

Abstract
A constitutive and dynamic interaction between tumor cells and their surrounding stroma is a prerequisite for tumor invasion and metastasis. Fibroblasts and myofibroblasts (collectively called cancer associated fibroblasts, CAFs) often represent the major cellular components of tumor stroma. Tumor cells secret different growth factors which induce CAFs proliferation and differentiation, and, consequently, CAFs secrete different chemokines, cytokines or growth factors which induce tumor cell invasion and metastasis. In this study we showed here that CAFs from breast cancer surgical specimens significantly induced the invasion of breast cancer cells in vitro. Most interestingly, the novel multiple tyrosine kinase inhibitor Dovitinib significantly blocked the CAFs-induced invasion of breast cancer cells by, at least in part, inhibition of the expression and secretion of CCL2, CCL5 and VEGF in CAFs. Inhibition of PI3K/Akt/mTOR signaling could be responsible for the effects of Dovitinib, since Dovitinib antagonized the promoted phosphorylated Akt after treatment with PDGF, FGF or breast cancer cell-conditioned media. Treatment with Dovitinib in combination with PI3K/Akt/mTOR signaling inhibitors Ly294002 or RAD001 resulted in additive inhibition of cell invasion. This is the first in vitro study to show that the multiple tyrosine kinase inhibitor has therapeutic activities against breast cancer metastasis by targeting both tumor cells and CAFs.
Keywords: breast cancer, cancer-associated fibroblasts, chemokines, Dovitinib, FGF, invasion, PDGF
Introduction
Triple negative breast cancer, an aggressive variant of breast cancer, is characterized by lack of expression of the estrogen receptor (ER), progesterone receptors (PRs) and the human epidermal growth factor receptor (c-erbB2, HER-2) that are commonly observed in other breast cancer subtypes. This sub-entity of breast cancer is characteristically aggressive with high recurrence, metastasis, and mortality rates. Treatment options are very limited except with the traditional cytotoxic chemotherapy.1 The systematic evaluation of different randomized clinical trials indicated that combining targeted agents with chemotherapy in triple negative breast cancer produced only modest gains in progression-free survival.2 Therefore, a better understanding of the molecular basis of breast cancer progression and development of novel molecular targeted therapy is required for the better treatment of this breast cancer subtype.
It is gradually becoming clear that the fibroblasts, the main component of tumor stroma, are prominent modifiers of breast cancer progression. These cells, which may be recruited locally from the normal tissues adjacent to the primary tumor or from bone marrow are called cancer associated fibroblasts (CAFs).3-6 Recently experimental evidence indicates that fibroblasts protect cancer cells against stress by reducing reactive oxygen species (ROS) production, apoptosis, autophagy, and senescence in epithelial cancer cells through providing high-energy fuels and precursors for biomass to adjacent cancer.7,8 It was also shown that CAFs support the malignant properties of cancer spreading by secreting soluble factors such as pro-angiogenic factors, matrix metalloproteinases (MMPs), cytokines as well as chemokines and growth factors.9 Among these cytokines, CCL2 and CCL5, which are known inflammatory mediators, were demonstrated to play important role in the mediation of the interaction between CAFs and breast cancer cells beyond SDF-110-13
Dovitinib (formerly CHIR-258, TKI-258, Novartis pharmaceuticals) is an investigational new inhibitor of multiple tyrosine kinases that has in vitro inhibitory activity against basic fibroblast growth factor receptor (bFGFR), platelet-derived growth factor receptor (PDGFR), and vascular endothelial growth factor receptor (VEGFR) kinases with IC50 values of approximately 10 nM. Further in vivo and in vitro data indicate that this drug blocked PDGFR/FGFR/VEGFR signaling in advanced melanoma,4 pancreatic cancer,14 breast carcinoma,15 urothelial carcinoma,16 impaired tumor growth, angiogenesis, and metastasis by effects on tumor cells, endothelial cells, and pericytes in vitro.14,15
Due to the important role of PDGFs and FGFs in the supportive effect of CAFs for the breast tumor progression through the autocrine or paracrine fashion,17-19 we hypothesized that targeting PDGFR and FGFR signaling by Dovitinib could block the cross-talk of CAFs-tumor cell and inhibit cell invasion of breast cancer. We selected an in vitro transwell chamber model for co-culture of breast cancer cells with CAFs and investigation of breast cancer cell invasion in this study. The concomitant change of cytokines/chemokines and the intracellular downstream signaling of these growth factors were also examined.
Results
Tyrosine kinase inhibitor Dovitinib inhibited the breast cancer invasion and antagonized the invasion-promoting effect of CAFs
For investigation whether the interaction between tumor cells and CAFs could result in enhanced invasion of breast cancer cells, we isolated at first the CAFs from breast tumor specimens obtained at surgery from patients with invasive breast cancer (n = 5) according to the method described in the Materials and Methods. A representative of the isolated CAFs in in vitro culture was shown (Fig. 1A).
Figure 1.
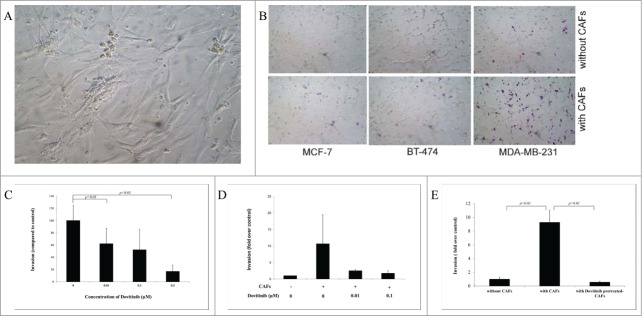
Dovitinib inhibited the breast cancer invasion and antagonize the invasion promoting-effect of CAFs. (A) One example of isolated CAFs from patient samples (B) Enhanced invasion ability of breast cancer cells MCF-7, BT-474 and MDA-MB-231 through co-culture with CAFs. Human breast cancer CAFs were seeded in 24-well-plate and cultured in serum-free medium for 3 d Breast cancer cells suspended in serum-free media were added into the inserts either with CAFs or with only serum-free medium in the bottom chamber. Invasion assay was performed as described in the Materials and Methods. Non-invaded cells were removed from the top surface of the insert by scrubbing with cotton tip swabs. 18 h later, the membranes of the inserts with invaded cells were fixed, stained, mounted on slides, and counted under light microscope. (C) Dose-dependently inhibited invasion ability of MDA-MB-231 cells after treatment with Dovitinib. Breast cancer cells MDA-MB-231 were pre-treated with Dovitinib (0.01, 0.1, 0.5 μM) for 2 days, suspended in cell culture medium, and added into the inserts with cell culture medium in the bottom chamber. Invasion assay was performed as described in the Materials and Methods. (D) Pre-treatment of MDA-MB-231 cells with Dovitinib led to inhibited invasion in the co-culture system. CAFs were seeded in 24-well-plate and cultured in serum-fee medium for 3 d Breast cancer cells MDA-MB-231 were pre-treated with Dovitinib (0.01, 0.1 μM) for 2 days, suspended in serum-free medium, and added into the inserts either with CAFs or with only serum-free medium in the bottom chamber. Invasion assay was performed as described in the Materials and Methods. (E) Pre-treatment of CAFs with Dovitinib led to inhibited invasion in the co-culture system. CAFs were seeded in 24-well-plate and pre-treated with Dovitinib (0.01 μM) for 1 day. MDA-MB-231 cells were suspended in serum-free medium, and added into the inserts either with CAFs or with only serum-free medium in the bottom chamber. Invasion assay was performed as described in the Materials and Methods.
We tested the invasive ability of non-aggressive breast cancer cells MCF-7, moderately aggressive breast cancer cells BT-474, and highly aggressive breast cancer cells MDA-MB-231 by co-culture of these cells with the CAFs using the BD BioCoatTM Martrigel Invasion Chambers. MCF-7 cells and BT-474 cells showed almost no invaded cells, MDA-MB-231 several invaded cells under our experimental conditions when serum-free cell culture medium was used in the bottom chambers. Significant more invaded cells were observed for all of the 3 breast cancer cell lines when CAFs were co-cultured in the bottom chambers, suggesting the CAFs promoted the invasion of breast cancer cells (Fig. 1B). The most invasive breast cancer cell line MDA-MB-231 was selected therefore for further investigations.
Inhibitory effect of the Dovitinib on the breast cancer cell invasion and its blocking effect on CAFs-mediated invasion promotion were quantitatively determined. MDA-MB-231 cells were treated with different concentrations of Dovitinib, and then added to the chambers for the invasion assay. Dovitinib treatment resulted in a dose-dependent reduction of invasion ability of MDA-MB-231 cells in the absence of CAFs (Fig. 1C). As next, invasion assay was performed in the presence or absence of Dovitinib either with CAFs or with serum-free medium in the bottom chamber (non-contact co-culture). With CAFs in the invasion system, the invasion of MDA-MB-231 cells was enhanced dramatically, while this effect of CAFs was antagonized by pre-treatment of MDA-MB-231 cells with Dovitinib (Fig. 1D). These experiments demonstrated clearly that Dovitinib exerted the inhibitory effect on invasion of MDA-MB-231 cells in the presence or absence of CAFs.
From the experiments with non-contact co-culture system we could suppose that the CAFs support the cell invasion possibly by secreting soluble factors. The effect of Dovitinib on the invasion of MDA-MB-231 cells in the co-culture system was possibly contributed by blocking of the secretion of those soluble factors by CAFs. To clarify this question, CAFs were plated in the 24-well plate and cultured in the presence of Dovitinib, Dovitinib was then washed out and invasion assay was carried out with the CAFs co-cultured in the bottom chamber which were either pre-treated with Dovitinib or not pre-treated. The result showed clearly that Dovitinib pre-treated CAFs lost the ability to stimulate the invasion of MDA-MB-231 cells, so that the invasion rate of MDA-MB-231 cells under the co-culture with Dovitinib pre-treated CAFs was reduced to the similar level as that without co-culture with CAFs (Fig. 1E).
Application of Dovitinib in the MDA-MB-231 cells, CAFs, or the co-culture of the both types of cells resulted in the inhibited production of chemokines CCL2, CCL5 and VEGF
MDA-MB-231 cells, CAFs cells were cultured separately or together through non- contact co-culture in 24-well plates for 2 d in the presence or absence of Dovitinib. Cells were then harvested for the mRNA isolation, cell supernatants were collected for the ELISA measurement of different chemokines and VEGF.
Co-culture of the MDA-MB-231 cells with CAFs resulted in a dramatically increased production of chemokines CCL2 and CCL5 in the cell supernatants in comparison to chemokine production by the 2 types of cells alone. The application of Dovitinib antagonized the enhanced production of these 2 chemokines significantly (Fig. 2A). Similarly, mRNA expression level of CCL2 and CCL5 was much higher in CAFs or CAFs in co-culture with MDA-MB-231 cells than in MDA-MB-231 cells, whereas Dovitinib application resulted in significant reduction of mRNA expression level of these chemokines especially in CAFs as determined with real-time PCR (Fig. 2B).
Figure 2.
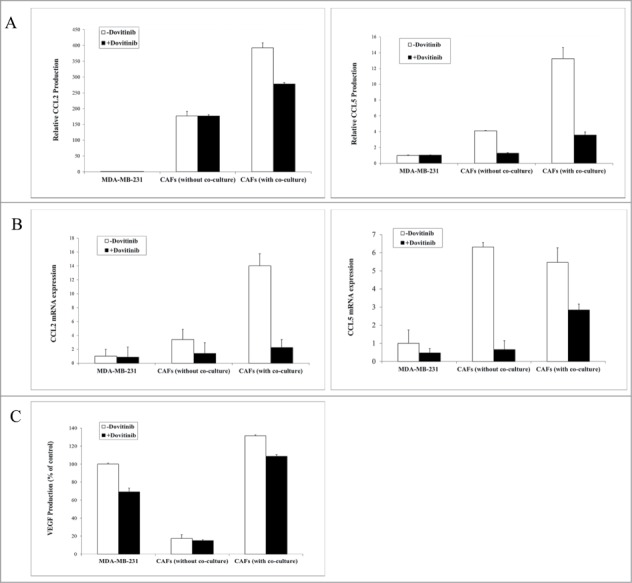
Application of Dovitinib in the MDA-MB-231 cells, CAFs, or the co-culture of the both types of cells resulted in the inhibited production of chemokines CCL2, CCL5 and VEGF. (A–C) 2 × 104 CAFs and 2 × 105 MDA-MB-231 cells were cultured alone in 24-well-plate or co-cultured with BD BioCoatTM Martrigel Invasion Chambers in serum-fee medium for 2 d in the presence or absence of Dovitinib (0.1 μM). After incubation, cell supernatants of MDA-MB-231 cells, CAFs, and co-culture were collected and served for ELISA measurement, cells were lysed for total RNA isolation. A: ELISA measurement of CCL2 and CCL5 production; B: Real-time PCR determination of CCL2 and CCL5 expression in MDA-MB-231 cells, CAFs, CAFs in co-culture with breast cancer cells; C: ELISA measurement of VEGF production.
VEGF is one of the important growth factors which are involved not only in breast cancer angiogenesis, but also in tumor metastasis and progression. Also, VEGFR belongs to the specific inhibitory targets of Dovitinib and this growth factor was therefore investigated with ELISA. Similar to the experimental result about the chemokines CCL2 and CCL5 production, VEGF concentration was much higher in supernatants of CAFs than in that of MDA-MB-231 cells, which was dramatically reduced by the treatment with Dovitinib (Fig. 2C).
Dovitinib antagonized the PDGF/FGF stimulated MDA-MB-231 cell invasion and chemokine production of CAFs and MDA-MB-231 cells
Dovitinib binds to the ATP-binding site and inhibits the tyrosine kinase activity of PDGFR and FGFR with IC50 of 10 nM in vitro.20 The inhibitory effect of Dovitinib on PDGFR was detected in MDA-MB-231 cells by checking the change of PDGFß mRNA expression after treatment. PDGFß mRNA expression level was enhanced by 2-fold when MDA-MB-231 cells were cultured with CAFs-conditioned media. However, it was inhibited to the basic level by Dovitinib treatment (Fig. 3A). Then as next we tried to answer the question if the inhibitory effect of Dovitinib on PDGFR played an important role on the breast cancer cell invasion. Again with invasion chamber assay we could demonstrate that human recombinant PDGF and FGF promoted MDA-MB-231 cells invasion dramatically, whereas the cell invasion was reduced significantly if Dovitinib was added to the culture media (Fig. 3B). Our experiments indicated that the CAFs may promote the breast cancer cell invasion by stimulating PDGFß/FGF signaling. The inhibitory effect of Dovitinib on this signaling contributed therefore to its inhibitory effect on cell invasion.
Figure 3.
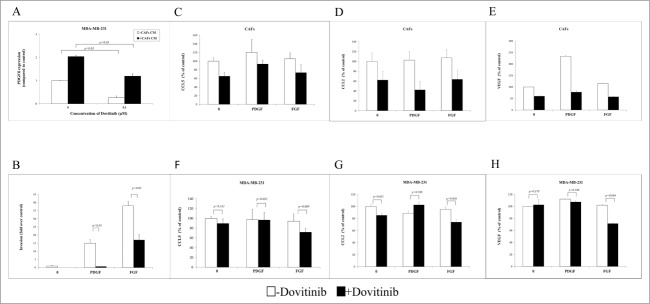
Dovitinib antagonized the PDGF/FGF stimulated MDA-MB-231 cell invasion and chemokine production of CAFs and MDA-MB-231 cells. (A) Dovitinib inhibited the PDGFß expression of MDA-MB-231 cells stimulated with CAFs-conditioned media.CAFs were cultured in serum-free medium for 3 d Cell culture media was collected for further experiment. MDA-MB-231 cells were cultured with serum-free medium or CAFs-conditioned media in the presence of Dovitinib (0.1 μM) for 2 d MDA-MB-231 cells were lysed and PDGFß expression was measured with real-time PCR. Each value presented is the mean ± SD of 3 parallel experiments. (B) Dovitinib antagonized the PDGF/FGF stimulated cell invasion of MDA-MB-231 cells.MDA-MB-231 cells were treated with Dovitinib (0.1 μM) for 2 d Cell invasion assay was performed in serum-free medium, serum-free medium including 10 ng/ml PDGF, or serum-free medium including 10 ng/ml FGF respectively. Each value presented is the mean ± SD of 3 parallel experiments. (C–H) Dovitinib treatment resulted in reduced CCL5, CCL2 and VEGF production in CAFs and MDA-MB-231 cells.CAFs (C-E) or MDA-MB-231 cells (F-H) were treated with PDGF (10 ng/ml) or FGF (10 ng/ml) in the presence or absence of Dovitinib (0.1 μM) for 2 d Cell supernatants were collected; CCL5, CCL2 or VEGF concentration was measured with ELISA.
To investigate if the inhibitory effect of Dovitinib on PDGF/FGF signaling was contributed to its effect on the MDA-MB-231 cell invasion and chemokines/cytokines secretion of CAFs, cell supernatants from MDA-MB-231 cells or CAFs treated with these growth factors in the presence or absence of Dovitinib were collected and chemokine/cytokines concentration was measured by ELISA. CCL5/CCL2/VEGF production was increased by the PDGF/FGF treatment in CAFs, whereas it decreased significantly by Dovitinib treatment (Figs. 3C–E). To our surprise, this effect of Dovitinib was not significant in MDA-MB-231 cells in the case the cells were treated with PDGF (Figs. 3F–H).
Dovitinib impaired Akt signaling in CAFs and led to additive inhibitory effect on cell invasion by combination with PI3K/Akt/mTOR signaling inhibitor Ly294002 or RAD001
PI3K/Akt/mTOR signaling is the common downstreaming signaling of growth factors PDGF and FGF.21,22 The Akt pathway also stimulates the transcriptional induction of VEGF which is then secreted and acts in a paracrine manner to stimulate its cognate receptor VEGF-R in endothelial cells. This in turn activates PI3K and Akt with a variety of consequences.23
We investigated therefore the Akt signaling change in response to Dovitinib treatment in MDA-MB-231 and CAFs respectively. Surprisingly, we could not detect significant protein expression change of the components of this signaling with Western blot in MDA-MB-231 cells (data not shown); instead, we could show the enhanced Akt phosphorylation in CAFs in the presence of PDGF or MDA-MB-231 cells-conditioned media, which was antagonized by Dovitinib treatment significantly (Fig. 4A). This result prompted us to investigate if simultaneous inhibition of PI3K/Akt/mTOR with their inhibitors could enhance the invasion inhibitory effect of Dovitinib. As expected, pre-treatment of CAFs with Akt inhibitor Ly294002 or mTOR inhibitor RAD001 resulted in a significant cell invasion reduction as detected with invasion chamber assay. This effect of Akt/mTOR inhibitor was intensified if the cells were pre-treated with the combination of Akt/mTOR inhibitor and Dovitinib (Fig. 4B).
Figure 4.
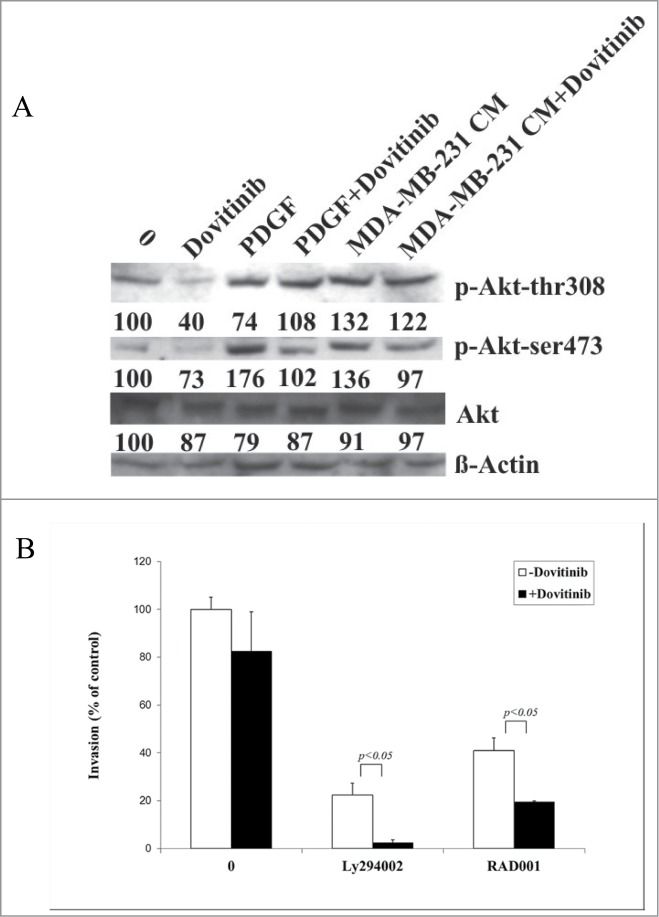
Dovitinib impaired Akt signaling in CAFs and led to enhanced inhibitory effect on cell invasion by combination with PI3K/Akt/mTOR signaling inhibitors Ly294002 or RAD001. (A) CAFs were cultured in serum-free RPMI-1640 media, serum-free RPMI-1640 media supplemented with 10 ng/ml PDGF, or MDA-MB-231 conditioned media (CM) in the presence or absence of Dovitinib for 24 h respectively. Total protein was extracted; the expression of phosphorylated Akt (T308) and phosphorylated Akt (S473) was determined by Western blot analysis. The relative intensity of each band shown under the band was quantified using PhoretixTM ID Quantifier software, normalized to ß-actin and expressed as percentage of control. (B) CAFs were seeded in 24-well-plate and cultured in serum-fee medium in the presence of Ly294002 (20 μM), RAD001 (10 nM), Ly294002 (20 μM)+Dovitinib (0.1 μM), or RAD001(10 nM)+Dovitinib (0.1 μM), respectively for 3 d MDA-MB-231 cells were suspended in serum-free media and added into the inserts either with or without CAFs in the bottom wells. Transwell® invasion assay was performed as described in the Materials and Methods. Each value presented is the mean ± SD of 3 parallel experiments.
Discussion
Most of current treatments for breast cancer target primarily carcinoma cells, while the pro-carcinogenic actions of the tumor microenvironment were not taken into consider. The fact that tumor-associated fibroblasts present some tumor features and are genetically stable and also different from normal fibroblasts make them to be important potential targets of anti-cancer therapy. With the increasing understanding of epithelial-stromal biochemical interactions and molecular reciprocal heterotypic signaling, targeting breast carcinomas and/or stromal fibroblasts will be new strategies for the development of new therapeutic regimens.
Here we demonstrated for the first time the possibility to use Dovitinib to target the breast cancer motility through interference with tumor supportive effects of CAFs. This might provide a complementary approach to conventional treatments that target the cancer cells themselves. The multiple tyrosine kinase inhibitor Dovitinib might become a promising agent for treatment of metastased breast cancer.
It has been reported that treatment of tumor-bearing mice with VEGF receptor inhibitors such as sunitinib led to enhanced distant metastasis.24,25 However, Dovitinib was shown to prevent lung metastasis in an in vivo xenograft model of hepatocellular carcinoma,26 reduce the 4T1 tumor-induced lung metastases after long-term treatment and lymph node metastases in an orthotopic tumor model when treatment was initiated early enough.15 In correlation with these in vivo data, we observed with our in vitro chamber assay that Dovitinib inhibited cell invasion of breast cancer dose-dependently. Importantly, Dovitinib antagonized the enhanced cell invasion through co-culture of CAFs with MDA-MB-231 cells (Fig. 1). The molecular mechanism(s) responsible for Dovitinib -inhibited metastasis was thereafter investigated.
Inflammation could be a causal role for many malignant diseases, including breast cancer. The different inflammatory mediators that are involved in this disease include cells, cytokines and chemokines. Of these, many studies have addressed the involvement and roles of the inflammatory chemokines CCL2 (MCP-1) and CCL5 (RANTES) in breast malignancy. These two chemokines were shown to mediate many types of tumor-promoting cross-talks between the tumor cells and cells of the tumor microenvironment: CCL2 and CCL5 which are expressed by cells of the tumor microenvironment osteoblasts and mesenchymal stem cells play a role in breast metastatic processes. In addition, both chemokines act directly on the tumor cells to promote their pro-malignancy phenotype, by increasing their migratory and invasion-related properties.13 In line with these knowledge, we observed in this study that the CCL2 and CCL5 were 2 of chemokines which were secreted from CAFs and mediated invasion-promoting effect of CAFs. Inhibition of the secretion of CCL2 and CCL5 and thereby inhibiting the cross talk between CAFs and breast cancer cells might be one of the mechanisms by which Dovitinib blocked the invasion-promoting effect of CAFs. Since we demonstrated clearly that Dovitinib application resulted in reduced CCL2 and CCL5 expression from the CAFs and the production in cell culture media with ELISA analysis and real time PCR. Besides CCL2 and CCL5, other chemokines/cytokines may also be involved in this process including VEGF as shown by our data (Fig. 2).
Paracrine PDGF signaling is commonly observed in epithelial cancers, where it triggers stromal recruitment and may be involved in epithelial–mesenchymal transition, thereby affecting tumor growth, angiogenesis, invasion, and metastasis.27 By using a genetically engineered mouse model of cervical carcinogenesis, pharmacological blockade of PDGF receptor signaling with the clinically approved kinase inhibitors slowed progression of premalignant cervical lesions in this model. Inhibition of stromal PDGF receptors reduced proliferation and angiogenesis in cervical lesions through a mechanism involving suppression of expression of the angiogenic FGF2 and the epithelial cell growth factor FGF-7 by cancer-associated fibroblasts. The possible functional model of PDGF and FGFs in epithelial tumors could be: PDGF expressed by cancerous epithelia evidently stimulate PDGFR-expressing stroma to up-regulate FGFs, promoting angiogenesis and epithelial proliferation, elements of a multi-cellular signaling network that elicits functional capabilities in the tumor microenvironment.28 Dovitinib has almost equal in vitro inhibitory activity against bFGF and PDFG receptor kinases with IC50 values of approximately 10 nM.29 In accordance with the previous pre-clinical and clinical data, interruption of the PDGF signaling by Dovitinib led to reduced breast cancer cell invasion in our in vitro test model (Figs. 3A and B). Interestingly, Dovitinib inhibited the PDGF-stimulated chemokine secretion of CAFs but had no significant effect in MDA-MB-231 cells, while it inhibited FGF2-stimulated chemokine secretion of both CAFs and MDA-MB-231 cells significantly (Figs. 3C–E and F–H). Our result was supported by the reports that FGFR2 was identified as a breast cancer susceptibility gene by genome-wide association studies, especially in the mesenchymal and mesenchymal-like subtypes of triple-negative breast cancer.22,30 In connection with the data that mRNA expression of CCL5/CCL2 in CAFs was regulated by Dovitinib treatment, our data suggest the model for Dovitinib to block the supporting effect of CAFs to cell invasion: By inhibition of PDGF and FGF signaling in CAFs, Dovitinib indirectly downregulated the chemokines/cytokines secretion by CAFs, so that the supporting effect of CAFs on breast cancer cells was blocked and this resulted in the inhibition of cell invasion.
Some studies in the recent years indicate that chronological age of the tumor environment specifically fibroblasts is the most significant risk factor for human cancer development. Mammary tumors grown in a Cav-1–deficient tumor microenvironment (an in vivo model of accelerated host aging) were more than five-fold larger than tumors grown in a wild-type microenvironment.31 In vitro experiments showed that the senescent fibroblasts generated by overexpressing CDK inhibitors, such as p16 (INK4A), p19 (ARF) or p21 (WAF1/CIP1) also dramatically promoted breast tumor growth without any comparable increases in tumor angiogenesis.32 PI3K/Akt/mTOR is one of the most critical pathways involved in the cell aging. The prolonged activation of PI3K/Akt/mTOR pathway in post-mitotic cells will convert quiescence or arrest into senescence. In turn, aging cells due to their hyper-functions cause diseases of aging including cancer.33 In this context, anti-tumor effect of PI3K/Akt/mTOR pathway inhibitors was not only achieved by significantly inhibiting the tumor cell proliferation and division, but also by targeting tumor stroma, for example decreasing the stromal content, and reducing the levels of both vimentin and phospho-S6 (2 known markers for cell aging) in aging CAFs model.31
Recent publications demonstrated with in vitro experiments, mouse models and clinical trials that Dovitinib showed antitumor activity in FGFR-amplified breast cancer.34 The effect of Dovitinib observed included blocking breast cancer cell proliferation and growth, inhibiting tumor metastatic speed of breast cancer in vivo and in vitro models.15,35 Co-targeting of ErbB receptors enhanced those effect of Dovitinib, since this 2 receptor tyrosine kinases have same downstream signaling PI3K/Akt/mTOR and the anti-tumor effect resulted from the blockade of receptor tyrosine kinases by Dovitinib was shown mostly via PI3K/Akt/mTOR signaling inhibition.15,35 However, most of these investigations were focused on the effect of Dovitinib on tumor cells itself. We demonstrated for the first time that the inhibition effect of Dovitinib on the breast cancer metastasis is also possibly contributed by its inhibition effect on PI3K/Akt/mTOR signaling in CAFs (Fig. 4A). This notion was further supported by the experiment that pre-treatment of CAFs with the combination of PI3K/Akt inhibitor Ly294002 or mTOR inhibitor RAD001 with Dovitinib resulted in additive invasion inhibition of MDA-MB-231 cells (Fig. 4B). We provide a new evidence for the increasingly accepted view that both cancer and aging share the activation of a common signaling pathway, and targeting this pathway directly or indirectly will retard breast cancer progression.33,36 Our data suggest a new mechanism by which Dovitinib antagonizes the breast cancer metastasis.
In conclusion, Dovitinib treatment resulted in lowered breast cancer cell invasion by interruption of the breast cancer cells-CAFs interaction. This is the first study to show that multiple tyrosine kinase inhibitors exert effects on CAFs by interfering with their secretion of chemokines and growth factors which are crucial for tumor invasion and metastasis. We are the first to find the phospho-Akt expression in CAFs was correlated with the capacity of Dovitinib to promote cell invasion. Treatment with Dovitinib in combination with PI3K/Akt/mTOR signaling inhibitors Ly294002 or RAD001 resulted in additive inhibition of cell invasion. Targeting PI3K/Akt/mTOR signaling of CAFs will be a complementary way for breast cancer therapy and requires more research.
Materials and Methods
Materials
Dovitinib (TKI-258, CHIR-258) was purchased from Selleckchem (Munich, Germany); RAD001 was kindly provided by Novartis Pharmaceuticals (Basel, Switzerland). Both of the drugs was dissolved in dimethyl sulfoxide (DMSO) at a concentration of 10−3 M as a stock solution, then further diluted to the working concentration with cell culture media before use. Ly294002 was purchased from Biomol (Hamburg, Germany) and dissolved in DMSO at a concentration of 10−2 M as a stock solution; Platelet-derived growth factor (PDGF) and fibroblast growth factor (FGF) were from Invitrogen (Darmstadt, Germany).
Breast cancer tissue samples and isolation of cancer-associated fibroblasts (CAFs)
This study was approved by the Medical Ethics Committee of Berlin University Medicine. The breast tumor specimens were provided by the Breast Center of University Hospital Charité.
The isolation of CAFs from tumor tissues was carried out according to the method of Sadnolova et al..37 Briefly, a 0.5 cm3 sample of breast tissue was cut from the edge of the tumor following surgical resection. The samples were then placed in Dulbecco's Modified Eagle's Medium (DMEM; Sigma-Aldrich, Taufkirchen, Germany) supplemented with 1% penicillin/streptomycin (P/S, Gibco Invitrogen, Darmstadt, Germany) and 10% fetal bovine serum (FBS, Gibco Invitrogen, Darmstadt, Germany) referred to as basal medium, and immediately transported to the laboratory. The tissue was then finely minced into 1–2 mm3 fragments, washed twice in PBS supplemented with 1% P/S, 1.5 g/ml fungizone (Fischer Scientific, Schwerte, Germany), and disaggregated overnight in DMEM/Ham`s F-12 medium supplemented with 10% FCS, 1% P/S and 2.5 μg/ml Amphotericin, 0.1% collagenase III (Worthington Biochemical Corp., Troisdorf, Germany) at 37°C on a rotator. After 24 h incubation, the epithelial cells were separated from CAFs by differential centrifugation, CAFs were washed twice and plated in 35 mm dishes with DMEM/Ham`s F-12 medium supplemented with 10% FCS, 1% P/S and 2.5 μg/ml fungizone at 37°C in a humidified chamber containing 5% CO2. Media was changed every 2 d. When cells reached confluence they were passaged to a 25 cm2 flask by treating with 0.25% trypsin-25 mM EDTA (Gibco Invitrogen, Darmstadt, Germany) and agitating until cells begin to detach from the surface of the flask (passage 1; p1). P2 cells were moved to a 75 cm2 flask and then passaged 1:4. All experiments were performed with fibroblasts that have been cultured for 3–10 passages.
Cell culture and conditioned media
The human breast cancer cell lines MCF-7, BT-474 and MDA-MB-231 were obtained from European Collection of Cell Cultures (Salisburg, Wiltshire, United Kingdom), and cultured in RPMI-1640 medium supplemented with 10% FBS and 1% P/S (all were purchased from Biochrom KG, Berlin, Germany). Cells in logarithmic growth phase were used for further experiments.
For collection of tumor cell-conditioned media, MDA-MB-231 cells were washed 2 times with PBS and cultured for 4 d in RPMI-1640 medium supplemented with 1% (v/v) P/S. For collection of CAFs-conditioned media, CAFs cells were washed 2 times with PBS and cultured for 4 d in DMEM (low glucose) medium supplemented with 1% (v/v) P/S.
Transwell® invasion assay
Cell invasion assay was performed with BD BioCoatTM Martrigel Invasion Chambers with 8.0 μm pore inserts in 24-well plates (BD Biosciences, Bedford, USA) according to manufacturer's protocol with modifications: 500 μl of serum-free medium, 2 × 104 CAFs, or conditioned media form CAFs, were added respectively in triplicate to the lower compartments of chambers. Inserts were replaced and 1 × 106 MDA-MB-231 cells in 500 μl serum-free media were added into inserts. Chambers were incubated for 18 h in humidified tissue culture incubator. Non-invaded cells were removed from the top surface of the inserts by scrubbing with cotton tip swabs. The cells on the lower surface of the membrane were stained with Hemacolar® staining kit (Millipore, Schwalbach/Ts., Germany) and mounted on slides. The average number of invaded cells per chamber was determined by counting invaded cells from 3 fields of triplicate membranes under the microscope at 40–200 times magnifications depending on cell density.
Measurement of chemokines with ELISA
The production of chemokines CCL2, CCL5 and VEGF in the cell culture media by MDA-MB-231 cells and/or CAFs were measured with ELISA kits Quantikine® Human Rantes (CCL5), human CCL2/MCP-1, or Human VEGF respectively (R&D Systems, Wiesbaden-Nordenstadt, Germany) according to the protocols provided by the manufacturers.
Western blot analysis
Cells were lysed after culture and protein concentrations of whole cell lysates were measured using a BCA protein assay kit (Pierce, Bonn, Germany). Western blot analysis was performed as described previously.38 Antibodies against p-AKT (S473), p-AKT (T308) and AKT were from Cell Signaling Technology (New England Biolabs, Frankfurt am Main, Germany); antibody against ß-Actin was from Santa Cruz Biotechnology (Heidelberg, Germany).
Quantitative reverse-transcription polymerase chain reaction (real time RT-PCR)
Total RNA from either untreated cells or cells treated with Dovitinib was extracted using RNAeasy® mini kit (Qiagen, Hilden, Germany) following the manufacturer's instructions. Two μg total RNA was reverse transcribed in a 25 μl reaction volume using Oligo d(T) primers and M-MLV reverse transcriptase (Gibco Invitrogen, Darmstadt, Germany) according to the protocol of the manufacturer. Real time PCR for CCL2 or CCL5 was performed in triplicates in a 25 μl reaction containing 1 μl cDNA, 500 pmol of each primer by using QuantiFastTM SYBR® Green PCR master Mix Kit (Qiagen, Hilden, Germany). As an endogenous control, GAPDH was also amplified at the same time. The amplification was carried out at the ABI Prism 7700 sequence detection system. The amplification program was as followings: 50°C for 2 minutes, 95°C for 10 minutes, the samples were amplified for 45 cycles at 95°C for 15 seconds, followed by 60°C for 1 minute. All primers were designed and synthesized by TIB MOLBIOL (Berlin, Germany). The house keeper gene GAPDH was used as internal control gene. Sequences of primers used were summarized in Table 1.
Table 1.
Primers and their sequences used in the real-time PCR
| Primer | Sequence |
|---|---|
| PDGFß | 5′- GAG ATG CTG AGT GAC CAC TC-3′ 5′- CGA ATG GTC ACC CGA GTT TG-3′ (542 bp) |
| CCL5 | 5′-ATG TGC TGT ACC AAG GAG TTT-3′ 5′-TGC AGA GGA TCA AGA CAG CA-3′ (225 bp) |
| CCL2 | 5′-CAG CCA GAT GCA ATC AAT GCC-3′ 5′-TGG AAT CCT GAA CCC ACT TCT-3′ (190 bp) |
| GAPDH | 5′-CGG GAA GCT TGT CAT CAA TGG-3′ 5′-CAG TCC ATG CCA TCA CTG CC-3′ (357 bp) |
Statistical analysis
Results of the experimental studies were reported as mean±SD obtained from at least 3 experiments. Student`s t-test for paired samples was used to evaluate the statistical significance.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Reference
- 1. Griffths CL, Olin JL. Triple Negative Breast Cancer: a brief review of its characteristics and treatment options. J Pharm Pract 2012; 25:319-23; PMID:22551559; http://dx.doi.org/ 10.1177/0897190012442062 [DOI] [PubMed] [Google Scholar]
- 2. Gelmon K, Dent R, Mackey JR, Laing K, McLeod D, Verma S. Targeting triple-negative breast cancer: optimising therapeutic outcomes. Ann Oncol 2012; 23:2223-34; PMID:22517820; http://dx.doi.org/ 10.1093/annonc/mds067 [DOI] [PubMed] [Google Scholar]
- 3. Tlsty TD, Coussens LM. Tumor stroma and regulation of cancer development. Annu Rev Pathol 2006; 1:119-50; PMID:18039110; http://dx.doi.org/ 10.1146/annurev.pathol.1.110304.100224 [DOI] [PubMed] [Google Scholar]
- 4. Kim JB, Stein R, O'Hare MJ. Tumour-stromal interactions in breast cancer: the role of stroma in tumourigenesis. Tumour Biol 2005; 26:173-85; PMID:16006771 [DOI] [PubMed] [Google Scholar]
- 5. Wiseman BS, Werb Z. Stromal effects on mammary gland development and breast cancer. Science 2002; 296:1046-9; PMID:12004111; http://dx.doi.org/ 10.1126/science.1067431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mueller MM, Fusenig NE. Friends or foes - bipolar effects of the tumour stroma in cancer. Nat Rev Cancer 2004; 4:839-49; PMID:15516957 [DOI] [PubMed] [Google Scholar]
- 7. Lisanti MP, Martinez-Outschoorn U, Sotgia F. Oncogenes induce the cancer-associated fibroblast phenotype. Cell Cycle 2013; 12:2723-32; PMID:23860382; http://dx.doi.org/ 10.4161/cc.25695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Guido C, Menezes DW, Lin Z, Pestell RG, Howell A, Zimmers TA, Casimiro MC, Aquila S, Ando' S, Martinez-Outschoorn UE, et al. Mitochondrial fission induces glycolytic reprogramming in cancer-associated myofibroblasts, driving stromal lactate production, and early tumor growth. Oncotarget 2012; 3:798-810; PMID:22878233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Aboussekhra A. Role of cancer-associated fibroblasts in breast cancer development and prognosis. Int J Dev Biol 2011; 55:841-9; PMID:22161840; http://dx.doi.org/ 10.1387/ijdb.113362aa [DOI] [PubMed] [Google Scholar]
- 10. Molloy AP, Martin FT, Dwyer RM, Griffin TP, Murphy M, Barry FP, O'Brien T, Kerin MJ. Mesenchymal Sstem cell secretion of chemokines during differentiation into osteoblasts, and their potential role in mediating interactions with breast cancer cells. Int J Cancer 2009; 124:326-32; PMID:19003962; http://dx.doi.org/ 10.1002/ijc.23939 [DOI] [PubMed] [Google Scholar]
- 11. Nam JS, Kang MJ, Suchar AM, Shimamura T, Kohn EA, Michalowska AM, Jordan VC, Hirohashi S, Wakefield LM. Chemokine (C-C motif) ligand 2 mediates the prometastatic effect of dysadherin in human breast cancer cells. Cancer Res 2006; 66:7176-84; PMID:16849564; http://dx.doi.org/ 10.1158/0008-5472.CAN-06-0825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pinilla S, Alt E, Abdul Khalek FJ, Jotzu C, Muehlberg F, Beckmann C, Song YH. Tissue resident stem cells produce CCL5 under the influence of cancer cells and thereby promote breast cancer cell invasion. Cancer Lett 2009; 284:80-5; PMID:19427114; http://dx.doi.org/ 10.1016/j.canlet.2009.04.013 [DOI] [PubMed] [Google Scholar]
- 13. Soria G, Ben-Baruch A. The inflammatory chemokines CCL2 and CCL5 in breast cancer. Cancer Lett 2008; 267:271-85; PMID:18439751; http://dx.doi.org/ 10.1016/j.canlet.2008.03.018 [DOI] [PubMed] [Google Scholar]
- 14. Taeger J, Moser C, Hellerbrand C, Mycielska ME, Glockzin G, Schlitt HJ, Geissler EK, Stoeltzing O, Lang SA. Targeting FGFR/PDGFR/VEGFR impairs tumor growth, angiogenesis, and metastasis by effects on tumor cells, endothelial cells, and pericytes in pancreatic cancer. Mol Cancer Ther 2011; 10:2157-67; PMID:21885862; http://dx.doi.org/ 10.1158/1535-7163.MCT-11-0312 [DOI] [PubMed] [Google Scholar]
- 15. Dey JH, Bianchi F, Voshol J, Bonenfant D, Oakeley EJ, Hynes NE. Targeting fibroblast growth factor receptors blocks PI3K/AKT signaling, induces apoptosis, and impairs mammary tumor outgrowth and metastasis. Cancer Res 2010; 70:4151-62; PMID:20460524; http://dx.doi.org/ 10.1158/0008-5472.CAN-09-4479 [DOI] [PubMed] [Google Scholar]
- 16. Lamont FR, Tomlinson DC, Cooper PA, Shnyder SD, Chester JD, Knowles MA. Small molecule FGF receptor inhibitors block FGFR-dependent urothelial carcinoma growth in vitro and in vivo. Br J Cancer 2011; 104:75-82; PMID:21119661; http://dx.doi.org/ 10.1038/sj.bjc.6606016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pietras K, Pahler J, Bergers G, Hanahan D. Functions of paracrine PDGF signaling in the proangiogenic tumor stroma revealed by pharmacological targeting. PLoS Med 2008; 5:e19; PMID:18232728; http://dx.doi.org/ 10.1371/journal.pmed.0050019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mattila MM, Harkonen PL. Role of fibroblast growth factor 8 in growth and progression of hormonal cancer. Cytokine Growth Factor Rev 2007; 18:257-66; PMID:17512240; http://dx.doi.org/ 10.1016/j.cytogfr.2007.04.010 [DOI] [PubMed] [Google Scholar]
- 19. Reis-Filho JS, Simpson PT, Turner NC, Lambros MB, Jones C, Mackay A, Grigoriadis A, Sarrio D, Savage K, Dexter T, et al. FGFR1 emerges as a potential therapeutic target for lobular breast carcinomas. Clin Cancer Res 2006; 12:6652-62; PMID:17121884 [DOI] [PubMed] [Google Scholar]
- 20. Renhowe PA, Pecchi S, Shafer CM, Machajewski TD, Jazan EM, Taylor C, Antonios-McCrea W, McBride CM, Frazier K, Wiesmann M, et al. Design, structure-activity relationships and in vivo characterization of 4-amino-3-benzimidazol-2-ylhydroquinolin-2-ones: a novel class of receptor tyrosine kinase inhibitors. J Med Chem 2009; 52:278-92; PMID:19113866; http://dx.doi.org/ 10.1021/jm800790t [DOI] [PubMed] [Google Scholar]
- 21. Ehnman M, Östman A. Therapeutic targeting of platelet-derived growth factor receptors in solid tumors. Expert Opin Investig Drugs 2014; 23:211-26; PMID:24206431; http://dx.doi.org/ 10.1517/13543784.2014.847086 [DOI] [PubMed] [Google Scholar]
- 22. Fearon AE, Gould CR, Grose RP. FGFR signalling in women's cancers. Int J Biochem Cell Biol 2013; 45:2832-42; PMID:24148254; http://dx.doi.org/ 10.1016/j.biocel.2013.09.017 [DOI] [PubMed] [Google Scholar]
- 23. Chin YR, Toker A. Function of Akt/PKB signaling to cell motility, invasion and the tumor stroma in cancer. Cell Signal 2009; 21:470-6; PMID:19110052; http://dx.doi.org/ 10.1016/j.cellsig.2008.11.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ebos JM, Lee CR, Cruz-Munoz W, Bjarnason GA, Christensen JG, Kerbel RS. Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis. Cancer Cell 2009; 15:232-9; PMID:19249681; http://dx.doi.org/ 10.1016/j.ccr.2009.01.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Paez-Ribes M, Allen E, Hudock J, Takeda T, Okuyama H, Vinals F, Inoue M, Bergers G, Hanahan D, Casanovas O. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell 2009; 15:220-31; PMID:19249680; http://dx.doi.org/ 10.1016/j.ccr.2009.01.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Huynh H, Chow PK, Tai WM, Choo SP, Chung AY, Ong HS, Linnartz R, Shi MM. Dovitinib demonstrates antitumor and antimetastatic activities in xenograft models of hepatocellular carcinoma. J Hepatol 2012; 56:595-601; PMID:22027573; http://dx.doi.org/ 10.1016/j.jhep.2011.09.017 [DOI] [PubMed] [Google Scholar]
- 27. Ostman A, Augsten M. Cancer-associated fibroblasts and tumor growth–bystanders turning into key players. Curr Opin Genet Dev 2009; 19:67-73; PMID:19211240; http://dx.doi.org/ 10.1016/j.gde.2009.01.003 [DOI] [PubMed] [Google Scholar]
- 28. Pietras K, Pahler J, Bergers G, Hanahan D. Functions of paracrine PDGF signaling in the proangiogenic tumor stroma revealed by pharmacological targeting. PLoS.Med 2008; 5:e19; PMID:18232728; http://dx.doi.org/ 10.1371/journal.pmed.0050019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lopes de Menezes DE, Peng J, Garrett EN, Louie SG, Lee SH, Wiesmann M, Tang Y, Shephard L, Goldbeck C, Oei Y, et al. CHIR-258: a potent inhibitor of FLT3 kinase in experimental tumor xenograft models of human acute myelogenous leukemia. Clin Cancer Res 2005; 11:5281-91; PMID:16033847 [DOI] [PubMed] [Google Scholar]
- 30. Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, Pietenpol JA. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest 2011; 121:2750-67; PMID:21633166; http://dx.doi.org/ 10.1172/JCI45014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mercier I, Camacho J, Titchen K, Gonzales DM, Quann K, Bryant KG, Molchansky A, Milliman JN,Whitaker-Menezes D, Sotgia F, et al. Caveolin-1 and accelerated host aging in the breast tumor microenvironment: chemoprevention with rapamycin, an mTOR inhibitor and anti-aging drug. Am J Pathol 2012; 181:278-93; PMID:22698676; http://dx.doi.org/ 10.1016/j.ajpath.2012.03.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Capparelli C, Chiavarina B, Whitaker-Menezes D, Pestell TG, Pestell RG, Hulit J, Andò S, Howell A, Martinez-Outschoorn UE, Sotgia F, et al. CDK inhibitors (p16/p19/p21) induce senescence and autophagy in cancer-associated fibroblasts, "fueling" tumor growth via paracrine interactions, without an increase in neo-angiogenesis. Cell Cycle 2012; 11:3599-610; PMID:22935696; http://dx.doi.org/ 10.4161/cc.21884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Blagosklonny MV. Rapalogs in cancer prevention: anti-aging or anticancer? Cancer Biol Ther 2012; 13:1349-54; PMID:23151465; http://dx.doi.org/ 10.4161/cbt.22859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Andre F, Bachelot T, Campone M, Dalenc F, Perez-Garcia JM, Hurvitz SA, Turner N, Rugo H, Smith JW, Deudon S, et al. Targeting FGFR with dovitinib (TKI258): preclinical and clinical data in breast cancer. Clin Cancer Res 2013; 19:3693-702; PMID:23658459; http://dx.doi.org/ 10.1158/1078-0432.CCR-13-0190 [DOI] [PubMed] [Google Scholar]
- 35. Issa A, Gill JW, Heideman MR, Sahin O, Wiemann S, Dey JH, Hynes NE. Combinatorial targeting of FGF and ErbB receptors blocks growth and metastatic spread of breast cancer models. Breast Cancer Res 2013; 15:R8; PMID:23343422; http://dx.doi.org/ 10.1186/bcr3379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Anisimov VN, Zabezhinski MA, Popovich IG, Piskunova TS, Semenchenko AV, Tyndyk ML, Yurova MN, Rosenfeld SV, Blagosklonny MV. Rapamycin increases lifespan and inhibits spontaneous tumorigenesis in inbred female mice. Cell Cycle 2011; 10:4230-6; PMID:22107964; http://dx.doi.org/ 10.4161/cc.10.24.18486 [DOI] [PubMed] [Google Scholar]
- 37. Sadlonova A, Novak Z, Johnson MR, Bowe DB, Gault SR, Page GP, Thottassery JV, Welch DR, Frost AR. Breast fibroblasts modulate epithelial cell proliferation in three-dimensional in vitro co-culture. Breast Cancer Res 2005; 7:R46-R59; PMID:15642169; http://dx.doi.org/ 10.1186/bcr949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Liu H, Zang C, Fenner MH, Liu D, Possinger K, Koeffler HP, Elstner E. Growth inhibition and apoptosis in human Philadelphia chromosome-positive lymphoblastic leukemia cell lines by treatment with the dual PPARalpha/gamma ligand TZD18. Blood 2006; 107: 3683-92; PMID:16403907; http://dx.doi.org/ 10.1182/blood-2005-05-2103 [DOI] [PubMed] [Google Scholar]