klatt P, Flores JM, Weill J-C, Blasco MA, Serrano M. 'Super p53' mice exhibit enhanced DNA damage response, are tumor resistant and age normally. EMBO J 2002; 21:6225-35; PMID:12426394; http://dx.doi.org/ 10.1093/emboj/cdf595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Venkatachalam S, Shi Y-P, Jones SN, Vogel H, Bradley A, Pinkel D, Donehower LA. Retention of wild-type p53 in tumors from p53 heterozygous mice: reduction of p53 dosage can promote cancer formation. EMBO J 1998; 17:4657-67; PMID:9707425; http://dx.doi.org/ 10.1093/emboj/17.16.4657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Sah VP, Attardi LD, Mulligan GJ, Williams BO, Bronson RT, Jacks T. A subset of p53-deficient embryos exhibit exencephaly. Nat Genet 1995; 10:175-80; PMID:7663512; http://dx.doi.org/ 10.1038/ng0695-175 [DOI] [PubMed] [Google Scholar]
- 89. Armstrong JF, Kaufman MH, Harrison DJ, Clarke AR. High-frequency developmental abnormalities in p53-deficient mice. Curr Biol 1995; 5:931-6; PMID:7583151; http://dx.doi.org/ 10.1016/S0960-9822(95)00183-7 [DOI] [PubMed] [Google Scholar]