

Abstract
Cellular senescence happens in 2 steps: cell cycle arrest followed, or sometimes preceded, by gerogenic conversion (geroconversion). Geroconvesrion is a form of growth, a futile growth during cell cycle arrest. It converts reversible arrest to irreversible senescence. Geroconversion is driven by growth-promoting, mitogen-/nutrient-sensing pathways such as mTOR. Geroconversion leads to hyper-secretory, hypertrophic and pro-inflammatory cellular phenotypes, hyperfunctions and malfunctions. On organismal level, geroconversion leads to age-related diseases and death. Rapamycin, a gerosuppressant, extends life span in diverse species from yeast to mammals. Stress–and oncogene-induced accelerated senescence, replicative senescence in vitro and life-long cellular aging in vivo all can be described by 2-step model.
Keywords: aging, cell cycle arrest, gerogenic conversion, mTOR, oncogenic trsnformation
Introduction
Defined as irreversible cell cycle arrest, cellular senescence is difficult to link to age-related diseases, which terminate our life span. If anything, cell cycle arrest per se should protect against atherosclerosis, hypertension, organ fibrosis, visceral adiposity, benign tumors and cancer. And why calorie restriction and rapamycin, which inhibit proliferation, extend life span. Something is missing. Indeed, aging is not just cell cycle arrest.1-18 In analogy, although cell cycle progression is important in carcinogenesis, we do not define cancer as cell cycle progression. For one, intestinal and bone marrow progenitor cells proliferate faster than tumor cells. And the cancer cell cycle can be easily arrested by p21, which is not even a tumor suppressor. A cell can be proliferating but not cancerous. Similarly, a cell can be arrested but not senescent. Even permanently-arrested cells (such as neurons) are not necessarily senescent. Here we will define the essence of senescence, as it had been done for cancer.
The essence of cancer is oncogenic transformation, driven by oncogenes and antagonized by tumor suppressors.19-31 Similarly, the essence of senescence is gerogenic conversion driven by gerogenes and antagonized by gerosuppressors. (There is an overlap between oncogenes and gerogenes18,32). But let us start from the beginning.
Two-types of Cell Cycle Arrest
Growth factors, hormones, cytokines and nutrients activate Ras/Raf/MEK/ERK and PI3K/Akt/mTOR signaling pathways.33-36 In cancer cells, these pathways are constitutively activated. These pathways (MAPK/mTOR, for brevity) stimulate cellular mass growth coupled with cell cycle progression (Fig. 1A).
Quiescence: Without GF, the MAPK/mTOR network is deactivated (Fig. 1B). Cell cycle comes to a halt. The quiescent cell neither grows nor cycles. Yet the cell retains the proliferative potential: re-addition of GF causes activation of MAPK/mTOR, cell mass growth, cell cycle progression, mitosis and cell proliferation. In quiescent cells, mTOR is deactivated, levels of pS6, cyclin D1, p21 and p16 are all low.37 “Everything is off." Contact inhibition also causes quiescence-like arrest. Contact inhibition in confluent culture inhibits mTOR and MAPK pathways.38,39 Cells neither grow in size, nor cycle and can restart proliferation after splitting.
Hyper-mitogenic type of arrest (Fig. 1C). Cell cycle can be arrested by CDK inhibitors such as p21 and p16. In this case, the cell cycle is blocked but mTOR and MAPK are still active (Fig. 1C). In futile attempt to overcome the block, growth-promoting pathways push a cell to become hypertrophic, hyper-active, hyper-functional and secondary signal-resistant and β-Gal-positive.40 Cyclin D1 goes above the roof. It is like pushing the brakes and the gas simultaneously. (In contrast, in quiescence the motor is off: normal parking).40 Activation of mTOR, when the cell cycle is blocked, leads to senescent morphology, including loss of the ability to re-start proliferation.41 This mTOR-driven process is gerogenic conversion.18
Figure 1.
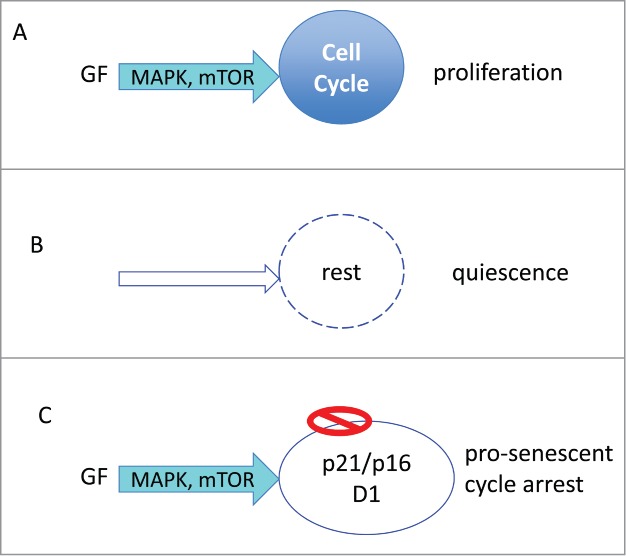
Proliferation versus arrest. (A) Proliferation: Growth factors (GF) activate MAPK and mTOR pathways, driving cell growth (in size) and cell cycle. Cellular growth is balanced by cell division. (B) Quiescence: In the absence of GF, cell growth and cycle are at rest. (C) Pro-senescent cycle arrest: When the cycle cycle is blocked by p21 or p16, the growth promoting pathway (MAPK/mTOR) causes growth in size (hypertrophy).
Depending on whether a cell is proliferating or arrested, mTOR drives either growth or geroconversion. Geroconversion is a form of growth, when actual growth is restricted.42 It leads to cellular hyperfunctions, hypertrophy and compensatory signal-resistance and lysosomal hyperfunction (β-Gal-positivity). Rapamycin slows down geroconversion.43
p21- and p16 -Induced Senescence
To test 2-step hypothesis experimentally, we first employed the simplest model of senescence: p21- or p16-induced senescence. In HT-p21 and HT-p16 cells, expressing IPTG-inducible p21 and p16, respectively, cell cycle and mTOR can be manipulated separately.41,43,44. Arrest can be caused by p21 or p16. During 2 days, cells are arrested but still can resume proliferation, when p21 (or p16) is switched off. After 3–4 days, p21/p16-arrested cells lose proliferative potential. If (after 3–4 days) p21 is switched off, cells cannot divide.45 The proliferative/mitotic potential is lost. What drives this conversion from initial arrest to irreversible state?
In proliferating HT-p21/p16 cells, mTOR is activated. When p21 (or p16) is induced, the cell cycle is arrested but mTOR is still active (Fig. 2A). It drives cellular mass growth (growth in size) leading to a large and flattened morphology. This hypertrophy is counterbalanced by increased lysosomal activity (lysosomal hyperfunction), manifested by β-Gal-staining. Loss of mitotic competence is one of manifestations of senescent phenotype.45
Figure 2.
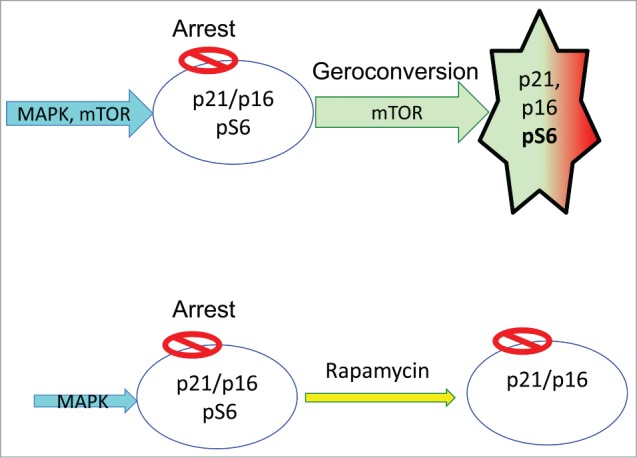
Senescence vs. quiescence.
Rapamycin suppresses geroconversion (Fig. 2B), so the arrested cells retain the proliferative potential. They can re-start proliferation, if cell-cycle arrest is abrogated. Importantly, rapamycin does not abrogate p21-or p16-induced arrest. Cells are still arrested but when p21 or p16 is switched off, they can restart proliferation.43-45
Rapamycin by itself slows down cell cycle. It slows growth (in proliferating cells) and geroconversion (in arrested cells). Literally, rapamycin slows down time. Geroconversion takes 3 times longer.43
Not only rapamycin but also any condition that deactivates mTOR in turn suppresses geroconversion.38,46-48 In particular, hypoxia, starvation and contact inhibition all suppress geroconversion.38,47-49 As we will discuss, this can explain how quiescent cells remain quiescent in the organism for so long.
Gerogenes and Gerosuppressors
Conditions that activate mTOR accelerate geroconversion. Therefore, we can predict genes that are gerogenes and gerosuppressors. Gerogenes activate the growth-promoting mTOR pathway.32,50,51 They include growth factor receptors (IGF-1, insulin, EGF, ErbB), Ras, Raf, Mek, PI-3K, Akt and other growth-promoting oncogenes. Gerosuppressors antagonize mTOR pathway and include PTEN, TSC1/2, AMPK, and other tumor-suppressors. This was discussed in detail.32,50-52 Gerogenes are oncogenes that promote cellular mass growth in cancer and senescence. One of surprising predictions is that p53, a tumor-suppressor that inhibits mTOR, will suppress geroconversion.50
p53-Induced Senescence
Like many tumor-suppressors such as PTEN, p53 can suppress cancer in part by inhibiting mTOR.50,53,54 p53 induces senescence because p53 is an extremely potent inducer of cell cycle arrest.55,56 When the cell cycle is arrested by p53, mTOR drives geroconversion,47,57,58 (Fig. 3, green arrow). But in some cell types, at high levels p53 also inhibits mTOR and cellular mass growth (Fig. 3, yellow arrow). When p53 is capable to inhibit mTOR, it induces reversible arrest (quiescence), instead of senescence.47,57-60 For example, when cell cycle is arrested by ectopic p21, then induction of p53 can inhibit geroconversion in p21-arrested cells.47 In some types, while p21 and p16 caused senescence, their combination with p53 caused quiescence.47 In MEFs, p53 does not inhibit mTOR,57 so it does not prevent geroconversion during replicative senescence. As a powerful inducer of arrest, p53 is involved in stress-induced, oncogene-induced and replicative senescence.6,9,55,56,61-63 p53 “induces” senescence by causing cell cycle arrest, while failing to inhibit mTOR.57,58 Noteworthy, rapamycin may partially substitute for p53 in mice.64-67
Figure 3.
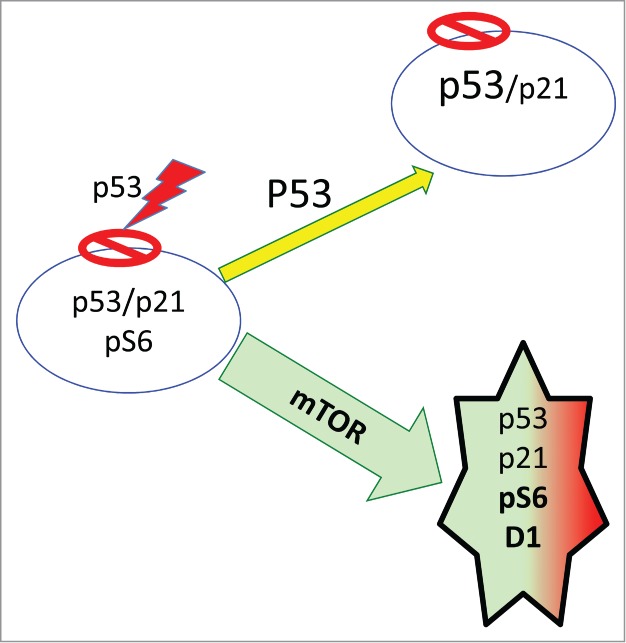
The dual role of p53 in senescence. P53 causes Arrest that is followed by geroconversion (green arrow). Yet, at very high levels, p53 can inhibit mTOR, suppressing geroconversion (yellow arrow) and leading to quiescence.
Stress-induced Senescence
In most cases, stress-induced senescence is p53-dependent (Fig. 3). Stresses such as acute DNA damage induce p53-dependent arrest, while mTOR drives geroconversion from arrest to senescence (Fig. 4A). Rapamycin and serum withdrawal decelerate geroconversion, (Fig. 4B) during stress-induced senescence.68
Figure 4.
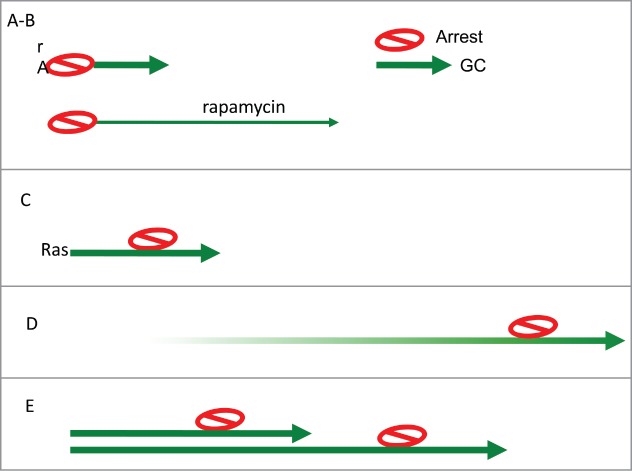
Arrest-Geroconversion model. Schematic representation of types of senescence (Arrest- red stop sign. Geroconversion – green arrow). (A) Typical arrest-induced senescence. DNA damage-induced senescence. CDK (p21 and 16)–induced senescence. (B) In the presence of rapamycin: geroconversion is slowed down and extended. (C) Oncogene-induced senescence. Oncogenes such as Ras empower growth, cause arrest and then empower geroconversion. (D) Replicative senescence of human cells in culture. Telomere shortening during cell proliferation eventually causes Arrest. Then geroconversion ensures senescence. (E) Replicative senescence of rodent cells in culture. Ovestumulation of mTOR by mitogen/nutrient/oxygen rich medium causes cellular hypertrophy.
Oncogene-induced Senescence
Oncogene-induced senescence is caused by oncogenes (Ras, Raf, MEK, AKT) that activate MAPK/mTOR pathways and also induce p53, p21 or p1669-74 These oncogenes are gerogenes, which cause 2 processes: cell cycle arrest and geroconversion. For example, Ras promotes growth, leading to hypertrophy (pro-gerogenic conversion,Fig. 4C). This pro-gerogenic conversion induces p53-dependent arrest.69,70,71,75 During cell cycle arrest, Ras-activated mTOR pathway completes geroconversion to senescence (Fig. 4C). Rapamycin suppresses Ras-induced growth and geroconverion.76 In contrast, loss of p53 prevents cell cycle arrest, without affecting geroconversion. Geroconversion without cell cycle arrest is cancer.
Replicative Senescence in Human Cells
In replicative senescence, telomere shortening culminates in DNA damage response in human cells. Noteworthy, there is a correlation with longevity in vivo77-79 Replicative senescence of human cells is a variant of stress-induced senescence (Fig. 4D). The main peculiarity is that stress (telomere crisis) occurs after many rounds of cell divisions. After a definite number of divisions (Hayflick limit), the process can be described by 2-step model: arrest followed by geroconvesion (Fig. 4D). Rapamycin suppress replicative senescence in human fibroblasts.76
Replicative Senescence in Rodent Cells
This type of senescence reminds protracted “oncogene-induced” senescence (Fig. 4E), because it is telomere-independent.80 Primary rodent cells are overstimulated by serum, high glucose (DMEM contains 5 fold excess of glucose) and other nutrients and non-physiological oxygen levels. Under such overstimulation of mTOR, cells gradually undergo geroconversion. Pro-geroconversion triggers p53-dependent arrest. Continous geroconversion makes the process irreversible. Rapamycin prevents replicative senescence.81,82 Similarly, low mitogenic-conditions delay replicatiove senescence.83 In hypoxia, MEFs do not undergo senescence84 Not co-incidentally, hypoxia inhibits mTOR.
Yeast Replicative Senescence
Yeast and rodent replicative aging are comparable (Fig. 5E). Yeast mother cell becomes progressively hypertrophic before ceasing proliferation. Geroconversion triggers the arrest. “CR” and genetic TOR inhibition extend replicative lifespan.85
Chronologic yeast aging is a different phenomenon.86,87 It has no analogy to traditional senescence in mammals. Instead, it is identical to metabolic self-destruction of cancer cells88,89 Both yeast and mammalian “chronological aging” is inhibited by rapamycin.90 The reason is that the same pathways that drive geroconversion and organismal aging also increase glycolisis production.88,91
Emerging Summary
In cell culture, a senescent program includes 2 events: cell cycle arrest and geroconversion. Cell cycle arrest is ultimately caused by CDK inhibitors such as p21 and p16. Geroconversion is driven by growth-signaling pathways such as mTOR.
When the cell cycle is blocked, growth-promoting pathways drive geroconversion. Geroconversion can be viewed as a continuation of growth, a quasi-program of growth. Like cellular growth, gerconversion is slowed down by rapamycin. Geroconversion leads to hypertrophy, hyper-differentiation and hyperfunctions such as SASP.
The action of growth-promoting pathways causes the opposite reaction. For example, a cell cannot grow in size infinitively. Re-activation of lysosomes and autophagy counteracts protein synthesis and growth. Signal resistance counteracts cellular overstimulation. Hyperfunctions may eventually lead to malfunctions, hypertrophy to atrophy.
From Cell Culture to the Organism
Arrest and geroconversion are obligatory steps of cellular senescence, including stress-induced, oncogene-induced and replicative senescence (Fig. 4). In cell culture, geroconversion is a rapid step because in cell culture mTOR is overactivated, especially in cancer cells. When arrested for several days, cells become senescent due to rapid geroconversion. If mTOR is not inhibited by contact inhibition or by starvation, then geroconversion is automatic in arrested cells. Therefore, arrest is a key event in most models of senescence in vitro (if arrest occurs, then geroconversion follows). Not surprisingly, it is arrest that attracted all attention, so that senescence was defined as “permanent arrest."
In long-lived organisms, it is geroconversion that is the limiting event. A cell can be quiescent for years without becoming senescent. In the organism, the mTOR activity is low and a cell can be arrested (quiescent and contact inhibited) without undergoing geroconversion. Geroconversion can either follow or precede arrest (or both). For example, geroconversion of stem cells may initially increase their proliferation followed by delayed arrest (stem cell exhaustion).15,92-97 Similar geroconversion exhausts oocytes,98,99 leading to menopause.100
Permanent cell cycle arrest is not a crucial marker of senescence in the organism. Senescent cell can re-enter cell cycle: in some situations with devastating consequences for the organism. (One example is death of neurons upon S-phase re-entry in Alzheimer's disease. Another example is cancer.)
Cell cycle arrest per se cannot be linked to age-related diseases. For one, organismal aging is associated with tumors, atherosclerosis, fibrosis and other hyperplastic and hypertrophic (obesity) conditions, rather than with cessation of cellular proliferation. It is geroconversion that leads to diseases.101 Overstimulation of mTOR in post-development makes cells hyperfunctional and signal resistant (e.g., insulin resistant). Examples of hyperfunctions include oxidative burst by neutrophils or contraction by SMC. Macrophages are converted to foam cells, smooth muscle cells (SMC) become hypertrophic and calcificated, hepatocytes produce lipoproteins and pro-inflammatory factors, adipocytes are hyperactive. mTOR dependent geroconversion renders cells “ pathogenic, thus leading to atherosclerosis, for instance. Atherosclerosis, hypertension, hyper-aggregation of blood cells and hyper-coagulation can culminate in stroke and infarction (for instance) and organismal death.
Geroconversion creates pathogenic or gerogenic cells, leading to diseases and organismal aging. Gerogenic and pathogenic abilities are tightly linked. In fact, organismal aging is an increase of the probability of death. It is the sum of age-related diseases and disease-like conditions102-105 Inhibition of mTOR by calorie restriction or rapamycin decelerates geroconversion, and increases health–and life span. In other words deceleration of cellular geroconversion slows down organismal aging. The link between geroconversion and organismal aging (via diseases) I will discuss in forthcoming paper “Geroconversion: irresistible path to organismal aging”
From Gerontology to Medicine
Geroconversion renders cells gerogenic (the ability to cause organismal aging) and pathogenic (the ability to cause diseases) in the organism. In rare cases, (stress-induced and oncogene-induced senescence), geroconversion renders cells “typically senescent," resembling senescence in vitro.5,106-111 A striking example is oncogene-induced senescent nevi.112 Gerontologists are focused on these “typically” senescent cells.113Yet, these senescent cells are only a tip of the iceberg. Characterized by increased cellular functions (hyperfunctions), gerogenic cells are not necessarily resemble “in vitro senescent fibroblasts."
By feedback loops, cells from different organs/tissues over-stimulate each other.100,114 Paracrine geroconversion also takes place.115,116 Gradually geroconversion involves most cells in diverse tissues. The extend of geroconversion can range from slightly gerogenic to typically senescent cells. In general, heterogeneity is one of hallmarks of aging.117 We know the contribution of all gerogenic cells together. Gerogenic cells are pathogenic by causing hyperlipedemia, pro-inflammation, hypertension, cardiac and so on. Foam cells (in atherosclerotic plaques), tumor cells, hypertrophic adipocytes and calcificated SMC are pathogenic. In medicine, the effect of all gerogenic cells together is determined by alterations in blood biochemistry, cardiac and renal functions and other laboratory and functional tests. Yet, medical science misses geroconversion as the universal process, which initiates age-related pathology and alters laboratory tests. Medicine concerns with thousands genetic and external factors of human diseases such as genetic predisposition, smoking and wrong diet. Although smoking and fat food can accelerate pathology, this pathology is developed anyway without any hazards (such as smoking or fat food) because. There is an universal process – geroconversion. And geroconversion is a continuation of developmental growth.
Senescence in cell culture does not model all the complexity of organismal aging. Still as we discussed here, these in vitro models reveal the process of mTOR-dependent geroconversion. Geroconversion is applicable to the organism, explaining why we develop age-related pathology (and cosmetic problems) and eventually die.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1. Mathon NF, Lloyd AC. Cell senescence and cancer. Nature Rev Cancer 2001; 1:203-213; http://dx.doi.org/ 10.1038/35106045 [DOI] [PubMed] [Google Scholar]
- 2. Dulic V, Beney GE, Frebourg G, Drullinger LF, Stein GH. Uncoupling between phenotypic senescence and cell cycle arrest in aging p21-deficient fibroblasts. Mol Cell Biol 2000; 20:6741-6754; PMID:10958672; http://dx.doi.org/ 10.1128/MCB.20.18.6741-6754.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wang Y, Meng A, Zhou D. Inhibition of phosphatidylinostol 3-kinase uncouples H2O2-induced senescent phenotype and cell cycle arrest in normal human diploid fibroblasts. Exp Cell Res 2004; 298:188-196; PMID:15242773; http://dx.doi.org/ 10.1016/j.yexcr.2004.04.012 [DOI] [PubMed] [Google Scholar]
- 4. Campisi J. Suppressing cancer: the importance of being senescent. Science 2005; 309:886-887; PMID:16081723; http://dx.doi.org/ 10.1126/science.1116801 [DOI] [PubMed] [Google Scholar]
- 5. Kang TW, Yevsa T, Woller N, Hoenicke L, Wuestefeld T, Dauch D, Hohmeyer A, Gereke M, Rudalska R, Potapova A, et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature 2011; 479:547-551; PMID:22080947; http://dx.doi.org/ 10.1038/nature10599 [DOI] [PubMed] [Google Scholar]
- 6. Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell 2013; 153:1194-1217; PMID:23746838; http://dx.doi.org/ 10.1016/j.cell.2013.05.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mao Z, Ke Z, Gorbunova V, Seluanov A. Replicatively senescent cells are arrested in G1 and G2 phases. Aging (Albany NY) 2012; 4:431-435; PMID:22745179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Serrano M. Dissecting the role of mTOR complexes in cellular senescence. Cell Cycle 2012; 11:2231-2232; PMID:22714590; http://dx.doi.org/ 10.4161/cc.21065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Salama R, Sadaie M, Hoare M, Narita M. Cellular senescence and its effector programs. Genes Dev 2014; 28:99-114; PMID:24449267; http://dx.doi.org/ 10.1101/gad.235184.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tchkonia T, Zhu Y, van Deursen J, Campisi J, Kirkland JL. Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. J Clin Invest 2013; 123:966-972; PMID:23454759; http://dx.doi.org/ 10.1172/JCI64098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hu Z, Klein JD, Mitch WE, Zhang L, Martinez I, Wang XH. MicroRNA-29 induces cellular senescence in aging muscle through multiple signaling pathways. Aging (Albany NY) 2014; 6:160-175; PMID:24659628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. McCubrey JA, Demidenko ZN. Recent discoveries in the cycling, growing and aging of the p53 field. Aging (Albany NY) 2012; 4:887-893; PMID:23425920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. van Deursen JM. The role of senescent cells in ageing. Nature 2014; 509:439-446; PMID:24848057; http://dx.doi.org/ 10.1038/nature13193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Munoz-Espin D, Serrano M. Cellular senescence: from physiology to pathology. Nat Rev Mol Cell Biol 2014; 15:482-496; PMID:24954210; http://dx.doi.org/ 10.1038/nrm3823 [DOI] [PubMed] [Google Scholar]
- 15. Sousa-Victor P, Gutarra S, Garcia-Prat L, Rodriguez-Ubreva J, Ortet L, Ruiz-Bonilla V, Jardí M, Ballestar E, González S, Serrano AL, et al. Geriatric muscle stem cells switch reversible quiescence into senescence. Nature 2014; 506:316-321; PMID:24522534; http://dx.doi.org/ 10.1038/nature13013 [DOI] [PubMed] [Google Scholar]
- 16. Vaughan S, Jat PS. Deciphering the role of nuclear factor-kappaB in cellular senescence. Aging (Albany NY) 2011; 3:913-919; PMID:21990145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pichierri P, Ammazzalorso F, Bignami M, Franchitto A. The Werner syndrome protein: linking the replication checkpoint response to genome stability. Aging (Albany NY) 2011; 3:311-318; PMID:21389352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Blagosklonny MV. Cell cycle arrest is not yet senescence, which is not just cell cycle arrest: terminology for TOR-driven aging. Aging (Albany NY) 2012; 4:159-165; PMID:22394614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Vogelstein B, Kinzler KW. The multistep nature of cancer. Trends Genet. 1993; 9:138-141; PMID:8516849; http://dx.doi.org/ 10.1016/0168-9525(93)90209-Z [DOI] [PubMed] [Google Scholar]
- 20. Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nat Med 2004; 10:789-799; PMID:15286780; http://dx.doi.org/ 10.1038/nm1087 [DOI] [PubMed] [Google Scholar]
- 21. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell 2000; 100:57-70; PMID:10647931; http://dx.doi.org/ 10.1016/S0092-8674(00)81683-9 [DOI] [PubMed] [Google Scholar]
- 22. Hahn WC, Counter CM, Lundberg AS, Beijersbergen RL, Brooks MW, Weinberg RA. Creation of human tumour cells with defined genetic elements. Nature 1999; 400:464-468; PMID:10440377; http://dx.doi.org/ 10.1038/22780 [DOI] [PubMed] [Google Scholar]
- 23. Kriegl L, Vieth M, Kirchner T, Menssen A. Up-regulation of c-MYC and SIRT1 expression correlates with malignant transformation in the serrated route to colorectal cancer. Oncotarget 2012; 3:1182-1193; PMID:23045412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mao XG, Hutt-Cabezas M, Orr BA, Weingart M, Taylor I, Rajan AK, Odia Y, Kahlert U, Maciaczyk J, Nikkhah G, et al. LIN28A facilitates the transformation of human neural stem cells and promotes glioblastoma tumorigenesis through a pro-invasive genetic program. Oncotarget 2013; 4:1050-1064; PMID:23846349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yi KH, Axtmayer J, Gustin JP, Rajpurohit A, Lauring J. Functional analysis of non-hotspot AKT1 mutants found in human breast cancers identifies novel driver mutations: implications for personalized medicine. Oncotarget 2013; 4:29-34; PMID:23237847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hart JR, Vogt PK. Phosphorylation of AKT: a mutational analysis. Oncotarget 2011; 2:467-476; PMID:21670491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jiao Y, Killela PJ, Reitman ZJ, Rasheed AB, Heaphy CM, de Wilde RF, Rodriguez FJ, Rosemberg S, Oba-Shinjo SM, Nagahashi Marie SK, et al. Frequent ATRX, CIC, FUBP1 and IDH1 mutations refine the classification of malignant gliomas. Oncotarget 2012; 3:709-722; PMID:22869205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pekarsky Y, Croce CM. Is miR-29 an oncogene or tumor suppressor in CLL? Oncotarget 2010; 1:224-227; PMID:20936047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Luo W, Semenza GL. Pyruvate kinase M2 regulates glucose metabolism by functioning as a coactivator for hypoxia-inducible factor 1 in cancer cells. Oncotarget 2011; 2:551-556; PMID:21709315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ellis L, Ku SY, Ramakrishnan S, Lasorsa E, Azabdaftari G, Godoy A, Pili R. Combinatorial antitumor effect of HDAC and the PI3K-Akt-mTOR pathway inhibition in a Pten defecient model of prostate cancer. Oncotarget 2013; 4:2225-2236; PMID:24163230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cipriano R, Miskimen KL, Bryson BL, Foy CR, Bartel CA, Jackson MW. FAM83B-mediated activation of PI3K/AKT and MAPK signaling cooperates to promote epithelial cell transformation and resistance to targeted therapies. Oncotarget 2013; 4:729-738; PMID:23676467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Blagosklonny MV. Molecular damage in cancer: an argument for mTOR-driven aging. Aging (Albany NY) 2011; 3:1130-1141; PMID:22246147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Howell JJ, Manning BD. mTOR couples cellular nutrient sensing to organismal metabolic homeostasis. Trends Endocrinol Metab 2011; 22:94-102; PMID:21269838; http://dx.doi.org/ 10.1016/j.tem.2010.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Avruch J, Hara K, Lin Y, Liu M, Long X, Ortiz-Vega S, Yonezawa K. Insulin and amino-acid regulation of mTOR signaling and kinase activity through the Rheb GTPase. Oncogene 2006; 25:6361-6372; PMID:17041622; http://dx.doi.org/ 10.1038/sj.onc.1209882 [DOI] [PubMed] [Google Scholar]
- 35. Dazert E, Hall MN. mTOR signaling in disease. Curr Opin Cell Biol 2011; 23:744-55; PMID:21963299 [DOI] [PubMed] [Google Scholar]
- 36. Cornu M, Albert V, Hall MN. mTOR in aging, metabolism, and cancer. Curr Opin Genet Dev 2012; 23:53-6. [DOI] [PubMed] [Google Scholar]
- 37. Leontieva OV, Blagosklonny MV. M(o)TOR of pseudo-hypoxic state in aging: Rapamycin to the rescue. Cell Cycle 2014; 13:PMID:24496328 [DOI] [PubMed] [Google Scholar]
- 38. Leontieva OV, Demidenko ZN, Blagosklonny MV. Contact inhibition and high cell density deactivate the mammalian target of rapamycin pathway, thus suppressing the senescence program. Proc Natl Acad Sci U S A 2014; 111:8832-8837; PMID:24889617; http://dx.doi.org/ 10.1073/pnas.1405723111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sun P. Contact inhibition against senescence. Oncotarget 2014;5:7212-7213; PMID:25277173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Blagosklonny MV. Cell senescence and hypermitogenic arrest. EMBO Rep 2003; 4:358-362; PMID:12671679; http://dx.doi.org/ 10.1038/sj.embor.embor806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Demidenko ZN, Blagosklonny MV. Growth stimulation leads to cellular senescence when the cell cycle is blocked. Cell Cycle 2008; 7:3355-3361; PMID:18948731; http://dx.doi.org/ 10.4161/cc.7.21.6919 [DOI] [PubMed] [Google Scholar]
- 42. Blagosklonny MV, Hall MN. Growth and aging: a common molecular mechanism. Aging (Albany NY) 2009; 1:357-362; PMID:20157523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Demidenko ZN, Zubova SG, Bukreeva EI, Pospelov VA, Pospelova TV, Blagosklonny MV. Rapamycin decelerates cellular senescence. Cell Cycle 2009; 8:1888-1895; PMID:19471117; http://dx.doi.org/ 10.4161/cc.8.12.8606 [DOI] [PubMed] [Google Scholar]
- 44. Demidenko ZN, Blagosklonny MV. Quantifying pharmacologic suppression of cellular senescence: prevention of cellular hypertrophy versus preservation of proliferative potential. Aging (Albany NY) 2009; 1:1008-1016; PMID:20157583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Leontieva OV, Lenzo F, Demidenko ZN, Blagosklonny MV. Hyper-mitogenic drive coexists with mitotic incompetence in senescent cells. Cell Cycle 2012; 11:4642-4649; PMID:23187803; http://dx.doi.org/ 10.4161/cc.22937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Demidenko ZN, Shtutman M, Blagosklonny MV. Pharmacologic inhibition of MEK and PI-3K converges on the mTOR/S6 pathway to decelerate cellular senescence. Cell Cycle 2009; 8:1896-1900; PMID:19478560; http://dx.doi.org/ 10.4161/cc.8.12.8809 [DOI] [PubMed] [Google Scholar]
- 47. Demidenko ZN, Korotchkina LG, Gudkov AV, Blagosklonny MV. Paradoxical suppression of cellular senescence by p53. Proc Natl Acad Sci U S A 2010; 107:9660-9664; PMID:20457898; http://dx.doi.org/ 10.1073/pnas.1002298107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Leontieva OV, Natarajan V, Demidenko ZN, Burdelya LG, Gudkov AV, Blagosklonny MV. Hypoxia suppresses conversion from proliferative arrest to cellular senescence. Proc Natl Acad Sci U S A 2012; 109:13314-13318; PMID:22847439; http://dx.doi.org/ 10.1073/pnas.1205690109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhao H, Halicka HD, Li J, Darzynkiewicz Z. Berberine suppresses gero-conversion from cell cycle arrest to senescence. Aging (Albany NY) 5:623-636; PMID:23974852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Blagosklonny MV. Tumor suppression by p53 without apoptosis and senescence: conundrum or rapalog-like gerosuppression? Aging (Albany NY) 2012; 4:450-455; PMID:22869016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Blagosklonny MV. An anti-aging drug today: from senescence-promoting genes to anti-aging pill. Drug Disc Today 2007; 12:218-224; http://dx.doi.org/ 10.1016/j.drudis.2007.01.004 [DOI] [PubMed] [Google Scholar]
- 52. Blagosklonny MV. Aging: ROS or TOR. Cell Cycle 2008; 7:3344-3354; PMID:18971624; http://dx.doi.org/ 10.4161/cc.7.21.6965 [DOI] [PubMed] [Google Scholar]
- 53. Levine AJ, Feng Z, Mak TW, You H, Jin S. Coordination and communication between the p53 and IGF-1-AKT-TOR signal transduction pathways. Genes Dev 2006; 20:267-275; PMID:16452501; http://dx.doi.org/ 10.1101/gad.1363206 [DOI] [PubMed] [Google Scholar]
- 54. Feng Z, Levine AJ. The regulation of energy metabolism and the IGF-1/mTOR pathways by the p53 protein. Trends Cell Biol 2010; 20:427-434; PMID:20399660; http://dx.doi.org/ 10.1016/j.tcb.2010.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Itahana K, Dimri G, Campisi J. Regulation of cellular senescence by p53. Eur. J. Biochem 2001; 268:2784-2791; PMID:11358493; http://dx.doi.org/ 10.1046/j.1432-1327.2001.02228.x [DOI] [PubMed] [Google Scholar]
- 56. Beausejour CM, Krtolica A, Galimi F, Narita M, Lowe SW, Yaswen P, Campisi J. Reversal of human cellular senescence: roles of the p53 and p16 pathways. EMBO J 2003; 22:4212-4222; PMID:12912919; http://dx.doi.org/ 10.1093/emboj/cdg417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Korotchkina LG, Leontieva OV, Bukreeva EI, Demidenko ZN, Gudkov AV, Blagosklonny MV. The choice between p53-induced senescence and quiescence is determined in part by the mTOR pathway. Aging (Albany NY) 2010; 2:344-352; PMID:20606252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Leontieva O, Gudkov A, Blagosklonny M. Weak p53 permits senescence during cell cycle arrest. Cell Cycle 2010; 9:4323-4327; PMID:21051933; http://dx.doi.org/ 10.4161/cc.9.21.13584 [DOI] [PubMed] [Google Scholar]
- 59. Lane DP, Verma C, Fang CC. The p53 inducing drug dosage may determine quiescence or senescence. Aging (Albany NY) 2010; 2:748; PMID:21068468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Dulic V. Senescence regulation by mTOR. Methods Mol Biol 2013; 965:15-35; PMID:23296649; http://dx.doi.org/ 10.1007/978-1-62703-239-1_2 [DOI] [PubMed] [Google Scholar]
- 61. Itahana K, Campisi J, Dimri GP. Mechanisms of cellular senescence in human and mouse cells. Biogerontology 2004; 5:1-10; PMID:15138376; http://dx.doi.org/ 10.1023/B:BGEN.0000017682.96395.10 [DOI] [PubMed] [Google Scholar]
- 62. Dirac AM, Bernards R. Reversal of senescence in mouse fibroblasts through lentiviral suppression of p53. J Biol Chem 2003; 278:11731-11734; PMID:12551891; http://dx.doi.org/ 10.1074/jbc.C300023200 [DOI] [PubMed] [Google Scholar]
- 63. Napoli M, Girardini JE, Piazza S, Del Sal G. Wiring the oncogenic circuitry: Pin1 unleashes mutant p53. Oncotarget 2011; 2:654-656; PMID:21926448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Donehower LA. Rapamycin as longevity enhancer and cancer preventative agent in the context of p53 deficiency. Aging (Albany NY) 2012; 4:660-661; PMID:23128359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Comas M, Toshkov I, Kuropatwinski KK, Chernova OB, Polinsky A, Blagosklonny MV, Gudkov AV, Antoch MP. New nanoformulation of rapamycin Rapatar extends lifespan in homozygous p53-/- mice by delaying carcinogenesis. Aging (Albany NY) 2012; 4:715-722; PMID:23117593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Komarova EA, Antoch MP, Novototskaya LR, Chernova OB, Paszkiewicz G, Leontieva OV, Blagosklonny MV, Gudkov AV. Rapamycin extends lifespan and delays tumorigenesis in heterozygous p53+/- mice. Aging (Albany NY). 2012; 4:709-714; PMID:23123616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Leontieva OV, Novototskaya LR, Paszkiewicz GM, Komarova EA, Gudkov AV, Blagosklonny MV. Dysregulation of the mTOR pathway in p53-deficient mice. Cancer Biol Ther 2013; 14; PMID:24184801; http://dx.doi.org/ 10.4161/cbt.26947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Leontieva OV, Blagosklonny MV. DNA damaging agents and p53 do not cause senescence in quiescent cells, while consecutive re-activation of mTOR is associated with conversion to senescence. Aging (Albany NY) 2010; 2:924-935; PMID:21212465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Serrano M, Lim AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK1A. Cell 1997; 88:593-602; PMID:9054499; http://dx.doi.org/ 10.1016/S0092-8674(00)81902-9 [DOI] [PubMed] [Google Scholar]
- 70. Lin AW, Barradas M, Stone JC, van Aelst L, Serrano M, Lowe SW. Premature senescence involving p53 and p16 is activated in response to constitutive MEK/MAPK mitogenic signaling. Genes Dev 1998; 12:3008-3019; PMID:9765203; http://dx.doi.org/ 10.1101/gad.12.19.3008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Zhu JY, Woods D, McMahon M, Bishop JM. Senescence of human fibroblasts induced by oncogenic Raf. Genes Dev 1998; 12:2997-3007; PMID:9765202; http://dx.doi.org/ 10.1101/gad.12.19.2997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Peeper DS, Dannenberg JH, Douma S, te Riele H, Bernards R. Escape from premature senescence is not sufficient for oncogenic transformation by Ras. Nat Cell Biol 2001; 3:198-203; PMID:11175753; http://dx.doi.org/ 10.1038/35055110 [DOI] [PubMed] [Google Scholar]
- 73. Gitenay D, Lallet-Daher H, Bernard D. Caspase-2 regulates oncogene-induced senescence. Oncotarget 2014; 5:5845-5847; PMID:25114039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Vizioli MG, Santos J, Pilotti S, Mazzoni M, Anania MC, Miranda C, Pagliardini S, Pierotti MA, Gil J, Greco A. Oncogenic RAS-induced senescence in human primary thyrocytes: molecular effectors and inflammatory secretome involved. Oncotarget 2014; 5:8270-8283; PMID:25268744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Ferbeyre G, de Stanchina E, Lin AW, Querido E, McCurrach ME, Hannon GJ, Lowe SW. Oncogenic ras and p53 cooperate to induce cellular senescence. Mol Cell Biol 2002; 22:3497-3508; PMID:11971980; http://dx.doi.org/ 10.1128/MCB.22.10.3497-3508.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Kolesnichenko M, Hong L, Liao R, Vogt PK, Sun P. Attenuation of TORC1 signaling delays replicative and oncogenic RAS-induced senescence. Cell Cycle 2012; 11:2391-2401; PMID:22627671; http://dx.doi.org/ 10.4161/cc.20683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Li B, Reddy S, Comai L. Depletion of Ku70/80 reduces the levels of extrachromosomal telomeric circles and inhibits proliferation of ALT cells. Aging (Albany NY) 2011; 3:395-406; PMID:21512205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Vera E, Blasco MA. Beyond average: potential for measurement of short telomeres. Aging (Albany NY) 2012; 4:379-392; PMID:22683684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Louwen F, Yuan J. Battle of the eternal rivals: restoring functional p53 and inhibiting Polo-like kinase 1 as cancer therapy. Oncotarget 2013; 4:958-971; PMID:23948487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Russo I, Silver AR, Cuthbert AP, Griffin DK, Trott DA, Newbold RF. A telomere-independent senescence mechanism is the sole barrier to Syrian hamster cell immortalization. Oncogene 1998; 17:3417-3426; PMID:10030665; http://dx.doi.org/ 10.1038/sj.onc.1202261 [DOI] [PubMed] [Google Scholar]
- 81. Kolesnichenko M, Hong L, Liao R, Vogt PK, Sun P. Attenuation of TORC1 signaling delays replicative and oncogenic RAS-induced senescence. Cell Cycle 2012; 11:2391-401 this issue; PMID:22627671; http://dx.doi.org/ 10.4161/cc.20683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Pospelova TV, Leontieva OV, Bykova TV, Zubova SG, Pospelov VA, Blagosklonny MV. Suppression of replicative senescence by rapamycin in rodent embryonic cells. Cell Cycle 2012; 11:2402-2407; PMID:22672902; http://dx.doi.org/ 10.4161/cc.20882 [DOI] [PubMed] [Google Scholar]
- 83. Tang DG, Tokumoto YM, Apperly JA, Lloyd AC, Raff MC. Lack of replicative senescence in cultured rat oligodendrocyte precursor cells. Science 2001; 291:868-871; PMID:11157165; http://dx.doi.org/ 10.1126/science.1056780 [DOI] [PubMed] [Google Scholar]
- 84. Parrinello S, Samper E, Krtolica A, Goldstein J, Melov S, Campisi J. Oxygen sensitivity severely limits the replicative lifespan of murine fibroblasts. Nat Cell Biol 2003; 5:741-747; PMID:12855956; http://dx.doi.org/ 10.1038/ncb1024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Kaeberlein M, Powers RWr, K.K S, Westman EA, Hu D, Dang N, Kerr EO, Kirkland KT, Fields S, Kennedy BK. Regulation of yeast replicative life span by TOR and Sch9 in response to nutrients. Science 2005; 310:1193-1196; PMID:16293764; http://dx.doi.org/ 10.1126/science.1115535 [DOI] [PubMed] [Google Scholar]
- 86. Longo VD, Shadel GS, Kaeberlein M, Kennedy B. Replicative and chronological aging in Saccharomyces cerevisiae. Cell Metab 2012; 16:18-31; PMID:22768836; http://dx.doi.org/ 10.1016/j.cmet.2012.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Polymenis M, Kennedy BK. Chronological and replicative lifespan in yeast: do they meet in the middle? Cell Cycle 2012; 11:3531-3532; PMID:22951539; http://dx.doi.org/ 10.4161/cc.22041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Leontieva OV, Blagosklonny MV. Yeast-like chronological senescence in mammalian cells: phenomenon, mechanism and pharmacological suppression. Aging (Albany NY) 2011; 3:1078-1091; PMID:22156391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Kaeberlein M, Kennedy BK. A new chronological survival assay in mammalian cell culture. Cell Cycle 2012; 11:201-202; PMID:22214665; http://dx.doi.org/ 10.4161/cc.11.2.18959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Pan Y, Shadel GS. Extension of chronological life span by reduced TOR signaling requires down-regulation of Sch9p and involves increased mitochondrial OXPHOS complex density. Aging (Albany NY) 2009; 1:131-135; PMID:20157595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Wei M, Fabrizio P, Madia F, Hu J, Ge H, Li LM, Longo VD. Tor1/Sch9-regulated carbon source substitution is as effective as calorie restriction in life span extension. PLoS Genet 2009; 5:e1000467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Castilho RM, Squarize CH, Chodosh LA, Williams BO, Gutkind JS. mTOR mediates Wnt-induced epidermal stem cell exhaustion and aging. Cell Stem Cell 2009; 5:279-289; PMID:19733540; http://dx.doi.org/ 10.1016/j.stem.2009.06.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Gan B, DePinho RA. mTORC1 signaling governs hematopoietic stem cell quiescence. Cell Cycle 2009; 8:1003-1006; PMID:19270523; http://dx.doi.org/ 10.4161/cc.8.7.8045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Menendez JA, Vellon L, Oliveras-Ferraros C, Cufi S, Vazquez-Martin A. mTOR-regulated senescence and autophagy during reprogramming of somatic cells to pluripotency: a roadmap from energy metabolism to stem cell renewal and aging. Cell Cycle 2011; 10:3658-3677; PMID:22052357; http://dx.doi.org/ 10.4161/cc.10.21.18128 [DOI] [PubMed] [Google Scholar]
- 95. Hinojosa CA, Mgbemena V, Van Roekel S, Austad SN, Miller RA, Bose S, Orihuela CJ. Enteric-delivered rapamycin enhances resistance of aged mice to pneumococcal pneumonia through reduced cellular senescence. Exp Gerontol 2012; 47:958-965; PMID:22981852; http://dx.doi.org/ 10.1016/j.exger.2012.08.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Luo Y, Li L, Zou P, Wang J, Shao L, Zhou D, Liu L. Rapamycin enhances long-term hematopoietic reconstitution of ex vivo expanded mouse hematopoietic stem cells by inhibiting senescence. Transplantation 2014; 97:20-29; PMID:24092377; http://dx.doi.org/ 10.1097/TP.0b013e3182a7fcf8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Blagosklonny MV. Aging, stem cells, and mammalian target of rapamycin: a prospect of pharmacologic rejuvenation of aging stem cells. Rejuvenation Res 2008; 11:801-808; PMID:18729812; http://dx.doi.org/ 10.1089/rej.2008.0722 [DOI] [PubMed] [Google Scholar]
- 98. Zhang XM, Li L, Xu JJ, Wang N, Liu WJ, Lin XH, Fu YC, Luo LL. Rapamycin preserves the follicle pool reserve and prolongs the ovarian lifespan of female rats via modulating mTOR activation and sirtuin expression. Gene 2013; 523:82-87; PMID:23566837; http://dx.doi.org/ 10.1016/j.gene.2013.03.039 [DOI] [PubMed] [Google Scholar]
- 99. Adhikari D, Zheng W, Shen Y, Gorre N, Hamalainen T, Cooney AJ, Huhtaniemi I, Lan ZJ, Liu K. Tsc/mTORC1 signaling in oocytes governs the quiescence and activation of primordial follicles. Hum Mol Genet 2010; 19:397-410; PMID:19843540; http://dx.doi.org/ 10.1093/hmg/ddp483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Blagosklonny MV. Why men age faster but reproduce longer than women: mTOR and evolutionary perspectives. Aging (Albany NY) 2010; 2:265-273; PMID:20519781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Blagosklonny MV. Prospective treatment of age-related diseases by slowing down aging. Am J Pathol 2012; 181:1142-1146; PMID:22841821; http://dx.doi.org/ 10.1016/j.ajpath.2012.06.024 [DOI] [PubMed] [Google Scholar]
- 102. Blagosklonny MV. Validation of anti-aging drugs by treating age-related diseases. Aging (Albany NY) 2009; 1:281-288; PMID:20157517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Blagosklonny MV. Answering the ultimate question “what is the proximal cause of aging?” Aging (Albany NY) 2012; 4:861-877; PMID:23425777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Gems DH, de la Guardia YI. Alternative Perspectives on Aging in C. elegans: Reactive Oxygen Species or Hyperfunction? Antioxid Redox Signal 2013; 19:321-329; PMID:22870907; http://dx.doi.org/ 10.1089/ars.2012.4840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Gems D, Partridge L. Genetics of Longevity in Model Organisms: Debates and Paradigm Shifts. Annu Rev Physiol 2013;75:621-644; PMID:23190075; http://dx.doi.org/ 10.1146/annurev-physiol-030212-183712 [DOI] [PubMed] [Google Scholar]
- 106. Wang C, Jurk D, Maddick M, Nelson G, Martin-Ruiz C, von Zglinicki T. DNA damage response and cellular senescence in tissues of aging mice. Aging Cell 2009; 8:311-323; PMID:19627270; http://dx.doi.org/ 10.1111/j.1474-9726.2009.00481.x [DOI] [PubMed] [Google Scholar]
- 107. De Cecco M, Jeyapalan J, Zhao X, Tamamori-Adachi M, Sedivy JM. Nuclear protein accumulation in cellular senescence and organismal aging revealed with a novel single-cell resolution fluorescence microscopy assay. Aging (Albany NY) 2011; 3:955-967; PMID:22006542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Bhat R, Crowe EP, Bitto A, Moh M, Katsetos CD, Garcia FU, Johnson FB, Trojanowski JQ, Sell C, Torres C. Astrocyte senescence as a component of Alzheimer's disease. PLoS One 2012; 7:e45069; PMID:22984612; http://dx.doi.org/ 10.1371/journal.pone.0045069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Berkenkamp B, Susnik N, Baisantry A, Kuznetsova I, Jacobi C, Sorensen-Zender I, Broecker V, Haller H, Melk A, Schmitt R. In vivo and in vitro analysis of age-associated changes and somatic cellular senescence in renal epithelial cells. PLoS One 2014; 9:e88071; PMID:24505380; http://dx.doi.org/ 10.1371/journal.pone.0088071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Burd CE, Sorrentino JA, Clark KS, Darr DB, Krishnamurthy J, Deal AM, Bardeesy N, Castrillon DH, Beach DH, Sharpless NE. Monitoring tumorigenesis and senescence in vivo with a p16(INK4a)-luciferase model. Cell 2013; 152:340-351; PMID:23332765; http://dx.doi.org/ 10.1016/j.cell.2012.12.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Georgakopoulou EA, Tsimaratou K, Evangelou K, Fernandez Marcos PJ, Zoumpourlis V, Trougakos IP, Kletsas D, Bartek J, Serrano M, Gorgoulis VG. Specific lipofuscin staining as a novel biomarker to detect replicative and stress-induced senescence. A method applicable in cryo-preserved and archival tissues. Aging (Albany NY) 2013; 5:37-50; PMID:23449538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Tran S, Rizos H. Human nevi lack distinguishing senescence traits. Aging (Albany NY) 2013; 5:98-99; PMID:23518674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Jeyapalan JC, Sedivy JM. How to measure RNA expression in rare senescent cells expressing any specific protein such as p16Ink4a. Aging (Albany NY) 2013; 5:120-129; PMID:23454889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Blagosklonny MV. TOR-centric view on insulin resistance and diabetic complications: perspective for endocrinologists and gerontologists. Cell Death Dis 2013; 4:e964; PMID:24336084; http://dx.doi.org/ 10.1038/cddis.2013.506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Nelson G, Wordsworth J, Wang C, Jurk D, Lawless C, Martin-Ruiz C, von Zglinicki T. A senescent cell bystander effect: senescence-induced senescence. Aging Cell 2012; 11:345-349; PMID:22321662; http://dx.doi.org/ 10.1111/j.1474-9726.2012.00795.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Acosta JC, Banito A, Wuestefeld T, Georgilis A, Janich P, Morton JP, Athineos D, Kang TW, Lasitschka F, Andrulis M, et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat Cell Biol 2013; 15:978-990; PMID:23770676; http://dx.doi.org/ 10.1038/ncb2784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Bahar R, Hartmann CH, Rodriguez KA, Denny AD, Busuttil RA, Dolle ME, Calder RB, Chisholm GB, Pollock BH, Klein CA, et al. Increased cell-to-cell variation in gene expression in ageing mouse heart. Nature 2006; 441:1011-1014; PMID:16791200; http://dx.doi.org/ 10.1038/nature04844 [DOI] [PubMed] [Google Scholar]