

The master regulators p53 and nuclear factor (NF)-κB are transcription factors that are activated in response to a broad range of environmental stressors. Upon activation, both proteins accumulate in the nucleus and bind to consensus DNA motifs near genes, thereby regulating gene expression. In addition, p53 and NF-κB have both been implicated in cancer. p53 is a potent tumor suppressor that is dysfunctional in >90% of human cancers, and NF-κB is constitutively activated in several types of human cancers.1 Fitting with their opposing consequences in tumorigenesis, many studies in human cancer cell lines show that p53 and NF-κB have divergent functions in the stress response. For example, p53 activation results in apoptosis, cell cycle arrest, and senescence, whereas NF-κB activation enhances cell survival. Furthermore, p53 and NF-κB inhibit each other, in part by competing for essential co-factors.1 However, the story is far more complicated, as some studies report that p53 and NF-κB can work together, suggesting that p53/NF-κB crosstalk may be context-dependent.2
Whereas p53 is widely studied in the context of cancer cells, far less is understood about p53 function and p53/NF-κB crosstalk in primary cells, particularly immune cells. This is a key issue as tissue-resident leukocytes, macrophages in particular, serve as sentinels that sense and respond on the front-line to stress signals both from the environment and from the host, including tumors. In this vein, our recent report in Cancer Research3 shed light on a previously uncharted territory, namely the role of p53/NF-κB interactions in normal and tumor-associated macrophages (TAMs). Surprisingly, we found that the p53/NF-κB relationship is not inhibitory in a macrophage setting – instead, these 2 factors cooperate in response to genotoxic stress to induce a host of pro-inflammatory genes. Remarkably, the kinetics of this cooperative p53/NF-κB-driven gene expression is unlike a typical p53-induced response, occurring very rapidly – within 1 hour of cell challenge. Greatly enriched among the p53/NF-κB-regulated gene targets are pro-inflammatory cytokines and chemokines, such as interleukin (IL)-6 and CXCL1. Further, we found that conditioned media from p53/NF-κB co-activated macrophages induced neutrophil migration, suggesting physiologic significance. Taken together, these results define a novel role for p53, placing it as a co-regulator with NF-κB of rapid immune responses of macrophages to environmental stressors.
After surveying several different types of cells, including cancer cell lines and primary human and murine cells, we found, interestingly, that cooperative p53/NF-κB-dependent pro-inflammatory gene expression is a phenomenon that appears to be unique to primary human monocytes and macrophages. While the reasons for this specificity are unclear, this may suggest that immune factors co-expressed by these human innate myeloid cells may ‘reprogram’ p53 to serve a unique role as amplifier of the stress response.
Extrapolating our findings on the role of p53 in normal macrophage gene expression to the context of tumors, we next evaluated the role of p53 in TAMs. In recent years, it has been shown that macrophages and other immune cells that infiltrate tumors can promote tumorigenesis by conditioning the tumor microenvironment with secreted factors. For example, TAMs secrete cytokines such as IL-6 that promote tumor cell proliferation and angiogenesis.4 Using an established in vitro TAM model system, namely culture of macrophages in conditioned media from cancer cells (i.e., tumor-conditioned macrophages [TCMs]5), we found that p53 protein is up-regulated in TCMs compared to normal macrophages. Consistent with increased p53 function, expression of classical p53 target genes such as CDKN1A and BBC3 was increased in TCMs. Moreover, suggesting that tonic p53 activity in TAMs may drive basal IL-6 secretion, we found increased p53 binding to the IL-6 promoter in TCMs, and demonstrated that basal IL-6 secretion by these cells was suppressible by the p53 inhibitor pifithrin-α. Taken together, our findings suggest a novel feed-forward paracrine loop in the tumor microenvironment whereby tumor-released factors activate p53 in local macrophages, in turn driving them to express cytokines that may further promote tumorigenesis, potentially both by direct effects on the tumor and also by promoting the infiltration of additional tumor-modifying immune cells such as neutrophils (Fig. 1). This said, given that the cocktail of p53-dependent macrophage-released factors is likely complex and that p53 may possibly exert additional effects upon TAMs (e.g., apoptosis), the net biological effect of p53-driven cytokine/chemokine expression on the tumor microenvironment – whether anti- or pro-tumor – is currently unclear and clearly warrants further investigation.
Figure 1.
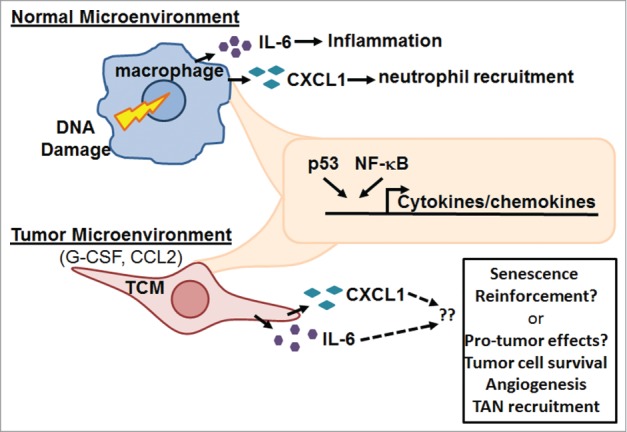
Schematic of p53 and NF-B interactions in human macrophages. Activation of p53 and NF-κB in normal and tumor-conditioned macrophages (TCMs) results in the induction of pro-inflammatory cytokines. Biological outcomes likely depend on the tissue microenvironment. In a normal microenvironment, cytokines result in inflammation; in a tumor microenvironment, enhanced tumor cell survival and tumor-associated neutrophil (TAN) recruitment may result.
In summary, we identify a novel role for p53 – rapid co-regulation with NF-κB of a subset of pro-inflammatory genes in response to genotoxic challenge. Remarkably, this network appears to be specific to human monocytes and macrophages. While further work is required, these findings may have important and far-reaching clinical implications, extending from tissue homeostasis to carcinogenesis. For example, they suggest that p53-sufficient TAMs might condition the tumor microenvironment in response to genotoxic chemotherapeutics, even in patients with tumors that bear mutant/non-functional p53. Secondly, in the non-cancer setting, they suggest the interesting possibility that p53 may drive aseptic tissue inflammation induced by genotoxic stress (e.g., radiation-induced lung injury). Despite years of study, the purview of p53 continues to grow with little sense of an end in sight.
References
- 1. Ak P, et al. . FASEB J 2010; 24:3643-52; PMID:20530750; http://dx.doi.org/ 10.1096/fj.10-160549 [DOI] [PubMed] [Google Scholar]
- 2. Schneider G, et al. . Biochim Biophys Acta 2011; 1815:90-103; PMID:20951769; http://dx.doi.org/ 10.1016/j.bbcan.2010.10.003 [DOI] [PubMed] [Google Scholar]
- 3. Lowe JM, et al. . Cancer Res 2014; 74:2182-92; PMID:24737129; http://dx.doi.org/ 10.1158/0008-5472.CAN-13-1070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Galdiero MR, et al. . J Cell Physiol 2013; 228:1404-12; PMID:23065796; http://dx.doi.org/ 10.1002/jcp.24260 [DOI] [PubMed] [Google Scholar]
- 5. Grugan KD, et al. . J Immunol 2012; 189:5457-66; PMID:23105143; http://dx.doi.org/ 10.4049/jimmunol.1201889 [DOI] [PubMed] [Google Scholar]