

Abstract
The hostile tumor microenvironment results in the generation of intracellular stresses including hypoxia and nutrient deprivation. In order to adapt to such conditions, the cell utilizes several stress-response mechanisms, including the attenuation of protein synthesis, the inhibition of cellular proliferation, and induction of autophagy. Autophagy leads to the degradation of cellular contents, including damaged organelles and mutant proteins, which the cell can then use as an alternate energy source. Two integral changes to the signaling milieu to promote such a response include inhibition of the mammalian target of rapamycin complex 1 (mTORC1) and phosphorylation of eIF2α. This review will describe how conditions found in the tumor microenvironment regulate mTORC1 as well as eIF2α, the downstream impact of these modifications, and the implications in tumorigenesis. We will then discuss the remarkable similarities and overlapping function of these 2 signaling pathways, focusing on the response to amino acid deprivation, and present a new model involving crosstalk between them based on our recent work.
Keywords: autophagy, eIF2α, integrated stress response, mTORC1, PP6C, unfolded protein response
Regulation of mTORC1
mTORC1 is a multiprotein complex containing the serine/threonine kinase mammalian target of rapamycin (mTOR), raptor, PRAS40 (proline-rich Akt substrate 40kDa), and mLST8.1-3 Unlike the related rictor-associated mTORC2 complex, mTORC1 is potently sensitive to the macrolide rapamycin and coordinates several signals regarding cellular nutrient status, including glucose, amino acids, and growth factor signaling. mTOR activity determines whether a cell is able to expand in size and divide or needs to conserve energy and induce autophagy (Fig. 1).
Figure 1.

mTORC1 signaling pathway. mTORC1 is a serine/theronine kinase complex that receives information regarding the nutrient status of the cell and orchestrates a pro-survival response to the conditions in the tumor microenvironment. In response to low glucose levels, intracellular AMP concentrations increase, leading to the activation of AMPK, which negatively regulates mTORC1 through activation of the TSC complex. In response to amino acid deprivation, the Rag-GTPases are unable to associate with mTORC1, preventing its activation. mTORC1 inhibition limits cellular energy use by regulating cell size and proliferation through S6K and 4EBP1, respectively. Additionally, mTORC1 inhibition induces autophagy to produce an alternative energy source.
mTORC1 receives information regarding the glucose status of the cell indirectly from the AMP-activated protein kinase (AMPK) and amino acid status of the cell indirectly from the Rag family GTPases. AMPK is a serine/threonine kinase, structured as a heterotrimer consisting of α, β, and γ subunits.4 The α subunit contains the catalytic site as well as an activation loop, which regulates the enzyme through phosphorylation from several kinases, in particular LKB1. The β subunit acts as a hinge to the γ subunit, which contains an adenosine-binding pocket. ATP, ADP, and AMP all fit in the binding pocket, but only AMP causes a conformational change to the enzyme, protecting dephosphorylation of the activation loop in the α subunit and increasing AMPK activity.5 As a consequence of low glucose availability, intracellular ATP levels decrease and AMP levels rise, increasing AMP binding to AMPK.6 Active AMPK phosphorylates several effectors that promote cellular adaptation to glucose deprivation, one of which is TSC2 (tuberin), a member of tuberous sclerosis complex (TSC). TSC2 heterodimerizes with TSC1 (hamartin) and functions as a GAP for the small GTPase Rheb (Ras homolog enriched in the brain). As with other small GTPases, Rheb cycles between a GTP-loaded “on” state and a GDP-loaded “off” state.7 Rheb has been shown to directly bind to mTORC1 and promote its activity when GTP-loaded.8
Many signaling pathways, including the Akt and MAPK pathways, negatively regulate TSC activity by phosphorylating TSC2 and recruiting the protein 14-3-3, which disrupts its binding to Rheb.9 Additionally, hypoxia promotes TSC activity through the regulated in development and DNA damage 1 (REDD1) protein, which is transcriptionally upregulated by HIF1 and dissociates 14-3-3 from TSC2.10 In summary, in response to glucose deprivation, AMPK acts a negative regulator of mTORC1 by inducing GAP activity of TSC2 and deactivating Rheb.
However, mTORC1 is best recognized to be a fine sensor of intracellular amino acid status. The Rag GTPases exist as heterodimers of RagA or RagB with RagC or RagD. In the presence of amino acids, RagA and RagB are GTP-loaded by a multiprotein complex on the lysosomal membrane known as the ragulator.11,12 Additionally, leucyl-tRNA synthetase may also have a noncanonical function by acting as a GAP that promotes the hydrolysis of GTP-bound RagD.13 Rag dimers of GTP-loaded RagA/B and GDP-loaded RagC/D directly bind to the mTORC1 component raptor and translocate the mTORC1 complex to the lysosomal membrane, where Rheb has recently been found to localize.14 Therefore, in the presence of abundant amino acids and ATP, the active Rag complex localizes mTORC1 to active, GTP-bound Rheb on the lysosomal membrane, where it activates the kinase activity of mTOR. Conversely, amino acid deprivation profoundly decreases mTORC1 activity.
mTORC1 downstream pathways and cancer
Because of insufficient and aberrant vascularization in tumors, including leaky vessels, blind loops, high interstitial pressures, tumors often exist in a microenvironment marked by profound hypoxia and nutrient deprivation (reviewed in15). mTORC1 is integral for adaptation to the tumor microenvironment by assessing the conditions of the tumor microenvironment and regulating protein synthesis, cell size, proliferation, and autophagy. In fact, TSC1 and TSC2, which negatively regulate mTORC1 activity, were identified as tumor suppressor genes. Mutations in TSC1 or TSC2 cause the autosomal dominant tuberous sclerosis complex, a multi-system disease characterized with the formation of benign tumors including hamartomas and angiomyolipomas.16 Additionally, several cancers demonstrate markedly increased mTORC1 activity due to mutations in upstream regulators of mTORC1, including PTEN inactivation which leads to the subsequent activation of the PI3K/Akt axis.17 Interestingly, similar to TSC1/TSC2, PTEN is also mutated in a number of hamartoma syndromes.18
Because of a rapid proliferation rate, neoplastic cells are thought to be dependent on high rates of protein synthesis. mTORC1 regulates protein synthesis and proliferation through phosphorylation and inactivation of eukaryotic translation initiation factor 4E-binding protein 1 (4EBP1). 4EBP1 negatively regulates cap-dependent protein synthesis by competing with eukaryotic translation initiation factor 4G (eIF4G) for binding to the eukaryotic translation initiation factor 4E (eIF4E) which is rate limiting for translation. eIF4E directly binds to the 5′ mRNA cap and recruits eukaryotic translation initiation factor 4G (eIF4G) to mRNA, which acts as a scaffold for the pre-initiation complex. Phosphorylation of 4EBP1 causes its sequestration, increasing eIF4G binding to eIF4E, which promotes the formation of the pre-initiation complex and increases the rate of protein synthesis. Notably, phosphorylation of 4EBP1 also promotes cellular proliferation by augmenting the translation of mRNAs involved in cell cycle progression.19 In the absence of mTORC1 activity protein synthesis is markedly suppressed.
Similar to 4EBP1, phosphorylated S6 kinase (S6K) has several substrates that increase protein synthesis, such as the eukaryotic initiation factor 4B (eIF4B) and PDCD4. Phosphorylated eIF4B is recruited to the eIF4F translation pre-initiation complex, where it enhances the helicase activity of eIF4A, which is necessary for cap-dependent protein translation.20 Additionally, phosphorylated PDCD4, which inhibits eukaryotic translation initiation factor 4G (eIF4A) activity by preventing it from binding to the pre-initiation complex, is ubiquinated by β-TrCP and targeted for degradation.21,22 Notably, S6K also promotes an increase in cell size through phosphorylation of the ribosomal protein S6 and SKAR, a scaffolding protein that recruits S6K to the EJC on spliced mRNAs.23,24
Another critical pathway regulated by mTORC1 is macroautophagy (referred to as autophagy), a process involving the formation of double membrane vesicles that surround cytoplasmic contents and fuse with the lysosome for degradation, which provides a source of macromolecules during periods of starvation, as well as acts as a surveillance mechanism to rid the cells of potentially harmful proteins.25 Although its role in cancer is complicated, autophagy is a stress survival mechanism that appears to be important for the growth of many established tumors.26 In melanoma, for example, increased autophagy is noted in areas of deep invading tumors, and high rates of autophagy seem to correlate with poor patient prognosis27,28 suggesting that autophagy serves as a pro-survival mechanism which allows melanomas to adapt to a hostile microenvironment.28,29 In recently described “canonical” mTORC1 signaling, mTORC1 phosphorylates the proteins ULK1 and ATG13.30 Of note, ULK1 can also be directly phosphorylated by AMPK at a site distinct from mTORC1.31,32 When mTORC1 is inhibited, dephosphorylated ULK1 forms a complex with ATG13 and FIP200. A class III phosphoinositide-3-kinase complex, which includes the PI3K known as vacuolar protein sorting associated (VPS) 34, AMBRA1, and Beclin 1, then assembles on the ULK1/ATG13/FIP200 scaffold. ULK1 phosphorylates AMBRA1, which dissociates the VPS34 complex from the cytoskeleton, leading to its relocalization to the ER.33 The subsequent phosphatidylinositol 3-phosphate (PI3P) generated on the ER membrane then recruits a number of autophagy proteins involved in assembly of membranes.34,35 However, as discussed later, this model for mTORC1 regulation of autophagy is incomplete.
Regulation of eIF2α phosphorylation
Similar to the mTORC1 signaling pathway, the integrated stress response system coordinates several cues regarding the metabolic status of a cell and orchestrates an adaptive response through the phosphorylation of the α subunit of the eukaryotic initiation factor 2 (eIF2α) (Fig. 2). eIF2 is a GTPase that forms a ternary complex with GTP and Met-tRNA and assembles with the 40S ribosomal subunit, forming the 43S pre-initation complex. The 43S pre-initiation complex then scans down the mRNA until it base pairs with the start codon on the mRNA, where the GTPase activating protein (GAP) eIF5 then mediates the hydrolysis of GTP-bound eIF2, depositing the Met-tRNA at the P site on the ribosome.36,37 GDP-bound eIF2 then exits the ribosome where the guanine nucleotide exchange factor (GEF) eIF2B exchanges GDP with GTP, enabling eIF2 to bind to Met-tRNA and be used for another round of translation initiation.38 Phosphorylation of eIF2α at serine 51 causes eIF2 to act as a competitive inhibitor of its GEF activity, preventing the recycling of eIF2 and attenuating translation initiation.39
Figure 2.

The integrated stress response. eIF2α is phosphorylated by kinases that are activated in response to particular stresses. Amino acid deprivation activates GCN2, accumulated proteins in the ER activates PERK, single stranded RNA viruses activate PKR, and low heme concentrations activate HRI. Phosphorylated eIF2α leads to a decrease in cap-dependent protein synthesis and increases the transcripts containing upstream open reading frames (uORFS) such as the stress responsive transcription factor ATF4. Phosphorylated eIF2α has also been in inducing autophagy in response to amino acid deprivation.
eIF2α phosphorylation is regulated by 4 specific kinases that sense various cellular stresses. Two of the kinases, protein kinase R (PKR) and heme regulated inhibitor (HRI) kinase, have a specific role in phosphorylating eIF2α in response to dsRNA viral infections and low heme, respectively.40,41 One kinase that regulates eIF2α phosphorylation in response to conditions found in the tumor microenvironment is the PKR-like endoplasmic reticulum kinase (PERK). PERK becomes activated in response to increased protein burden in the endoplasmic reticulum, which occurs in hypoxic cells and in cells generating copious secretory proteins such as myeloma, in glucose deprivation, and with high Myc expression.42-47
In addition to PERK, the serine/threonine kinase general control nonderepressible 2 (GCN2) plays an integral role in transmitting nutrient status to eIF2α. GCN2 senses intracellular amino acid levels, and in the absence of amino acids phosphorylates eIF2α at serine 51. In Saccharomyces cerevisiae, the GCN2 homodimer contains a C-terminal HisRS domain with a sequence similar to histidyl-tRNA synthetase that binds to uncharged tRNAs.48 In the absence of uncharged tRNA binding, the C-terminal domain (CTD) adjacent to the HisRS binds to and inhibits the protein kinase (PK) domain.49 When cells are deprived of amino acids, the subsequent increase of uncharged tRNAs increases the probability of binding to GCN2, which disrupts the interaction between the CTD and PK domains. The PK domains are then free to autophosphorylate at threonine 887, which is critical for the full activation of the kinase.50 Such a mechanism has not yet been elucidated in mammalian systems.
Pathways activated by eIF2α and cancer
In addition to globally suppressing protein synthesis, eIF2α phosphorylation paradoxically enhances the translation of mRNAs containing upstream open reading frames (uORFs) in particular the transcription factor ATF4.51,52 ATF4 contains 2 uORFs that are both translated under basal conditions, due to efficient scanning and re-initiation of the 43S complex, leading to minimal translation of the ATF4 open reading frame. When eIF2α is phosphorylated, the upstream uORF is translated, but due to decreased efficiency in 43S turnover, the second uORF is skipped and translation increases at the ATF4 ORF.53
eIF2α plays a pivotal role in tumorigenesis by integrating the conditions of the tumor microenvironment and coordinating an adaptive response. In fact, histological staining has demonstrated that eIF2α is phosphorylated throughout several tumors.54,55 Additionally, eIF2α phosphorylation not only is necessary as an adaptive response to hypoxia and nutrient deprivation, but also promotes tumorigenesis.54-56 When expressed, ATF4 induces a number of genes involved in protein folding, amino acid metabolism, and reactive oxygen species response, and autophagy.57 In addition, eIF2α phosphorylation has been shown to lead to the inhibition of a cellular surveillance system known as nonsense mediated mRNA decay (NMD), which targets and degrades aberrantly expressed mRNA isoforms.54 Interestingly, NMD also targets a subset of endogenous transcripts for degradation including ATF4, implicating its role in coordinating the cellular response to stress.58
Unlike mTORC1, the exact mechanism by which eIF2α phosphorylation regulates autophagy is less delineated. However, our group has also demonstrated that inhibition of NMD in response to eIF2α phosphorylation induces autophagy in a mechanism that seems partly dependent on ATF4 stabilization.59 Additionally, ATF4 has been demonstrated to upregulate the expression of ATG5 and LC3B, both proteins involved in the elongation phase of autophagy.57,60 Interestingly, in S. cerevisiae, the ATF4 homolog GCN4 upregulates the expression of ATG1 and ATG13, homologs of the mTORC1 substrates ULK1 and ATG13, respectively.61
Links between the mTOR and eIF2α pathways
Although thought of as separate and independent pathways, mTORC1 signaling and the integrated stress response orchestrate remarkably similar adaptive mechanisms in response to the conditions of the tumor microenvironment. As mentioned above, glucose deprivation, ROS, amino acid deprivation, and hypoxia lead to the inhibition of mTORC1 and the phosphorylation of eIF2α, all though through separate upstream signaling networks. Additionally, eIF2α phosphorylation and mTORC1 inhibition both lead to the general attenuation of cap-dependent protein synthesis. Furthermore, eIF2α and mTORC1 co-coordinately regulate the translation of mRNAs which contain hallmark 5′ terminal oligopyrimidine tracts (5′TOPS), cis elements containing a cytosine followed by a stretch of 4 to 15 pyrimidines.62 The disruption of the pre-initiation complex in response to mTORC1 inhibition and eIF2α phosphorylation allows the proteins TIA-1 and TIAR to bind to 5′TOPS, causing dissociation of the mRNA from polysomes and subsequent localization to stress granules.63 Interestingly, 5′TOPS are unique to mRNAs encoding genes involved in protein synthesis, further enhancing the effects on translation attenuation in response to mTORC1 inhibition and eIF2α phosphorylation.64,65
As mentioned previously, both mTORC1 inhibition and eIF2α phosphorylation also have been demonstrated to induce autophagy. There are suggestions that in fact eIF2α phosphorylation plays a critical role in autophagy, as it has been reported that eIF2α phosphorylation is necessary for the induction of autophagy in response to amino acid deprivation.66 The observation that autophagy in response to amino acid deprivation requires the phosphorylation of eIF2α suggests that inhibition of mTORC1 and the canonical inhibition of ULK1/ULK2 phosphorylation is not sufficient for autophagy. While the coordinated inhibition of mTORC1 and phosphorylation of eIF2α are necessary for autophagy in response to amino acid deprivation, both pathways have been thought of as independent.57,59-61,66
However, the remarkably similar adaptive responses coordinated by mTORC1 and eIF2α signaling suggest potential crosstalk between these 2 important nutrient sensing pathways. Interestingly, the eIF2α effector ATF4 increases the expression of 4EBP1, augmenting 4EBP1 disinhibition in response to mTORC1 inhibition, indicating that eIF2α phosphorylation can affect mTORC1 function.67
In addition, TOR has been demonstrated to directly regulate eIF2α phosphorylation through GCN2 activation in S. cerevisiae.68 Specifically, TOR inhibits the serine threonine phosphatase SIT4 by enhancing its interaction with an inhibitory protein TAP42. In response to TOR inhibition, SIT4 becomes active and dephosphorylates GCN2 at serine 577 in the PK domain, which enhances binding of uncharged tRNAs to the HisRS domain.69
The human homologs of SIT4 and TAP42 have been identified as PP6C and IGBP/α4, respectively.70,71 PP6C is the catalytic subunit of the PP6 phosphatase, a member of the okadaic acid-sensitive PP2A family of serine/threonine phosphatases that include PP2A, PP4, and PP6.72 PP6C has been implicated in regulating several cellular process including DNA double-strand break repair, mitotic spindle formation, and inflammation.73,74 PP6C obtains its substrate specificity through binding to one of 3 regulatory subunits: PP6R1, PP6R2, and PP6R3.75 Our group recently identified a signaling pathway that communicates between mTORC1 and eIF2α.76 Specifically, we showed that inhibition of mTORC1 leads to activation of GCN2 and phosphorylation eIF2α in a mechanism dependent on the PP6C phosphatase (Fig. 3). Thus, depletion of PP6C, inhibition of PP6C through over-expression of IGBP/α4, deficiency of GCN2, or inability to phosphorylate eIF2α (due to eIF2α S51A/S51A knock-in alleles) resulted to blunted autophagy in response to mTORC1 inhibition. The finding that the PP6C/GCN2/eIF2α pathway is necessary for mTORC1 regulated autophagy is reinforced by our observation that in cells with constitutive activation of mTORC1 via loss of TSC, blunting eIF2α phosphorylation reduced basal autophagic activity. Furthermore, cells deficient of both ULK1 and ULK2 had increased, autophagy in response to rapamycin, albeit to a lesser degree than wild-type cells, that was blunted when eIF2α phosphorylation was reduced.76
Figure 3.
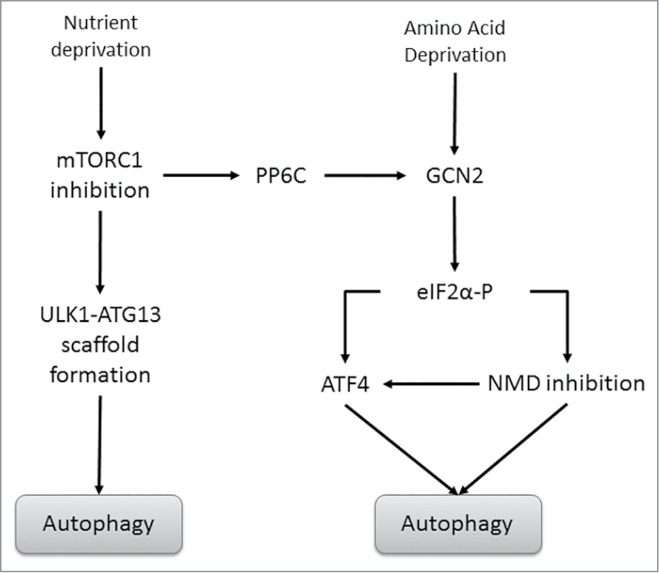
Canonical and non-canonical mTORC1-mediated regulation of autophagy. Under periods of nutrient deprivation, mTORC1 inhibition enables the formation of the ULK1/ATG13 scaffold complex, which allows for the initial nucleation steps of autophagy to occur. mTORC1 inhibition also activates the phosphatase PP6C. PP6C then associates with GCN2 in a complex with a PP6 regulatory protein, promoting its dephosphorylation and activation. Activated GCN2 then leads to the phosphorylation of eIF2α and induction of autophagy.
Interestingly, 2 independent groups sequenced the exomes of melanoma samples and identified PP6C as the driver mutation in approximately 10% of melanomas.77,78 A subsequent study indicated that specific mutations are prognostic in this disease.74 We found that several of the PP6C driver mutants found in melanoma cause disruption with the PP6C regulatory proteins and decrease their protein stability. Paradoxically these mutants stabilize wild-type PP6C and promote eIF2α phosphorylation and autophagy.
Together these findings definitively place eIF2α phosphorylation downstream of mTORC1 inhibition, and link 2 important mechanisms for sensing intracellular amino acid status. Remarkably our studies demonstrate that eIF2α phosphorylation in response to nutrient deprivation is necessary for induction of autophagy. We also suggest a novel function of PP6C in regulating autophagy, a role that may be hijacked in melanoma to promote tumorigenesis.
Implications of mTORC1 and eIF2α interactions
The effect of active mTOR in promoting cell growth and proliferation has led to mTOR inhibition as an approved therapy in renal cell cancer and breast cancer. However in general, mTOR inhibition has suboptimal effects in clinical trials. While there are several potential reasons for these negative trials, one hypothesis is that the concomitant induction of autophagy with rapamycin or rapalogs serve as a pro-survival mechanism for 3 dimensional tumors undergoing metabolic stress.79 Thus, a strategy to maintain the anti-tumor effects of mTOR inhibition but avoid the induction of autophagy is an attractive approach.
Indeed, even in the absence of blunting autophagy induction with mTOR inhibitors, the pharmacological inhibition of autophagy is a potential anti-cancer strategy. Over 40 clinical trials are actively testing the autophagy inhibitor chloroquine either by itself or in combination with chemotherapy. To date, these combinations have not demonstrated robust clinical activity. However chloroquine and its analogs are non-specific inhibitors of lysosomal acidification and late inhibitors of autophagy, often stimulating autophagosome biosynthesis.80 In addition, high concentrations of chloroquine are required to inhibit autophagy, and these may be difficult to achieve clinically.81 Other autophagy inhibitors identified in high-throughput screens, including PIK3C3 inhibitors, have multiple effects, and ATG4B and ATG7 inhibitors are in their infancy. There are currently no commercially available inhibitors of ULK1/2.
Our observation that inhibition of mTORC1 and disruption of canonical mTORC1 signaling through ULK1 is not sufficient to signal autophagy suggests there may be novel mechanisms to disrupt autophagy. The recent understanding of the link between mTORC1 and eIF2α phosphorylation suggests that pharmacological inhibition of PP6C, GCN2, or eIF2α should block autophagy and potentially synergize with mTOR inhibitors to promote cell death. Of note, small-molecule screens have identified inhibitors of GCN2, but their ability to modulate autophagy have not yet been investigated.82 Targeting PP6C would likely be more of a challenge as the protein lacks substrate specificity and phosphatase inhibitors tend to be very non-specific.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Hara K, Maruki Y, Long X, Yoshino K, Oshiro N, Hidayat S, Tokunaga C, Avruch J, Yonezawa K. Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell 2002; 110:177–89; PMID:12150926; http://dx.doi.org/ 10.1016/S0092-8674(02)00833-4 [DOI] [PubMed] [Google Scholar]
- 2.Kim DH, Sarbassov DD, Ali SM, Latek RR, Guntur KV, Erdjument-Bromage H, Tempst P, Sabatini DM. GbetaL, a positive regulator of the rapamycin-sensitive pathway required for the nutrient-sensitive interaction between raptor and mTOR. Mol cell 2003; 11:895–904; PMID:12718876; http://dx.doi.org/ 10.1016/S1097-2765(03)00114-X [DOI] [PubMed] [Google Scholar]
- 3.Vander Haar E, Lee SI, Bandhakavi S, Griffin TJ, Kim DH. Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40. Nat Cell Biol 2007; 9:316–23; PMID:17277771; http://dx.doi.org/ 10.1038/ncb1547 [DOI] [PubMed] [Google Scholar]
- 4.Hardie DG, Carling D, Carlson M. The AMP-activated/SNF1 protein kinase subfamily: metabolic sensors of the eukaryotic cell? Ann Rev Biochem 1998; 67:821–55; PMID:9759505; http://dx.doi.org/ 10.1146/annurev.biochem.67.1.821 [DOI] [PubMed] [Google Scholar]
- 5.Oakhill JS, Scott JW, Kemp BE. Structure and function of AMP-activated protein kinase. Acta Physiol 2009; 196:3–14; PMID:19245650; http://dx.doi.org/ 10.1111/j.1748-1716.2009.01977.x [DOI] [PubMed] [Google Scholar]
- 6.Xiao B, Sanders MJ, Underwood E, Heath R, Mayer FV, Carmena D, Jing C, Walker PA, Eccleston JF, Haire LF, et al.. Structure of mammalian AMPK and its regulation by ADP. Nature 2011; 472:230–3; PMID:21399626; http://dx.doi.org/ 10.1038/nature09932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yamagata K, Sanders LK, Kaufmann WE, Yee W, Barnes CA, Nathans D, Worley PF. rheb, a growth factor- and synaptic activity-regulated gene, encodes a novel Ras-related protein. J Biol Chem 1994; 269:16333–9; PMID:8206940 [PubMed] [Google Scholar]
- 8.Long X, Lin Y, Ortiz-Vega S, Yonezawa K, Avruch J. Rheb binds and regulates the mTOR kinase. Curr Biol 2005; 15:702–13; PMID:15854902; http://dx.doi.org/ 10.1016/j.cub.2005.02.053 [DOI] [PubMed] [Google Scholar]
- 9.Cai SL, Tee AR, Short JD, Bergeron JM, Kim J, Shen J, Guo R, Johnson CL, Kiguchi K, Walker CL. Activity of TSC2 is inhibited by AKT-mediated phosphorylation and membrane partitioning. J Cell Biol 2006; 173:279–89; PMID:16636147; http://dx.doi.org/ 10.1083/jcb.200507119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.DeYoung MP, Horak P, Sofer A, Sgroi D, Ellisen LW. Hypoxia regulates TSC1/2-mTOR signaling and tumor suppression through REDD1-mediated 14-3-3 shuttling. Genes Dev 2008; 22:239–51; PMID:18198340; http://dx.doi.org/ 10.1101/gad.1617608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bar-Peled L, Schweitzer LD, Zoncu R, Sabatini DM. Ragulator is a GEF for the rag GTPases that signal amino acid levels to mTORC1. Cell 2012; 150:1196–208; PMID:22980980; http://dx.doi.org/ 10.1016/j.cell.2012.07.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sekiguchi T, Hirose E, Nakashima N, Ii M, Nishimoto T. Novel G proteins, Rag C and Rag D, interact with GTP-binding proteins, Rag A and Rag B. J Biol Chem 2001; 276:7246–57; PMID:11073942; http://dx.doi.org/ 10.1074/jbc.M004389200 [DOI] [PubMed] [Google Scholar]
- 13.Han JM, Jeong SJ, Park MC, Kim G, Kwon NH, Kim HK, Ha SH, Ryu SH, Kim S. Leucyl-tRNA synthetase is an intracellular leucine sensor for the mTORC1-signaling pathway. Cell 2012; 149:410–24; PMID:22424946; http://dx.doi.org/ 10.1016/j.cell.2012.02.044 [DOI] [PubMed] [Google Scholar]
- 14.Sancak Y, Bar-Peled L, Zoncu R, Markhard AL, Nada S, Sabatini DM. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 2010; 141:290–303; PMID:20381137; http://dx.doi.org/ 10.1016/j.cell.2010.02.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gardner LB, Corn PG. Hypoxic regulation of mRNA expression. Cell Cycle 2008; 7:1916–24; PMID:18604161; http://dx.doi.org/ 10.4161/cc.7.13.6203 [DOI] [PubMed] [Google Scholar]
- 16.Rakowski SK, Winterkorn EB, Paul E, Steele DJ, Halpern EF, Thiele EA. Renal manifestations of tuberous sclerosis complex: Incidence, prognosis, and predictive factors. Kidney Int 2006; 70:1777–82; PMID:17003820; http://dx.doi.org/ 10.1038/sj.ki.5001853 [DOI] [PubMed] [Google Scholar]
- 17.Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer 2002; 2:489–501; PMID:12094235; http://dx.doi.org/ 10.1038/nrc839 [DOI] [PubMed] [Google Scholar]
- 18.Inoki K, Corradetti MN, Guan KL. Dysregulation of the TSC-mTOR pathway in human disease. Nat Genet 2005; 37:19–24; PMID:15624019; http://dx.doi.org/ 10.1038/ng1494 [DOI] [PubMed] [Google Scholar]
- 19.Dowling RJ, Topisirovic I, Alain T, Bidinosti M, Fonseca BD, Petroulakis E, Wang X, Larsson O, Selvaraj A, Liu Y, et al.. mTORC1-mediated cell proliferation, but not cell growth, controlled by the 4E-BPs. Science 2010; 328:1172–6; PMID:20508131; http://dx.doi.org/ 10.1126/science.1187532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Raught B, Peiretti F, Gingras AC, Livingstone M, Shahbazian D, Mayeur GL, Polakiewicz RD, Sonenberg N, Hershey JW. Phosphorylation of eucaryotic translation initiation factor 4B Ser422 is modulated by S6 kinases. EMBO J 2004; 23:1761–9; PMID:15071500; http://dx.doi.org/ 10.1038/sj.emboj.7600193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dorrello NV, Peschiaroli A, Guardavaccaro D, Colburn NH, Sherman NE, Pagano M. S6K1- and betaTRCP-mediated degradation of PDCD4 promotes protein translation and cell growth. Science 2006; 314:467–71; PMID:17053147; http://dx.doi.org/ 10.1126/science.1130276 [DOI] [PubMed] [Google Scholar]
- 22.Yang HS, Cho MH, Zakowicz H, Hegamyer G, Sonenberg N, Colburn NH. A novel function of the MA-3 domains in transformation and translation suppressor Pdcd4 is essential for its binding to eukaryotic translation initiation factor 4A. Mol Cel Biol 2004; 24:3894–906; PMID:15082783; http://dx.doi.org/ 10.1128/MCB.24.9.3894-3906.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ma XM, Yoon SO, Richardson CJ, Julich K, Blenis J. SKAR links pre-mRNA splicing to mTOR/S6K1-mediated enhanced translation efficiency of spliced mRNAs. Cell 2008; 133:303–13; PMID:18423201; http://dx.doi.org/ 10.1016/j.cell.2008.02.031 [DOI] [PubMed] [Google Scholar]
- 24.Ruvinsky I, Sharon N, Lerer T, Cohen H, Stolovich-Rain M, Nir T, Dor Y, Zisman P, Meyuhas O. Ribosomal protein S6 phosphorylation is a determinant of cell size and glucose homeostasis. Genes Dev 2005; 19:2199–211; PMID:16166381; http://dx.doi.org/ 10.1101/gad.351605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yorimitsu T, Klionsky DJ. Autophagy: molecular machinery for self-eating. Cell Death Differ 2005; 12(Suppl 2):1542–52; PMID:16247502; http://dx.doi.org/ 10.1038/sj.cdd.4401765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Degenhardt K, Mathew R, Beaudoin B, Bray K, Anderson D, Chen G, Mukherjee C, Shi Y, Gelinas C, Fan Y, et al.. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006; 10:51–64; PMID:16843265; http://dx.doi.org/ 10.1016/j.ccr.2006.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ma XH, Piao S, Wang D, McAfee QW, Nathanson KL, Lum JJ, Li LZ, Amaravadi RK. Measurements of tumor cell autophagy predict invasiveness, resistance to chemotherapy, and survival in melanoma. Clin Cancer Res 2011; 17:3478–89; PMID:21325076; http://dx.doi.org/ 10.1158/1078-0432.CCR-10-2372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sivridis E, Giatromanolaki A, Liberis V, Koukourakis MI. Autophagy in endometrial carcinomas and prognostic relevance of 'stone-like' structures (SLS): what is destined for the atypical endometrial hyperplasia? Autophagy 2011; 7:74–82; PMID:21099253; http://dx.doi.org/ 10.4161/auto.7.1.13947 [DOI] [PubMed] [Google Scholar]
- 29.Lazova R, Klump V, Pawelek J. Autophagy in cutaneous malignant melanoma. J Cutan Pathol 2010; 37:256–68; PMID:19615007; http://dx.doi.org/ 10.1111/j.1600-0560.2009.01359.x [DOI] [PubMed] [Google Scholar]
- 30.Ganley IG, Lam H, Wang J, Ding X, Chen S, Jiang X. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J Biol Chem 2009; 284:12297–305; PMID:19258318; http://dx.doi.org/ 10.1074/jbc.M900573200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, Vasquez DS, Joshi A, Gwinn DM, Taylor R, et al.. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 2011; 331:456–61; PMID:21205641; http://dx.doi.org/ 10.1126/science.1196371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 2011; 13:132–41; PMID:21258367; http://dx.doi.org/ 10.1038/ncb2152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Di Bartolomeo S, Corazzari M, Nazio F, Oliverio S, Lisi G, Antonioli M, Pagliarini V, Matteoni S, Fuoco C, Giunta L, et al.. The dynamic interaction of AMBRA1 with the dynein motor complex regulates mammalian autophagy. The J Cell Biol 2010; 191:155–68; PMID:20921139; http://dx.doi.org/ 10.1083/jcb.201002100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Axe EL, Walker SA, Manifava M, Chandra P, Roderick HL, Habermann A, Griffiths G, Ktistakis NT. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J Cell Biol 2008; 182:685–701; PMID:18725538; http://dx.doi.org/ 10.1083/jcb.200803137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Matsunaga K, Morita E, Saitoh T, Akira S, Ktistakis NT, Izumi T, Noda T, Yoshimori T. Autophagy requires endoplasmic reticulum targeting of the PI3-kinase complex via Atg14L. J Cell Biol 2010; 190:511–21; PMID:20713597; http://dx.doi.org/ 10.1083/jcb.200911141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chakrabarti A, Maitra U. Function of eukaryotic initiation factor 5 in the formation of an 80 S ribosomal polypeptide chain initiation complex. J Biol Chem 1991; 266:14039–45; PMID:1856230 [PubMed] [Google Scholar]
- 37.Paulin FE, Campbell LE, O'Brien K, Loughlin J, Proud CG. Eukaryotic translation initiation factor 5 (eIF5) acts as a classical GTPase-activator protein. Curr Biol 2001; 11:55–9; PMID:11166181; http://dx.doi.org/ 10.1016/S0960-9822(00)00025-7 [DOI] [PubMed] [Google Scholar]
- 38.Webb BL, Proud CG. Eukaryotic initiation factor 2B (eIF2B). Int J Biochem Cell Biol 1997; 29:1127–31; PMID:9438375; http://dx.doi.org/ 10.1016/S1357-2725(97)00039-3 [DOI] [PubMed] [Google Scholar]
- 39.Pavitt GD, Ramaiah KV, Kimball SR, Hinnebusch AG. eIF2 independently binds two distinct eIF2B subcomplexes that catalyze and regulate guanine-nucleotide exchange. Genes Dev 1998; 12:514–26; PMID:9472020; http://dx.doi.org/ 10.1101/gad.12.4.514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hovanessian AG. The double stranded RNA-activated protein kinase induced by interferon: dsRNA-PK. J Interferon Res 1989; 9:641–7; PMID:2481698; http://dx.doi.org/ 10.1089/jir.1989.9.641 [DOI] [PubMed] [Google Scholar]
- 41.Hunt T, Vanderhoff G, London IM. Control of globin synthesis: the role of heme. J Mol Biol 1972; 66:471–81; PMID:5037023; http://dx.doi.org/ 10.1016/0022-2836(72)90427-5 [DOI] [PubMed] [Google Scholar]
- 42.Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat Cell Biol 2000; 2:326–32; PMID:10854322; http://dx.doi.org/ 10.1038/35014014 [DOI] [PubMed] [Google Scholar]
- 43.Liu L, Wise DR, Diehl JA, Simon MC. Hypoxic reactive oxygen species regulate the integrated stress response and cell survival. J Biol Chem 2008; 283:31153–62; PMID:18768473; http://dx.doi.org/ 10.1074/jbc.M805056200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Scheuner D, Song B, McEwen E, Liu C, Laybutt R, Gillespie P, Saunders T, Bonner-Weir S, Kaufman RJ. Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Mol Cell 2001; 7:1165–76; PMID:11430820; http://dx.doi.org/ 10.1016/S1097-2765(01)00265-9 [DOI] [PubMed] [Google Scholar]
- 45.Tu BP, Ho-Schleyer SC, Travers KJ, Weissman JS. Biochemical basis of oxidative protein folding in the endoplasmic reticulum. Science 2000; 290:1571–4; PMID:11090354; http://dx.doi.org/ 10.1126/science.290.5496.1571 [DOI] [PubMed] [Google Scholar]
- 46.Hart LS, Cunningham JT, Datta T, Dey S, Tameire F, Lehman SL, Qiu B, Zhang H, Cerniglia G, Bi M, et al.. ER stress-mediated autophagy promotes Myc-dependent transformation and tumor growth. J Clin Invest 2012; 122:4621–34; PMID:23143306; http://dx.doi.org/ 10.1172/JCI62973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang D, Wengrod J, Gardner LB. Overexpression of the c-myc oncogene inhibits nonsense-mediated RNA decay in B lymphocytes. J Biol Chem 2011; 286:40038–43; PMID:21969377; http://dx.doi.org/ 10.1074/jbc.M111.266361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wek RC, Jackson BM, Hinnebusch AG. Juxtaposition of domains homologous to protein kinases and histidyl-tRNA synthetases in GCN2 protein suggests a mechanism for coupling GCN4 expression to amino acid availability. Proc Natl Acad Sci U S A 1989; 86:4579–83; PMID:2660141; http://dx.doi.org/ 10.1073/pnas.86.12.4579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dong J, Qiu H, Garcia-Barrio M, Anderson J, Hinnebusch AG. Uncharged tRNA activates GCN2 by displacing the protein kinase moiety from a bipartite tRNA-binding domain. Mol Cell 2000; 6:269–79; PMID:10983975; http://dx.doi.org/ 10.1016/S1097-2765(00)00028-9 [DOI] [PubMed] [Google Scholar]
- 50.Romano PR, Garcia-Barrio MT, Zhang X, Wang Q, Taylor DR, Zhang F, Herring C, Mathews MB, Qin J, Hinnebusch AG. Autophosphorylation in the activation loop is required for full kinase activity in vivo of human and yeast eukaryotic initiation factor 2alpha kinases PKR and GCN2. Mol Cell Biol 1998; 18:2282–97; PMID:9528799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, Ron D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell 2000; 6:1099–108; PMID:11106749; http://dx.doi.org/ 10.1016/S1097-2765(00)00108-8 [DOI] [PubMed] [Google Scholar]
- 52.Kozak M. Pushing the limits of the scanning mechanism for initiation of translation. Gene 2002; 299:1–34; PMID:12459250; http://dx.doi.org/ 10.1016/S0378-1119(02)01056-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lu PD, Harding HP, Ron D. Translation reinitiation at alternative open reading frames regulates gene expression in an integrated stress response. J Cell Biol 2004; 167:27–33; PMID:15479734; http://dx.doi.org/ 10.1083/jcb.200408003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang D, Zavadil J, Martin L, Parisi F, Friedman E, Levy D, Harding H, Ron D, Gardner LB. Inhibition of nonsense-mediated RNA decay by the tumor microenvironment promotes tumorigenesis. Mol Cell Biol 2011; 31:3670–80; PMID:21730287; http://dx.doi.org/ 10.1128/MCB.05704-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ye J, Kumanova M, Hart LS, Sloane K, Zhang H, De Panis DN, Bobrovnikova-Marjon E, Diehl JA, Ron D, Koumenis C. The GCN2-ATF4 pathway is critical for tumour cell survival and proliferation in response to nutrient deprivation. EMBO J 2010; 29:2082–96; PMID:20473272; http://dx.doi.org/ 10.1038/emboj.2010.81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bi M, Naczki C, Koritzinsky M, Fels D, Blais J, Hu N, Harding H, Novoa I, Varia M, Raleigh J, et al.. ER stress-regulated translation increases tolerance to extreme hypoxia and promotes tumor growth. EMBO J 2005; 24:3470–81; PMID:16148948; http://dx.doi.org/ 10.1038/sj.emboj.7600777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rouschop KM, van den Beucken T, Dubois L, Niessen H, Bussink J, Savelkouls K, Keulers T, Mujcic H, Landuyt W, Voncken JW, et al.. The unfolded protein response protects human tumor cells during hypoxia through regulation of the autophagy genes MAP1LC3B and ATG5. J Clin Invest 2010; 120:127–41; PMID:20038797; http://dx.doi.org/ 10.1172/JCI40027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gardner LB. Hypoxic inhibition of nonsense-mediated RNA decay regulates gene expression and the integrated stress response. Mol Cell Biol 2008; 28:3729–41; PMID:18362164; http://dx.doi.org/ 10.1128/MCB.02284-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wengrod J, Martin L, Wang D, Frischmeyer-Guerrerio P, Dietz HC, Gardner LB. Inhibition of nonsense-mediated RNA decay activates autophagy. Mol Cell Biol 2013; 33:2128–35; PMID:23508110; http://dx.doi.org/ 10.1128/MCB.00174-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Milani M, Rzymski T, Mellor HR, Pike L, Bottini A, Generali D, Harris AL. The role of ATF4 stabilization and autophagy in resistance of breast cancer cells treated with Bortezomib. Cancer Res 2009; 69:4415–23; PMID:19417138; http://dx.doi.org/ 10.1158/0008-5472.CAN-08-2839 [DOI] [PubMed] [Google Scholar]
- 61.Natarajan K, Meyer MR, Jackson BM, Slade D, Roberts C, Hinnebusch AG, Marton MJ. Transcriptional profiling shows that Gcn4p is a master regulator of gene expression during amino acid starvation in yeast. Mol Cell Biol 2001; 21:4347–68; PMID:11390663; http://dx.doi.org/ 10.1128/MCB.21.13.4347-4368.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hamilton TL, Stoneley M, Spriggs KA, Bushell M. TOPs and their regulation. Biochem Soc Trans 2006; 34:12–6; PMID:16246169; http://dx.doi.org/ 10.1042/BST0340012 [DOI] [PubMed] [Google Scholar]
- 63.Damgaard CK, Lykke-Andersen J. Translational coregulation of 5′TOP mRNAs by TIA-1 and TIAR. Genes Dev 2011; 25:2057–68; PMID:21979918; http://dx.doi.org/ 10.1101/gad.17355911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Iadevaia V, Caldarola S, Tino E, Amaldi F, Loreni F. All translation elongation factors and the e, f, and h subunits of translation initiation factor 3 are encoded by 5′-terminal oligopyrimidine (TOP) mRNAs. Rna 2008; 14:1730–6; PMID:18658124; http://dx.doi.org/ 10.1261/rna.1037108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Meyuhas O. Synthesis of the translational apparatus is regulated at the translational level. Eur J Biochem 2000; 267:6321–30; PMID:11029573; http://dx.doi.org/ 10.1046/j.1432-1327.2000.01719.x [DOI] [PubMed] [Google Scholar]
- 66.Talloczy Z, Jiang W, Virgin HWT, Leib DA, Scheuner D, Kaufman RJ, Eskelinen EL, Levine B. Regulation of starvation- and virus-induced autophagy by the eIF2alpha kinase signaling pathway. Proc Natl Acad Sci U S A 2002; 99:190–5; PMID:11756670; http://dx.doi.org/ 10.1073/pnas.012485299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yamaguchi S, Ishihara H, Yamada T, Tamura A, Usui M, Tominaga R, Munakata Y, Satake C, Katagiri H, Tashiro F, et al.. ATF4-mediated induction of 4E-BP1 contributes to pancreatic beta cell survival under endoplasmic reticulum stress. Cell Metab 2008; 7:269–76; PMID:18316032; http://dx.doi.org/ 10.1016/j.cmet.2008.01.008 [DOI] [PubMed] [Google Scholar]
- 68.Cherkasova VA, Hinnebusch AG. Translational control by TOR and TAP42 through dephosphorylation of eIF2alpha kinase GCN2. Genes Dev 2003; 17:859–72; PMID:12654728; http://dx.doi.org/ 10.1101/gad.1069003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Garcia-Barrio M, Dong J, Cherkasova VA, Zhang X, Zhang F, Ufano S, Lai R, Qin J, Hinnebusch AG. Serine 577 is phosphorylated and negatively affects the tRNA binding and eIF2alpha kinase activities of GCN2. J Biol Chem 2002; 277:30675–83; PMID:12070158; http://dx.doi.org/ 10.1074/jbc.M203187200 [DOI] [PubMed] [Google Scholar]
- 70.Bastians H, Ponstingl H. The novel human protein serine/threonine phosphatase 6 is a functional homologue of budding yeast Sit4p and fission yeast ppe1, which are involved in cell cycle regulation. J Cell Sci 1996; 109(Pt 12):2865–74; PMID:9013334 [DOI] [PubMed] [Google Scholar]
- 71.Di Como CJ, Arndt KT. Nutrients, via the Tor proteins, stimulate the association of Tap42 with type 2A phosphatases. Genes Dev 1996; 10:1904–16; PMID:8756348; http://dx.doi.org/ 10.1101/gad.10.15.1904 [DOI] [PubMed] [Google Scholar]
- 72.Ingebritsen TS, Cohen P. The protein phosphatases involved in cellular regulation. 1. Classification and substrate specificities. Eur J Biochem 1983; 132:255–61; PMID:6301824; http://dx.doi.org/ 10.1111/j.1432-1033.1983.tb07357.x [DOI] [PubMed] [Google Scholar]
- 73.Zeng K, Bastos RN, Barr FA, Gruneberg U. Protein phosphatase 6 regulates mitotic spindle formation by controlling the T-loop phosphorylation state of Aurora A bound to its activator TPX2. J Cell Biol 2010; 191:1315–32; PMID:21187329; http://dx.doi.org/ 10.1083/jcb.201008106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gold HL, Wengrod J, de Miera EV, Wang D, Fleming N, Sikkema L, Kirchhoff T, Hochman T, Goldberg JD, Osman I, et al.. PP6C hotspot mutations in melanoma display sensitivity to Aurora kinase inhibition. Mol Cancer Res 2014; 12:433–9; PMID:24336958; http://dx.doi.org/ 10.1158/1541-7786.MCR-13-0422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Stefansson B, Brautigan DL. Protein phosphatase 6 subunit with conserved Sit4-associated protein domain targets IkappaBepsilon. J Biol Chem 2006; 281:22624–34; PMID:16769727; http://dx.doi.org/ 10.1074/jbc.M601772200 [DOI] [PubMed] [Google Scholar]
- 76.Wengrod J, Wang D, Weiss S, Zhong H, Osman I, Gardner LB. Phosphorylation of eIF2alpha triggered by mTORC1 inhibition and PP6C activation is required for autophagy and is aberrant in PP6C-mutated melanoma. Sci Signal 2015; 8:ra27; PMID:25759478; http://dx.doi.org/ 10.1126/scisignal.aaa0899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hodis E, Watson IR, Kryukov GV, Arold ST, Imielinski M, Theurillat JP, Nickerson E, Auclair D, Li L, Place C, et al.. A landscape of driver mutations in melanoma. Cell 2012; 150:251–63; PMID:22817889; http://dx.doi.org/ 10.1016/j.cell.2012.06.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kong M, Ditsworth D, Lindsten T, Thompson CB. Alpha4 is an essential regulator of PP2A phosphatase activity. Mol Cell 2009; 36:51–60; PMID:19818709; http://dx.doi.org/ 10.1016/j.molcel.2009.09.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wander SA, Hennessy BT, Slingerland JM. Next-generation mTOR inhibitors in clinical oncology: how pathway complexity informs therapeutic strategy. J Clin Invest 2011; 121:1231–41; PMID:21490404; http://dx.doi.org/ 10.1172/JCI44145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yang ZJ, Chee CE, Huang S, Sinicrope FA. The role of autophagy in cancer: therapeutic implications. Mol Cancer Ther 2011; 10:1533–41; PMID:21878654; http://dx.doi.org/ 10.1158/1535-7163.MCT-11-0047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.McAfee Q, Zhang Z, Samanta A, Levi SM, Ma XH, Piao S, Lynch JP, Uehara T, Sepulveda AR, Davis LE, et al.. Autophagy inhibitor Lys05 has single-agent antitumor activity and reproduces the phenotype of a genetic autophagy deficiency. Proc Natl Acad Sci U S A 2012; 109:8253–8; PMID:22566612; http://dx.doi.org/ 10.1073/pnas.1118193109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Robert F, Williams C, Yan Y, Donohue E, Cencic R, Burley SK, Pelletier J. Blocking UV-induced eIF2alpha phosphorylation with small molecule inhibitors of GCN2. Chem Biol Drug Des 2009; 74:57–67; PMID:19519745; http://dx.doi.org/ 10.1111/j.1747-0285.2009.00827.x [DOI] [PMC free article] [PubMed] [Google Scholar]