

Abstract
Humans and primates are long-lived animals with long reproductive phases. One factor that appears to contribute to longevity and fertility in humans, as well as to cancer-free survival, is the transcription factor and tumor suppressor p53, controlled by its main negative regulator MDM2. However, p53 and MDM2 homologs are found throughout the metazoan kingdom from Trichoplacidae to Hominidae. Therefore the question arises, if p53/MDM2 contributes to the shaping of primate features, then through which mechanisms. Previous findings have indicated that the appearances of novel p53-regulated genes and wild-type p53 variants during primate evolution are important in this context. Here, we report on another mechanism of potential relevance. Human endogenous retrovirus K subgroup HML-2 (HERV-K(HML-2)) type 1 proviral sequences were formed in the genomes of the predecessors of contemporary Hominoidea and can be identified in the genomes of Nomascus leucogenys (gibbon) up to Homo sapiens. We previously reported on an alternative splicing event in HERV-K(HML-2) type 1 proviruses that can give rise to nuclear protein of 9 kDa (Np9). We document here the evolution of Np9-coding capacity in human, chimpanzee and gorilla, and show that the C-terminal half of Np9 binds directly to MDM2, through a domain of MDM2 that is known to be contacted by various cellular proteins in response to stress. Np9 can inhibit the MDM2 ubiquitin ligase activity toward p53 in the cell nucleus, and can support the transactivation of genes by p53. Our findings point to the possibility that endogenous retrovirus protein Np9 contributes to the regulation of the p53-MDM2 pathway specifically in humans, chimpanzees and gorillas.
Keywords: endogenous retrovirus, evolution, MDM2, Np9, p53, ubiquitylation
Introduction
During infection, retroviruses reverse-transcribe their RNA genomes into double-stranded DNA which is then inserted into the host cell genome to become a permanent part of it if fixed in the population. Although retroviruses typically infect somatic cells, some of them have managed to conquer cells of the germ line on numerous occasions during vertebrate evolution. The resulting, so-called endogenous retroviruses (ERVs) are transmitted like Mendelian genes. Most ERVs survived as sequence relics long after the original, exogenous retroviruses have gone extinct. In the human genome, HERV-derived DNA is scattered over ˜700,000 different loci and collectively makes up ∼8 % of the total cellular DNA,1,2 with the vast majority of it consisting of solitary long terminal repeats (LTRs) that are normally positioned at both ends of the proviral genome and bear regulatory sequences. Approximately 40 different groups of HERVs are currently on record.2-4 The number of loci varies widely between the different HERV groups and may count between a few and thousands. HERV transcripts were detected in every human tissue examined so far.5,6 The majority of these transcripts stems from HERV loci that cannot express proteins as the result of numerous mutations within protein coding sequences. Nevertheless, such non-coding RNAs may serve as regulatory RNAs.
It is now generally accepted that ERVs not only helped shape vertebrate genomes but exercise important functions in defined cell types through at least 2 manners: the regulation of cellular genes by neighboring LTRs and the activity of ERV-encoded proteins.2 An intriguing example of the latter is the ERVWE1/ERVW-1 gene locus encoding syncytin-1. This retroviral envelope-like protein has evolved to crucially contribute to fusion of trophoblasts into syncytiotrophoblasts during human placenta formation.7,8 Strikingly, some other HERV loci also harbour one or several intact ORFs that can code for the major retroviral proteins. Especially the HERV-K(HML-2) group bears loci with ORFs for Gag (group-specific antigen), (Prot)ease, (Pol)ymerase and (Env)elope proteins, as well as for some regulatory accessory proteins (see ahead).9,10
HERV-K(HML-2) is endowed with the most extensive coding capacity and in addition includes a number of HERV loci that are specific for humans.11-16 Intriguingly, recent work indicates that the expression of RNA and protein from HERV-K(HML-2) constitutes a marker for embryonic and induced pluripotent stem cells.17 HERV-K(HML-2) comes in 2 flavours: so-called type 1 proviruses lack a 292 bp sequence within the proviral pol-env gene boundary compared to HERV-K(HML-2) type 2 proviruses. Type 2 sequences bear an ORF, encoded by an mRNA resulting from an additional splicing of the envelope mRNA, that can translate into the accessory protein Rec, a functional homolog of the HIV Rev, Human T-cell leukemia Virus Rex, Mouse Mammary Tumor Virus Rem and Jaagsiekte Sheep Retrovirus Rej proteins, all of which export unspliced retroviral RNA from the nucleus. Type 1 sequences, by contrast, can give rise to the accessory protein Np9 (Nuclear protein of 9 kDa; see below).18,19 HERV-K(HML-2) type 1 homologous proviruses were identified in the genomes of orang-utan, gorilla, chimpanzee and human, thus appeared during primate evolution after the evolutionary split of Hominoidea from Old World primates; the latter harbour only type 2 proviruses.20
Transcription of both HERV-K(HML-2) type 1 and type 2 proviruses has been identified in the context of various human diseases including cancer,11 yet nothing is known about their functions in the healthy organism. However, since some functional ERVs are present exclusively in defined genera of Hominoidae or are even specific for humans,14 it is conceivable that their hypothetical normal cellular activities contribute to phenotypic shaping in at least some higher primates including humans. In this context, we recently reported transcription of HERV-K(HML-2) rec and np9 in various normal human tissues.21
Mouse Double Minute 2 homolog (MDM2) seems to function primarily as the major negative regulator of the p53 family of transcription factors, comprising p53, p63 and p73 plus several isoforms thereof. These control a large number of processes in normal somatic cells, stem cells and development. For example, p53 is activated by various stresses including DNA damage, hypoxia, hypo/hyperthermia, metabolite shortages and imbalances, inappropriate proliferation stimuli, and viral/bacterial/parasitic infections, to regulate a large number of genes that control, among other processes, the cell cycle, cell survival, senescence, autophagy, DNA repair, respiration, oxidative stress protection, metabolism, cell adhesion/motility, the cytoskeleton and endo/exosome compartments.22,23 p53 is important, at the level of the cell, for homeostasis, genomic stability and tumor suppression,23 and it is a determinant of longevity and female fertility at the level of the organism.24,25 p63 and p73 are crucial regulators of embryonic development, with p63 acting primarily in epithelial stem cell maintenance and differentiation, and p73 being important for the shaping, as well as the function, of the adult nervous and immune systems. In addition, p63 ensures the quality of female germ cells while p73 controls the quality of male germ cells. An overview on p63 and p73 is provided in refs.26,27 The regulation of the cell by the p53 family is highly complex, and some mechanisms have evolved to be specific for primates or even humans. These include, for example, the control of female fertility through the functionally distinct wild-type p53 variants p53–72P and p53–72R 28,29; the alteration of genes, through alterations of DNA promoter sequences, from p53-non-responsiveness to p53-responsiveness30; and the appearance of novel p53-regulated open reading frames.31
MDM2 is of central importance for restraining p53; it exerts its inhibitory effects through binding to the p53 family proteins p53 and p73 and, with much lower affinity, p63, with 2 major consequences: the ubiquitin-marking of p53 for degradation or nuclear exclusion, and the blocking of p53's and p73's functions as transcription factors.32 The importance of MDM2 as a negative regulator of p53 in humans is underscored, for instance, by the finding that individuals may have inherently different cellular levels of MDM2 as the result of a single nucleotide polymorphism in a regulatory sequence of the MDM2 gene, and that the genotype that causes increased MDM2 is associated with reduced activity of p53 and elevated cancer susceptibility.33 Here, we present findings that point to the possibility that the MDM2-p53 pathway is regulated in a human-specific manner by the endogenous retrovirus-encoded protein Np9. We furthermore examined primate genome sequences for np9-specific features and delineated independent mutation events leading to Np9-coding capacity restricted to potentially a few primate species.
Results and Discussion
Human ERV-K(HML-2) endogenous retroviruses
HERV-K(HML-2) type 1 sequences evolved from type 2 by a deletion of 292 bp within the proviral pol-env boundary that occurred in an ancestor of present-day Hominoidea. An additional mutation gave rise to a splice donor site immediately upstream of the 292 bp deletion34 (Figs. 1A, B). The resulting np9 (nuclear protein of 9 kDa) ORF (GenBank accession number: AF164609) consists of 3 exons which together encode the 74 amino acid residues (aa) protein Np9 with a predicted molecular weight of 8.7 kDa. The N-terminal 15 aa of Np9, encoded by the second exon, are identical with the N-terminus of the 87 aa Rec protein while the third exon of np9, beginning at nt 8118,34 translates into the unique 59 aa C-terminus in a reading frame different from rec. The function of Np9 is largely unknown.19,35-37
Figure 1.

Provirus structure of HERV-K(HML-2) and germ line integration during primate evolution. (A) Structure of the more ancient HERV-K(HML) type 2 provirus harbouring the rec open reading frame. Thick lines at each end indicate host cell genomic DNA. LTR, Long-terminal Repeat. SD, SA, splice donor and splice acceptor sites. gag, pro, pol, env, group specific antigen, protease, polymerase and envelope protein open reading frames. nt, nucleotides. aa, amino acid residues. (B) Structure of HERV-K(HML-2) type 1 provirus. A 292 bp deletion at the 5′-end of the env open reading frame defines type 1 proviruses. Another mutation (see Figure 2) generated a new splice donor site (SD; highlighted in black), resulting in an alternatively spliced mRNA with the ORF encoding the 74 aa residue Np9 protein (for further details, see main text). (C) Evolutionary relationships among groups of primates and selected primate species (adapted from66). Arrows depict the fixation of HERV-K(HML-2) type 1 and type 2 proviruses. myr, million years.
Evolution of a splice donor site resulting in Np9-coding capacity
Human endogenous retroviruses of the HERV-K(HML-2) group initially integrated into the primate germ line after the evolutionary split of Old World primates (Catarrhini) from New World primates (Platyrrhini).20 Previous results indicated that the mutational event leading to type 1 proviruses should have occurred after the evolutionary split of Hominoidea from Old World primates as type 1 proviruses were identified in human, chimpanzee, gorilla and orang-utan (gibbon not tested)20 (Fig. 1C). We here examined the meanwhile available genome sequences of various Old World and hominoid primate species specifically for presence of type 1 and type 2 provirus sequences. In accord with previous findings, we did not detect type 1 proviruses in Old World primates, specifically baboon and rhesus monkey. However, we detected 3 type 1 proviruses in the genome sequence of gibbon, 6 type 1 proviruses in the genome sequences of gorilla and orang-utan, 19 in chimpanzee and 17 in human (Fig. 2). Thus, this study identified type 1 proviruses also in the genome of gibbon. Type 1 proviruses therefore formed early and subsequently amplified in copy number during the evolution of Hominoidea.
Figure 2.

Evolution in Hominoidea of the np9 splice donor site 2 (SD2) crucial for Np9 protein expression. A subregion of HERV-K(HML-2) type 1 homologous sequences identified in various Hominoidea genome sequences is shown as a multiple alignment. For comparisons, corresponding regions of reference sequences HERV-K(HML-2.HOM) (type 2) (Genbank acc. no. AF074086; 15), the previously identified HERV-K103 provirus (type 1) (AF164611; 14) and a previously described np9 mRNA sequence.19 The 3′ end of the np9 exon 2 is located at nt 6495 and the 292 bp deletion, defining type 1 proviruses, starts at nt 6502 with respect to the HERV-K(HML-2.HOM) sequence. Approximate locations of HERV-K(HML-2) homologous type 1 sequences in the various primate genomes (see Materials and Methods) are indicated by sequence names left from actual sequences as ‘species abbreviation_chromosome no._start position_end position’. The critical GA–GT mutation generating the 5′ end of np9 intron 2 is indicated by a horizontal line and the number of sequences displaying GT, relative to the total number of type 1 provirus sequences, is given below each of the multiple alignments.
A second mutational event was required for splicing of np9 mRNA, specifically, generation of a splice donor site (SD2) for splicing of np9 exons 2 and 3 due to a mutation GA–GT at nucleotide 6495 with respect to the previously reported HERV-K(HML-2.HOM) provirus sequence (Genbank acc. no. AF074086;15). Examination of type 1 proviral sequences in the various Hominoidea genome sequences revealed that gibbon and orang-utan type 1 provirus sequences do not yet harbour the specific GA–GT mutation. However, 3 out of 6 type 1 proviruses in gorilla harboured GT instead of GA at that position, and the same was observed for 15 out of 19 type 1 proviruses in chimpanzee and 12 out of 17 type 1 proviruses in human (Fig. 2). Therefore, type 1 proviruses potentially being spliced to np9 mRNA, due to a GA–GT mutation, appeared for the first time in gorilla, and such mutated type 1 proviruses subsequently amplified in copy number up to the numbers now observed in chimpanzee and human. Thus, among the HERV-K(HML-2) homologous sequences-harbouring primates, only human, chimpanzee and gorilla can, in principle, encode Np9 protein due to presence of proviruses with required splice signals.
Expression of Np9 protein
In contrast to np9 transcript which may be fairly abundant, Np9 protein is typically present in small amounts only, in most cell types analyzed so far 19,35,37 However, we and others had used rabbit polyclonal antisera that produced, in addition to weak Np9 signals, a greater number of unspecific signals in western immunoblottings.19,36 In order to gain more information on the expression of Np9 protein, we here initially developed and tested a panel of rat monoclonal antibodies directed against the central portion of Np9. Antibodies 22E4 and 10B1 were the most sensitive and specific (see Materials and Methods section, and below).
Our previous work had indicated that Np9 protein levels may rise in cells when these are exposed to certain forms of stress.38 To investigate this in more detail, human Tera-1 embryonic carcinoma cells which were known to robustly express np9 transcript but produce almost no detectable Np9 protein under normal growth conditions,38 were chosen as the primary study cell type. Initially, Tera-1 cultures were either mock-treated or exposed to various stresses, including the inhibition of transcription of nucleolar rDNA by ActD, DNA damage by ADR or etoposide, inhibition of 26S proteasome by epoxomicin, DNA and RNA damage by the anti-metabolite 5-FU, oxidative stress by hydrogen peroxide, DNA crosslinking by mitomycin C or UV irradiation, and the depletion of growth factors by serum withdrawal. As expected, almost all of these stresses increased the cellular levels of the stress-response transcription factor and tumor suppressor p53 that is wild-type in Tera-1 cells, and simultaneously raised the levels of the p53-controlling ubiquitin ligase MDM2 whose gene is subject to transactivation by p53 (Fig. 3A). With respect to Np9 expression, rat monoclonal anti-Np9 antibody 22E4 (and similarly, 10B1; not shown) gave rise to 4 signals of 24, 16.5, 15.5 and 12.5 kDa in western blottings, respectively. Of these, the 24 and 15.5 kDa signals were identified as probably unspecific, i.e., unlikely to originate from proteins translated from np9 transcripts (see ahead).
Figure 3.

Expression of Np9 protein in response to various stresses. (A) Proteasome inhibition causes accumulation of Np9. Exponentially growing Tera-1 cultures were either mock-treated or exposed to actinomycin D (ActD; 10 nM), adriamycin (ADR; 0.34 μM), epoxomicin (Epoxo; 0.4 μM), etoposide (Eto, 10 μM), 5-fluorouracil (5-FU; 375 μM), H2O2 (0.4 mM); mitomycin C (Mito; 3 μM); serum withdrawal (SW) and UV light (200 J/m2). Total protein extracts were prepared after 24 h, and 15 μg protein per lane was analyzed by western blotting, using monoclonal anti-MDM2 antibody 3G9 (1:2,000), monoclonal anti-p53 antibody DO-1 (1 : 2,000), and rat monoclonal anti-Np9 antibody 22E4 (1:5). (B) np9 transcripts sensitive to np9-specific siRNA produce 12.5 kDa and a 16.5 kDa Np9 proteins. Tera-1 cells were transfected for 24 h with scrambled siRNA (C=control; 20 nM) or a mixture of 4 np9 siRNAs (5 nM each). After transfection, the cells were treated with 2 concentrations of epoxomicin for another 24 h. Western blot analysis was as in (A). (C) H1299 cells negative for np9 give rise to a single signal of 12.5 kDa upon transfection for 24 h with plasmid pCMV-np9 containing the np9 ‘reference’ ORF (left panel). Tera-1 cells transfected with pCMV-np9 produce an Np9 signal of similar size as the endogenous Np9 produced under epoxomicin, and this signal is further increased in response to epoxomicin (right panel). Western blots were performed as in (A). (D) Dose-dependent increase in Tera-1 cells of Np9 expression after 24 h of treatment with increasing, sub-micromolar concentrations of epoxomicin. Western blot was performed as in A; β-actin was detected with monoclonal anti-β-actin antibody (1 : 10,000). (E) Late accumulation of Np9 in Tera-1 cells compared to MDM2 and p53, various times after the exposure to epoxomicin (0.4 μM).
Remarkably, only one of the tested stresses, the inhibition of the 26S proteasome by epoxomicin, induced Np9 protein expression to high levels (Fig. 3A). Most studies typically employ, in order to inhibit the 26S proteasome, the peptide aldehyde MG132. However, this compound is less specific and generates more off-target effects than epoxomicin from Actinomycetes, the most specific and potent natural proteasome inhibitor known to date.39 Nonetheless, MG132 generated Np9 bands with similar kinetics as epoxomicin (data not shown). Two Np9 signals were obtained, a major signal of 12.5 kDa and a minor signal of 16.5 kDa (Fig. 3A). To examine whether these bands were indeed translated from np9 transcripts, Tera-1 cells were treated either with scrambled (control) siRNAs or with a mixture of 4 np9 siRNAs (5 nM of each per transfection; see Materials and Methods) prior to the treatment with epoxomicin. np9 siRNA strongly reduced the 12.5 and 16.5 kDa signals but failed to affect the 24 and 15.5 kDa signals (Fig. 3B). Thus, inhibition of proteasomal protein degradation by epoxomicin increased the levels of Np9 protein, which was mainly represented by a major 12.5 kDa form (hereafter referred to as Np9) and a minor 15.5 kDa form (designated Np9-α) in Tera-1 cells.
In accord with these findings, when human H1299 lung adenocarcinoma cells were transiently transfected with a plasmid harbouring the np9 ‘reference’ ORF (19; GenBank accession number: AF164609), a single band of 12.5 kDa was produced. Moreover, when Tera-1 cells were transfected with control vector or np9 expression vector, and were treated or not treated with epoxomicin, ectopic Np9 and endogenous Np9 both gave rise to bands of similar molecular weight (12.5 kDa), and importantly, proteasome inhibition caused an increase not only of the endogenous Np9 levels but also of the transfected Np9 levels (Fig. 3C). Thus, Np9 of the predicted molecular weight of 8.7 kDa migrated at 12.5 kDa in SDS-PAGE and western immunoblots performed with our newly developed rat anti-Np9 antibodies, and accumulated when proteasomal degradation was inhibited. This accumulation of Np9 was dependent of the dose of epoxomicin, with as little as 50 nM of the compound sufficient to cause a rise in the level of Np9, as well as, of p53 and MDM2 which both are known to be degraded through the proteasome pathway (Fig. 3D). However, when we examined the accumulation of Np9 in the presence of 0.4 μM epoxomicin over time, it turned out that while MDM2 accumulated at a constant rate over the time course of 24 h, Np9 consistently became visible only after ˜16 h and was present at high levels only after ˜24 h (Fig. 3E). This suggests that Np9 protein is produced at comparatively low rates and is rapidly degraded in Tera-1 cells, such that it accumulates to detectable levels only after relatively long times of proteasome inhibition. Inspection of peripheral blood mononuclear cells (PBMC) and non-cycling human diploid fibroblasts (HDF) from healthy volunteers revealed that, in contrast to Tera-1 cells, the very low levels of Np9 expressed in PBMC could not be raised by proteasome inhibition, while HDF constantly expressed some Np9 protein regardless of proteasome inhibition (Fig. S1). Thus, Np9 seems to be stable in dense cultures of mostly contact-inhibited HDF.
Np9 binds to MDM2
We had observed that the RING-type E3 ubiquitin ligase MDM2 that was known to be able to ubiquitin-mark proteins including p53 for proteasomal degradation, accumulated in Tera-1 cells along with Np9 in response to epoxomicin (see, for example, Fig. 3A, D, E). We therefore entertained the possibility that Np9 might interact with, and be a substrate of, MDM2. Our previous searches for binding partners of Np9 had identified, among others, the Promyelocytic Leukemia Zinc Finger (PLZF) protein40 and the Ligand of Numb X (LNX) protein.35 PLZF is a transcriptional repressor important for the self-renewal and maintenance of spermatogonial stem cells,41 whereas LNX like MDM2 acts as a RING-type E3 ubiquitin ligase. It marks Numb, a negative regulator of transcription factor Notch, for proteasomal degradation.42 To address whether Np9 and MDM2 interact, Tera-1 cells were first treated with proteasome inhibitor to accumulate endogenous Np9 and MDM2. Np9 was then immunoprecipitated from the total cell extracts. In accord with Np9 and MDM2 interacting in vivo, MDM2 precipitated along with Np9 (Fig. 4A). Moreover, when human H1299 lung adenocarcinoma cells (p53- and Np9-negative; low levels of endogenous MDM2) were transfected with a combination of plasmids producing HA-tagged Np9 and MDM2, HA-Np9 also co-precipitated MDM2 (data not shown). To corroborate this finding and interrogate whether the interaction was direct, GST pull-down assays with full-length Np9 fused to GST and with in vitro-translated35 S-labeled full-length MDM2 were performed. Like LNX, but unlike Numb or Notch43,35 S-labeled MDM21–491 was retained by GST-Np91–74 (Fig. 4B). MDM2 did not bind to GST alone. Finally, to narrow down the interaction domains, further GST pull-downs were employed. Incubation of 35S-labeled Np9 with GST-coupled fragments of MDM2 revealed that Np9 mainly contacts the central acidic and zinc finger domains of MDM2 (aa residues 222–366) but also, weakly, the C-terminal RING domain of MDM2 (Fig. 4C). Np9 failed to bind to the N-terminal domain of MDM2 that is the interaction domain for p53.44 Conversely,35S-labeled MDM2 bound to the C-terminus of Np9 (Fig. 4D) that we showed previously is also contacted by the ubiquitin ligase LNX.35 Combined, these results suggest that Np9 can interact with MDM2 in vitro and in vivo.
Figure 4.
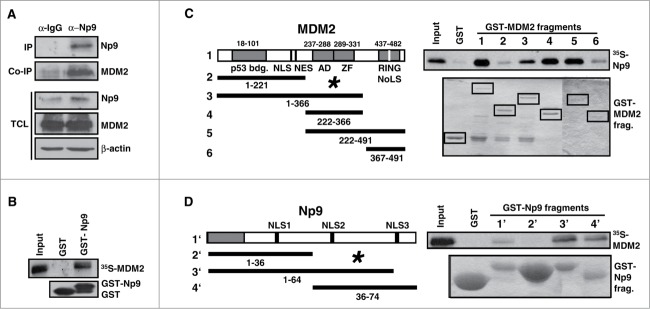
Np9 associates with MDM2. (A) Np9 coprecipitates MDM2 from transfected H1299 cell extracts. Cells were transfected with expression plasmids pCMV-np9 and pcmdm2. At 24 h after transfection, cell extracts were incubated with irrelevant IgG antibody or with anti-Np9 antibody 22E4 (2 μg/IP). Precipitates were analyzed by western blotting. Proteins were detected as specified in the legend of Figure 2. IP, immunoprecipitation. Co-IP, co-immunoprecipitation. TCL, total cell lysate. (B) GST pull-down analysis. Lower panel: Equal load of GST and GST-Np9 on beads. Upper panel: In vitro translated 35S-labeled full-length human MDM2 was retained by GST-Np9 but not GST alone. Input shows 10 % of total 35S-MDM2. (C) Radiolabeled Np9 binds to the central acidic and zinc finger domain of MDM2 in GST pull-down assays. (D) Radiolabeled MDM2 associates with the C-terminal half of Np9. The indicated MDM2 fragments (1–6) and Np9 fragments (1′–4′) were fused to GST and incubated with in vitro translated 35S-Np9 or 35S-MDM2. While labeled protein failed to bind to GST alone, 35S-Np9 was retained primarily by GST-MDM2 fragments 3–5, and 35S-MDM2 was retained mostly by GST-Np9 fragments 3 and 4. Asterisks mark main interaction domains. p53-bdg., p53 binding domain. NLS, nuclear localization signal. NES, nuclear export signal. AD, acidic domain. ZF, zinc finger domain. RING is the domain important for ubiquitin ligase activity, intra/inter-molecular protein interactions and interaction with RNA. NoLS, nucleolar localization domain. Numbers denote amino acid residues.
Np9 can regulate MDM2 function
MDM2 acts as an ubiquitin ligase toward many interacting proteins including p53. As a result, these proteins are either functionally modulated through multiple mono-ubiquitylations of lysine residues, or are degraded by the 26S proteasome upon multiple poly-ubiquitylations of lysine residues. Since i) the stability of MDM2 itself is regulated by ubiquitylation32; ii) both MDM2 and Np9 accumulated in response to proteasome inhibition (see Fig. 3D, E); and iii) Np9 and MDM2 bind to each other, we hypothesized that the cellular levels of Np9 might be regulated in part by MDM2. To address this, Tera-1 cells were transfected with control siRNA or MDM2 siRNA for the duration of 24 h, after which the levels of MDM2, p53 and Np9 were determined by western blotting. While MDM2 knockdown resulted in an increase of the cellular level of p53, as expected, it failed to increase the Np9 level detectably (Fig. 5A). We then examined whether Np9 can be a substrate of in vivo ubiquitylation by MDM2. For this purpose, we transfected H1299 cells harbouring low levels of endogenous MDM2 with combinations of plasmids expressing Np9 and MDM2, and included in the transfections a plasmid that produced an HA-tagged ubiquitin. At 24 h after transfection, proteasomal degradation was inhibited for another 4 h to prevent the degradation of ubiquitylated proteins. Np9 was then immunoprecipitated from cell extracts that had been denatured to preclude the detection of co-precipitating ubiquitylated proteins rather than ubiquitylated Np9 itself. Figure 5B (HA-ub, upper panel) documents that HA-ubiquitylated Np9 could be precipitated from the extracts even in the absence of ectopic MDM2 (lane 2) while the expression of ectopic MDM2 increased the level of HA-ubiquitylated Np9 (lane 3). Thus, in accordance with Np9 accumulating in response to proteasome inhibition, these findings suggest that Np9 is ubiquitylated in the cell. Consistent with our previous finding showing that Np9 is regulated by the ubiquitin ligase LNX,19 which is a RING-type ubiquitin ligase like MDM2, Np9 ubiquitylation is probably mediated by several ubiquitin ligases, of which MDM2 may be one.
Figure 5.

Effect of MDM2 on the expression level and modification of Np9. (A) MDM2 knockdown increases p53 level but fails to cause accumulation of Np9. Proteasome inhibition increases the expression of Np9 to higher levels when MDM2 has been knocked down. Tera-1 cells were transfected with control siRNA (20 nM; C) or MDM2 siRNA (20 nM) for 24 h. Mock-transfected Tera-1 cultures treated with epoxomicin (100 nM) for 24 h served as positive control for Np9 expression. Western blot analysis was performed as detailed in the legend of Figure 2. (B) In vivo HA-ubiquitylation of Np9 in the presence of ectopic MDM2. H1299 cells harbouring low levels of MDM2 were transfected with pCMV-np9, pcmdm2 and pCMV-HA-ub producing HA-tagged ubiquitin, as indicated. At 24 h after transfection, proteasomal degradation was inhibited by MG132 (10 μM) for another 4 h. Np9 was then immunoprecipitated from denatured cell extracts with a mixture of anti-Np9 antibody 22E4 and our polyclonal anti-Np9 antiserum (2 μg/IP). Western blot analysis was done as before (Figure 2). Monoclonal anti-HA antibody was used at 1 : 1,000. IP, immunoprecipitation. TCL, total cell lysate.
MDM2 itself has a short half-life in many cells due to being ubiquitin-marked for degradation. Next we investigated whether the novel MDM2 interaction partner Np9 may affect the ubiquitylation of MDM2. To this end, H1299 cells were again transfected with combinations of plasmids expressing MDM2, Np9 and HA-ubiquitin. Then, MDM2 was immunoprecipitated from denatured cell extracts and was inspected for HA-ubiquitylation as before. Slower-migrating MDM2 molecules indicative of ubiquitylation were detectable in western blots, and anti-HA antibody detected HA-ubiquitin covalently linked to MDM2 (Fig. 6A, lanes 3 and 4). Notably, co-expressed Np9 affected the ubiquitylation of MDM2 only weakly (Fig. 6A, lane 4). In striking contrast, when similar experiments were carried out with p53 as a substrate of MDM2, it turned out that, while MDM2 ubiquitylated p53 readily as expected, Np9 inhibited this ubiquitylation almost completely (Fig. 6B, lanes 3 and 4). Consistently, this inhibition of ubiquitin modification was reflected by a reduction in the intensity of the slower migrating western blot signals produced by anti-p53 antibody (Fig. 6B, see panel p53LE).
Figure 6.

For figure legend, see page 10.
Next, we wished to examine the effect of Np9 on p53 modification in the absence of ectopic HA-ubiquitin. We transfected H1299 cells with plasmids producing MDM2, p53 and Np9, and then analyzed, by western blotting, cytoplasmic and nuclear fractions in order to obtain information on the compartment in which the modification of p53 by MDM2, and the impact of Np9 on it, takes place. As before, Np9 inhibited the modification of p53 by MDM2 (Fig. 6C, lanes 1 and 2). Although a small fraction of Np9 was located in the cytoplasm, the modification of p53, and its inhibition by Np9, predominantly occurred in the nucleus where most of the MDM2 and Np9 resided (Fig. 6C, lanes 5 and 6). In a second study, we examined the modification of p53 in Tera-1 cells transfected with control siRNA or np9 siRNA, and upon induction or no induction of Np9 by epoxomicin. While MDM2 accumulated under epoxomicin to equal levels regardless of Np9, there was consistently more modified p53 in the absence of Np9 (Fig. 6D), in accord with the notion that Np9 inhibits the modification of p53. Finally and along the same line, when H1299 cells were transfected with combinations of plasmids producing p53, MDM2 and Np9, the level of p53 was cut approximately in half when ectopic MDM2 was present, as expected, but was restored when Np9 was coexpressed (Fig. 6E). Collectively, these findings suggest that Np9 can modify the activity of MDM2 toward p53.
These findings also raised the question how Np9 interferes with the ubiquitylation of p53 by MDM2. Active MDM2 is either a homodimer or, through the interaction of their RING domains, part of a heterodimer with its close relative MDMX (MDM4).32 Although Np9 primarily contacted the central acidic and zinc finger domain of MDM2 but not the RING domain and the N-terminal domain which are the interaction domains for MDMX and p53, respectively (see Fig. 4C), it was conceivable that Np9 interfered with the bindings through mechanisms other than direct competition, as for instance, through the induction of conformational changes. To address this, coimmunoprecipitation assays were performed. H1299 cells were transfected with plasmids producing HA-MDMX or Flag-p53, plus combinations of plasmids expressing MDM2 and Np9 (Fig. S2). Immunoprecipitation of HA-MDMX and Flag-p53 brought down MDM2, as expected. Importantly, the presence of increasing amounts of Np9 failed to interfere with the coprecipitation of MDM2 with HA-MDMX (Fig. S2A) and Flag-p53 (Fig. S2B), suggesting that Np9 does not affect p53 modulation by MDM2 through the inhibition of the binding of MDM2 to HA-MDMX or Flag-p53. Flag-p53 was able to bring down Np9 together with MDM2, pointing to the possibility that these proteins exist in the cell as a ternary complex. However, whether complex formation affects the enzymatic activity of MDM2 toward p53, and if so, what the underlying mechanism are, awaits further study.
Np9 can affect the function of p53 as a transcription factor
p53 acts as a transactivating transcription factor whose function in cells is restrained by MDM2.32 In light of the findings outlined above, we hypothesized that Np9 may support p53 as a gene transactivator. To begin to investigate this, we first transfected human U2OS osteosarcoma cells (wild-type p53 and MDM2 proficient) i) with a control reporter plasmid carrying defective p53 recognition elements (p53RE) in front of a luciferase gene, ii) with a reporter carrying functional p53RE, or iii) with a reporter carrying the p53-responsive p21 promoter. As expected, the transfection procedure alone was sufficient to induce a p53 response, but only with plasmids harbouring the functional p53REs. Notably, coexpression of Np9 resulted in a significantly increased p53 response, consistent with the notion that Np9 can support p53 as a transcriptional activator (Fig. 7A). Next, we cotransfected plasmid expressing MDM2 along with the p53-responsive reporter plasmids, after we had determined, in prior transfections, the minimal MDM2 plasmid quantity required to elicit a significant reduction of the p53 response. Again, Np9 was able to overcome this repression (Fig. 7B).
Figure 7.

Effect of Np9 on p53-induced gene transcription. (A) Np9 supports expression from a p53-responsive reporter plasmid. Luciferase gene reporter plasmids controlled by 15 copies of a mutated p53 response element (MG15-luc), 13 copies of the unmutated sequence (PG13-luc), or the p53-responsive p21 promoter, were transfected into p53-proficient U2OS cells. pCMV-np9 was cotransfected or not cotransfected. Cell extracts were prepared for standard luciferase assay at 24 h after transfection. T bars denote the standard deviations of the means derived from at least 3 transfections. P-values were determined with the Student's t-test (2-tailed). (B) Np9 can overcome the inhibitory effect of MDM2 on p53-mediated transactivation. Transfections of U2OS cells were performed as specified in A, only that this time pcmdm2 plasmid was included where indicated. (C) Efficiency of Np9 and p19ARF in suppressing the inhibitory effect of MDM2 on p53-mediated transactivation. Again, U2OS cells were transfected with plasmids producing the indicated proteins. The quantities of the plasmids expressing Np9 and p14ARF were adjusted according to the expression levels of HA-Np9 and HA-p14ARF in standard western blotting to achieve approximately equal levels of expression (Np9: 0.8, 1.6 and 2.4 μg. p14ARF: 0.5, 1.0 and 1.5 μg.) (D) Knockdown of np9 affects transcript levels from the p53-regulated genes KITLG and p21. HDFs were transfected with control siRNA (20 nM) or a mixture of 4 np9 siRNAs (5 nM each) for 24 h. Total RNA was prepared, reversely transcribed into cDNA, and subjected to quantitative PCR with primers specific for KITLG or p21. The diagrams show the results of 2 experiments; each experiment was performed in triplicate. T bars denote the standard deviations of the means, P-values were determined using Student's t-test (2-tailed). n.s., not significant.
The tumor suppressor p14ARF is a well-studied protein which like Np9 binds to MDM2's central acidic and zinc finger domain to inhibit MDM2's negative effect on p53-induced transactivation.48 To compare the effects of Np9 and p14ARF in this reporter assay, we first determined the relative expression levels of tagged Np9 and p14ARF by standard western blotting and then transfected U2OS cells as before (see Fig. 7A, B) but included adjusted amounts of plasmids expressing Np9 or p14ARF to approximately equal levels. In this assay, both p14ARF and Np9 suppressed the inhibition of p53 by MDM2 in a dose-dependent manner, although p14ARF was somewhat more efficient in this respect, in particular at low plasmid concentrations (Fig. 7C; compare gray and black columns).
Finally, we asked whether the knockdown by siRNA of np9 in HDF would yield an effect on endogenous p53-regulated genes. We chose to quantify, by qRT-PCR, the transcript levels from the KITLG (KIT ligand; stem cell factor) and p21 genes which happen to be the only known, so-called default p53-regulated genes in the cell, meaning that they are p53-inducible in most tested tissues and in response to most kinds of p53-activating stress.45 The results of 2 independent experiments, each performed in triplicate, are summarized in Figure 7D. Consistent with Np9 supporting p53, knockdown of np9 reduced the KITLG transcript level significantly. A similar trend was observed with the p21 transcript level. Combined, these data suggest that Np9 can aid p53 as a transcription factor. One mechanism may be the inhibition of MDM2.
MDM2-like proteins are around for more than 1 billion years.46 Paralogs of MDM2 and its relative MDMX (MDM4) can be found in all Euteleostomi (bony vertebrates). Altogether, these molecules have undergone substantial evolutionary change. However, the MDM2 proteins from Homo sapiens and Pan troglodytes (Chimpanzee), primates that harbour np9-encoding endogenous retrovirus (this study), are completely identical in their N-terminal domain that associates with p53 (aa 50–104), as well as in their zinc finger domain (aa 299–328) located within the central portion of MDM2 with which Np9 primarily associates. Only the acidic domain (aa 243–301) next to the zinc finger domain, that is also located within the Np9-interacting portion of the protein, exhibits a 2% variation in Pan troglodytes compared to Homo sapiens.47 This acidic domain also contains several serine residues that are targeted by the stress-induced cell cycle checkpoint kinases Chk1 and Chk2.48 Whether the amino acid variations, or perhaps the phosphorylations of the serine residues, affect the interaction of MDM2 and Np9 remains to be seen. In any event appears the central acidic and zinc finger domain of MDM2 to be a hub for the contact with cellular proteins that regulate the p53-MDM2 pathway.49 The already mentioned p14ARF protein binds there and like Np9 inhibits the MDM2-mediated ubiquitylation of p53.48 Similarly, the ribosomal proteins L5, L11 and L23 that leave the nucleolus in response to nucleolar or ribosomal stress, bind MDM2's central domain to activate p53.49 Perhaps of note in this context, Np9 can be localized in the nucleolus as well 35 and may thus be a novel member of the nucleolus-based p53-regulatory protein family.
HERV-K(HML-2) sequences are transcribed in many human normal tissues,21 yet strong protein expression, formation of retroviral particles and the induction of anti-ERV protein immune responses seem to be predominantly observed with stem cells, stem-like progenitor cells and germ cell-derived cancers, as well as some other types of cancers.11 A notable exception is human diploid fibroblasts which, as shown here, produce Np9 protein in culture. The human Tera-1 cells used throughout most of this study are malignant testicular germ cells consisting almost entirely of undifferentiated, so-called embryonic carcinoma (EC) cells. These are regarded as cancer stem cell-like cells and are related to the pluripotent embryonic stem (ES) cells that constitute the inner cell mass of the early embryo blastocyst.50 Tera-1 cells are nullipotent, i.e., they have lost their capacity to differentiate in response to stimuli such as retinoic acid. The reason that germ cell tumor-derived cells including Tera-1, express high levels of endogenous retroviral transcripts is perhaps due to the altogether low levels of repressive epigenetic methylation marks in these cells, and/or the presence of HERV-K(HML-2) specific activating transcription factors.51,52 Although some Np9 protein was detectable in unstressed Tera-1 cells, most of it was unstable and accumulated only upon proteasome inhibition. Intriguing recent findings by Fuchs and colleagues have documented that elevated transcript levels of np9 (as well as from other ERV genes) constitute markers for various pluripotent stem cells.17 However, these authors do not report of any Np9 protein in these cells. It is thus conceivable that the Np9/proteasome pathway has evolved, inter alia, as a sensor for natural proteasome inhibitors specifically in human, chimpanzee and gorilla. Proteasome inhibitors are widely regarded as powerful natural environmental toxins produced mostly by Actinomycetes. Chronic reduction in 26S proteasome activity has been linked, for instance, to a reduction in sperm count and quality in mice and rats.53,54
Alternatively or in addition, Np9 might fulfil functions in stem and progenitor cells in human, chimpanzee and gorilla that are independent of the proteasome. In this context it is perhaps important to note that among the first proteins discovered by us as interaction partners of Np9 were the Promyelocytic Leukemia Zinc Finger protein (PLZF) and the related Testicular Zinc Finger protein (TZF) (40,55; and unpublished results). Although originally implicated in cancers, these proteins are now recognized to control the self-renewal of spermatogonial stem cells.41,56 The p53-MDM2 pathway which we here have shown to be targeted by Np9 as well, seems to be an important regulator of stem cell number.57 Moreover, the p53-MDM2 connection appears to be intact and operational in embryonic stem cells.58 Thus, Np9 may have a role in stem cells by regulating the PLZF/TZF and p53/MDM2 axes specifically in few higher primates.
Materials and Methods
Analysis of HERV-K(HML-2) homologues in primate genome sequences
We identified HERV-K(HML-2) homologous sequences in primate genome sequences by similarity using BLAT at the UCSC Genome Browser and the UCSC Table Browser.59-61 The following primate genome sequences, as provided by the UCSC Genome Browser, were probed using the HERV-K(HML-2.HOM) sequence (GenBank acc. no. AF074086;15) excluding the LTR portions: papAnu2 (Papio anubis; baboon); rheMac3 (Macaca mulatta; rhesus monkey); nomLeu3 (Nomascus leucogenys; white-cheeked gibbon); ponAbe2 (Pongo pygmaeus abelii; orang- utan); gorGor3 (Gorilla gorilla gorilla; gorilla); panTro4 (Pan troglodytes; chimpanzee). Genomic sequences corresponding to chromosome coordinates of matching genome regions, plus 1 kb of upstream and downstream flanking sequence, were retrieved and multiply aligned using MAFFT and the FFT-NS-i iterative refinement option.62 Multiple alignments were further optimized by hand and proviral portions in the alignment relevant for this study were analyzed in detail. A previously generated multiple alignment of HERV-K(HML-2) sequences extracted from the hg18/NCBI Build 36.1 human genome reference sequence (for instance, see 63) was used in the course of this analysis.
Cell cultures and transfections
Tera-1 cells were grown at 37°C and in a 5% CO2 atmosphere in McCoy's 5a medium, while H1299 and U2OS cells were maintained in DMEM (Gibco), all supplemented with 10% FCS and antibiotics. Peripheral blood mononuclear cells (PBMC) were isolated from 20 ml of whole blood donated by healthy volunteers, using standard Ficoll gradient centrifugation. PBMC were cultured at 37°C, 5% CO2, for up to 48 h in RPMI medium (Sigma) containing 10% FCS and antibiotics. Human diploid fibroblasts (HDF) were grown and maintained as reported by us previously.64 For transient transfection of DNA, cells were seeded to reach 60–70% confluency at transfection and were then transfected with jetPEI reagent (Polyplus, Illkirch, France), following the manufacturer's instructions. Transfection of siRNA (20 nM) was performed with the HiPerFect transfection reagent (Qiagen), as recommended by the supplier. The following siRNAs were used:
MDM2: UCAUCGGACUCAGGUACAUTT (sense)
AUGUACCUGAGUCCGAUGATT (antisense)
np9-1: CCAUCGGAGAUGCAAAGAATT (sense)
UUCUUUGCAUCUCCGAUGGGT (antisense)
np9-2: CAUCGGAGAUGCAAAGAAATT (sense)
UUUCUUUGCAUCUCCGAUGGG (antisense)
np9-3: GCAAGAGAGAUCAGAUUGUTT (sense)
ACAAUCUGAUCUCUCUUGCTT (antisense)
np9-4: GAAGUAGACAUAGGAGACUTT (sense)
AGUCUCCUAUGUCUACUUCTT (antisense)
control: UUCUCCGAACGUGUCACGUTT (sense)
ACGUGACACGUUCGGAGAATT (antisense)
Plasmids, reagents and antibodies
pCMV-np9 was constructed by PCR-cloning of the np9 sequence (with np9 proviral region being identical to the corresponding sequence portions in GenBank accession number: AF164609) into expression plasmid pCMV-pA. The various GST-MDM2 and GST-Np9 constructs were obtained by PCR-cloning into vector pGEX-4T1 (Amersham). Cloning details will be provided on request. pcDNA3-HA-ubiquitin was purchased from Addgene. pcmdm2, pCMV-p53, p21-luc, MG15-luc and PG13-luc have been used by us before.64,65 Epoxomicin was purchased from Enzo (Farmingdale, USA), and MG132 from Sigma, as were the β-actin monoclonal antibody AC-15, the peroxidase-conjugated secondary anti-mouse and anti-rabbit antibodies, and the monoclonal anti-glutathione-s-transferase antibody (clone GST-2). The secondary anti-rat antibody was provided by one of us (EK). The anti-retinoblastoma monoclonal antibody was obtained from BD PharMingen, and the anti-α-tubulin monoclonal antibody from abcam. The monoclonal anti-MDM2 antibody 3G9 was from Millipore. The irrelevant monoclonal anti-HRS3 IgG was kindly provided by Michael Pfreundschuh (Internal Medicine I, Homburg, Germany). The monoclonal anti-HA antibody was from Covance, and the monoclonal anti-p53 antibody DO-1 was purchased from Santa Cruz Biotechnology. For production of anti-NP9 monoclonal antibodies, a peptide encompassing amino acids L16QVYPTAPKRQRPSRTGHDDDGG32 of human Np9 of HERK-K(HML-2), coupled to ovalbumin, was used to immunize Lou/C rats according to standard protocol. Clones designated 22E4 and 10B1 (rat IgG2a) that reacted in western blots with endogenous Np9 in Tera-1 cells and with transfected Np9-HA in HEK293-T cells were stably subcloned and used for further analysis. Details on the development of the Np9 antibodies will be provided on request by EK ( kremmer@helmholtz-muenchen.de).
Immunoprecipitation
Coimmunoprecipitation of Np9 and MDM2 was performed according to our recently published, detailed protocol.65 For the study of in vivo ubiquitylation of Np9, MDM2 and p53, the proteins were immunoprecipitated from denatured cell extracts.65 In brief, H1299 cells were transfected with the relevant plasmids. After 24 h, cells were treated with 10 µM MG132 for another 4 h. Cells were then washed in cold PBS; 1/10 of the cells was saved as input control. The rest was lysed in 400 µl TBS-lysis buffer (1% SDS in TBS) per 10 cm-dish at 95°C for 5 min. Lysates were squeezed repeatedly through a 23 Gauge needle and vortexed vigorously for 10 seconds. 800 µl TBS-Triton buffer (1.5% Triton X-100 in TBS) per 400 µl lysate was added and mixed prior to incubation with 100 µl of a 1 : 1 mix of protein G and protein A sepharose 4 Fast Flow (GE Healthcare), for 1 h on a rotating wheel at 4°C (pre-clearing). Samples were centrifuged for 5 min at max speed and supernatant was incubated with 100 µl of a 1 : 1 mix of protein G and protein A sepharose 4 Fast Flow pre-conjugated with 4 µg of the indicated antibody, for at least 4 h at 4°C on a rotating wheel. Samples were washed 3 times in 1 ml cold TBS mix (1 part TBS-lysis buffer plus 2 parts TBS-Triton buffer); beads were resuspended in 30 µl of 95°C SDS-sample buffer (100 mM Tris-HCl (pH 6.8), 100 mM DTT, 4 % SDS, and 20 % glycerol) and were boiled for 10 min. The proteins were separated by SDS-PAGE, immobilized on PVDF membrane (Immobilon P, Millipore) and detected by the indicated antibodies.65
Western blot analysis
Cells were lysed in standard SDS lysis buffer (100 mM Tris-HCl pH 6.8, 4% SDS and 20% glycerol) at 100°C. Fifteen microgram of total cellular protein was run on an 8–14% SDS polyacrylamide gel and then transferred to a PVDF membrane (Immobilon-P, Millipore). The membrane was submersed in PBS supplemented with 5% (w/w) dry milk powder, and was incubated overnight with antibodies as indicated in the figure legends. The bound primary antibodies were detected by incubating the membranes for 1 h with a peroxidase-conjugated secondary anti-mouse (1 : 10,000), anti-rabbit (1 : 2,000) or anti-rat (1 : 10,000) antibodies. Signals were detected by the Thermo Scientific ECL western blotting substrate, as recommended by the manufacturer.
GST pull-down assay
GST pull-down analyses were performed by immobilizing equal amounts of GST, GST-MDM2, GST-MDM2 deletion mutants, or GST-Np9, respectively, on Gluthation-Sepharose beads (GE-Healthcare), following the protocol published by us recently.65 In brief, the beads were washed 5 times with GST low salt buffer (50 mM Tris/HCl, 200 mM NaCl, 0.8 mM EDTA, 0.1% NP40, 1 mM PMSF, 10 µg/ml aprotinin) and incubated with equal amounts of 35S-Np9 or 35S-MDM2 (in vitro translated). The in vitro translation was carried out with the TNT-T7 Coupled Reticulocyte Lysate System (Promega), according to the manufacturer's protocol, with 1 µg plasmid and 35S radiolabeled cysteine and methionine (Tran35S-Label™, MP Biomedicals). After overnight incubation at 4°C, all probes were washed 5 times with GST high salt buffer (50 mM Tris/HCl, 500 mM NaCl, 0.8 mM EDTA, 0.1% NP40). GST protein complexes were eluted from the sepharose by adding SDS-sample buffer (100 mM Tris-HCl (pH 6.8), 100 mM DTT, 4% SDS, and 20% glycerol) and by boiling samples for 10 min. The proteins were separated by SDS-PAGE, immobilized on PVDF membrane (Immobilon-P, Millipore) and visualized by autoradiography. GST-proteins were detected with the monoclonal anti-glutathione-s-transferase antibody (clone GST-2).
Quantitative Reverse Transcription-PCR
Total cellular RNA was prepared as detailed by us recently.65 Cells were lysed in solution D (236.4 g guanidium thiocyanate in 293 ml water, 17.6 ml 0.75 M sodium citrate pH 7.0, and 26.4 ml 10% sarcosyl, 0.72% 2-mercaptoethanol). Lysate was harvested and 0.1 ml of 2 M sodium acetate pH 4, 0.1 ml of water-saturated phenol (Roth), and 0.2 ml of chloroform-isoamylalcohol (49 : 1) were added, mixed, and cooled on ice for 15 min. After centrifugation (10000 g, 20 min, 4°C), the aqueous phase was collected and precipitated with isopropanol at −20°C over night. After a further centrifugation (10,000 g, 20 min, 4°C), RNA was dissolved in solution D and precipitated with isopropanol at −20°C for 1 h. The pellet was washed in 70% ethanol and dissolved in DEPC-water. The RNA was digested with RNase-free DNase I (Roche) for 60 min at 37°C, and 4 µg was used for the first-strand cDNA synthesis with SuperScript™III (Invitrogen, USA) as specified by the supplier. Quantitative RT-PCR analysis for KITLG, p21, and gapdh was performed with the LightCycler® FastStart DNA Master SYBR Green I from Roche. The conditions were: KITLG primers; TA: 58°C; final concentration of primers: 0.5 µM; final MgCl2 concentration: 2 mM; p21 primers; TA: 62°C; final concentration of primers: 0.5 µM; final MgCl2 concentration: 2 mM. The following primers were employed:
KITLG: TCTGCAGGAACTGTGTGACT (forward)
TGGGTTCTGGGCTCTTGAAT (reverse)
p21: GGCGGCAGACCAGCATGACAGATT (forward)
ATGAAGCCGGCCCACCCAACCTC (reverse)
gapdh: TGGTATCGTGGAAGGACTCATGAC (forward)
AGTCCAGTGAGCTTCCCGTTCAGC (reverse)
Reporter gene assay
H1299 cells or U2OS cells were seeded into 24-well dishes at approximately 2 × 105 cells per dish. After 24 h, cells were transfected with jetPEI transfection reagent (Polyplus, Illkirch, France), as recommended by the supplier. At the time points indicated in the figure legends, cells were harvested, cell extracts were prepared and luciferase activity was determined with the Luciferase Assay System (Promega), as specified by the manufacturer.
Statistical analysis
P-values were determined using the paired Student's t-test. P-values <0.05 were considered significant.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Michael Pfreundschuh (Internal Medicine I, University Medical Center, Homburg, Germany) for materials, and the Mayer and Roemer Lab members for lively discussions.
Funding
This work was supported by grants from the DFG to JM and HOMFOR to FG and KR.
Author Contributions
KH and KK carried out all lab experiments except antibody development and the in vivo ubiquitylation assays; EK developed the Np9 antibodies; MB and FG tested the Np9 antibodies; JM analyzed np9 evolution, wrote the evolution part of the paper; KR performed the in vivo ubiquitylation assays, conceived and supervised the study, and wrote the paper.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- 1.Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, et al.. Initial sequencing and analysis of the human genome. Nature 2001; 409:860-921; PMID:11237011; http://dx.doi.org/ 10.1038/35057062 [DOI] [PubMed] [Google Scholar]
- 2.Stoye JP. Studies of endogenous retroviruses reveal a continuing evolutionary saga. Nat Rev Microbiol 2012; 10:395-406; PMID:22565131 [DOI] [PubMed] [Google Scholar]
- 3.Blomberg J, Benachenhou F, Blikstad V, Sperber G, Mayer J. Classification and nomenclature of endogenous retroviral sequences (ERVs): problems and recommendations. Gene 2009; 448:115-23; PMID:19540319; http://dx.doi.org/ 10.1016/j.gene.2009.06.007 [DOI] [PubMed] [Google Scholar]
- 4.Mayer J, Blomberg J, Seal RL. A revised nomenclature for transcribed human endogenous retroviral loci. Mob DNA 2011; 2:7; PMID:21542922; http://dx.doi.org/ 10.1186/1759-8753-2-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Flockerzi A, Ruggieri A, Frank O, Sauter M, Maldener E, Kopper B, Wullich B, Seifarth W, Müller-Lantzsch N, Leib-Mösch C, et al.. Expression patterns of transcribed human endogenous retrovirus HERV-K(HML-2) loci in human tissues and the need for a HERV Transcriptome Project. BMC Genomics 2008; 9:354; PMID:18664271; http://dx.doi.org/ 10.1186/1471-2164-9-354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Seifarth W, Frank O, Zeilfelder U, Spiess B, Greenwood AD, Hehlmann R, Leib-Mösch C. Comprehensive analysis of human endogenous retrovirus transcriptional activity in human tissues with a retrovirus-specific microarray. J Virol 2005; 79:341-52; PMID:15596828; http://dx.doi.org/ 10.1128/JVI.79.1.341-352.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mallet F, Bouton O, Prudhomme S, Cheynet V, Oriol G, Bonnaud B, Lucotte G, Duret L, Mandrand B. The endogenous retroviral locus ERVWE1 is a bona fide gene involved in hominoid placental physiology. Proc Natl Acad Sci USA 2004; 101:1731-6; PMID:14757826; http://dx.doi.org/ 10.1073/pnas.0305763101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dupressoir A, Lavialle C, Heidmann T. From ancestral infectious retroviruses to bona fide cellular genes: role of the captured syncytins in placentation. Placenta 2012; 33:663-71; PMID:22695103; http://dx.doi.org/ 10.1016/j.placenta.2012.05.005 [DOI] [PubMed] [Google Scholar]
- 9.Oja M, Peltonen J, Blomberg J, Kaski S. Methods for estimating human endogenous retrovirus activities from EST databases. BMC Bioinformatics 2007; 8 Suppl 2:S11; PMID:17493249; http://dx.doi.org/ 10.1186/1471-2105-8-S2-S11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Perot P, Mugnier N, Montgiraud C, Gimenez J, Jaillard M, Bonnaud B, Mallet F. Microarray-based sketches of the HERV transcriptome landscape. PLoS One 2012; 7:e40194; PMID:22761958; http://dx.doi.org/ 10.1371/journal.pone.0040194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ruprecht K, Mayer J, Sauter M, Roemer K, Mueller-Lantzsch N. Endogenous retroviruses and cancer. Cell Mol Life Sci 2008; 65:3366-82; PMID:18818873; http://dx.doi.org/ 10.1007/s00018-008-8496-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kitamura Y, Ayukawa T, Ishikawa T, Kanda T, Yoshiike K. Human endogenous retrovirus K10 encodes a functional integrase. J Virol 1996; 70:3302-6; PMID:8627815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schommer S, Sauter M, Krausslich HG, Best B, Mueller-Lantzsch N. Characterization of the human endogenous retrovirus K proteinase. J Gen Virol 1996; 77 (Pt 2):375-9; PMID:8627242; http://dx.doi.org/ 10.1099/0022-1317-77-2-375 [DOI] [PubMed] [Google Scholar]
- 14.Barbulescu M, Turner G, Seaman MI, Deinard AS, Kidd KK, Lenz J. Many human endogenous retrovirus K (HERV-K) proviruses are unique to humans. Curr Biol 1999; 9:861-8; PMID:10469592; http://dx.doi.org/ 10.1016/S0960-9822(99)80390-X [DOI] [PubMed] [Google Scholar]
- 15.Mayer J, Sauter M, Racz A, Scherer D, Mueller-Lantzsch N, Meese E. An almost intact human endogenous retrovirus K on human chromosome 7. Nat Genet 1999; 21:257-8; PMID:10080172; http://dx.doi.org/ 10.1038/6766 [DOI] [PubMed] [Google Scholar]
- 16.Tonjes RR, Czauderna F, Kurth R. Genome-wide screening, cloning, chromosomal assignment, and expression of full-length human endogenous retrovirus type K. J Virol 1999; 73:9187-95; PMID:10516026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fuchs NV, Loewer S, Daley GQ, Izsvak Z, Lower J, Lower R. Human endogenous retrovirus K (HML-2) RNA and protein expression is a marker for human embryonic and induced pluripotent stem cells. Retrovirology 2013; 10:115; PMID:24156636; http://dx.doi.org/ 10.1186/1742-4690-10-115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lower R, Tonjes RR, Korbmacher C, Kurth R, Lower J. Identification of a Rev-related protein by analysis of spliced transcripts of the human endogenous retroviruses HTDV/HERV-K. J Virol 1995; 69:141-9; PMID:7983704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Armbruester V, Sauter M, Krautkraemer E, Meese E, Kleiman A, Best B, Roemer K, Mueller-Lantzsch N. A novel gene from the human endogenous retrovirus K expressed in transformed cells. Clin Cancer Res 2002; 8:1800-7; PMID:12060620 [PubMed] [Google Scholar]
- 20.Mayer J, Meese E, Mueller-Lantzsch N. Human endogenous retrovirus K homologous sequences and their coding capacity in Old World primates. J Virol 1998; 72:1870-5; PMID:9499038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schmitt K, Heyne K, Roemer K, Meese E, Mayer J. HERV-K(HML-2) rec and np9 transcripts not restricted to disease but present in many normal human tissues. Mobile DNA 2015; 6:4; accepted; PMID:25750667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vousden KH, Lane DP. p53 in health and disease. Nat Rev Mol Cell Biol 2007; 8:275-83; PMID:17380161; http://dx.doi.org/ 10.1038/nrm2147 [DOI] [PubMed] [Google Scholar]
- 23.Vousden KH, Prives C. Blinded by the Light: The Growing Complexity of p53. Cell 2009; 137:413-31; PMID:19410540; http://dx.doi.org/ 10.1016/j.cell.2009.04.037 [DOI] [PubMed] [Google Scholar]
- 24.Hu W, Zheng T, Wang J. Regulation of Fertility by the p53 Family Members. Genes Cancer 2011; 2:420-30; PMID:21779510; http://dx.doi.org/ 10.1177/1947601911408892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Feng Z, Lin M, Wu R. The Regulation of Aging and Longevity: A New and Complex Role of p53. Genes Cancer 2011; 2:443-52; PMID:21779512; http://dx.doi.org/ 10.1177/1947601911410223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moll UM, Slade N. p63 and p73: roles in development and tumor formation. Mol Cancer Res 2004; 2:371-86; PMID:15280445 [PubMed] [Google Scholar]
- 27.Candi E, Dinsdale D, Rufini A, Salomoni P, Knight RA, Mueller M, Krammer PH, Melino G. TAp63 and DeltaNp63 in cancer and epidermal development. Cell Cycle 2007; 6:274-85; PMID:17264681; http://dx.doi.org/ 10.4161/cc.6.3.3797 [DOI] [PubMed] [Google Scholar]
- 28.Pietsch EC, Humbey O, Murphy ME. Polymorphisms in the p53 pathway. Oncogene 2006; 25:1602-11; PMID:16550160; http://dx.doi.org/ 10.1038/sj.onc.1209367 [DOI] [PubMed] [Google Scholar]
- 29.Whibley C, Pharoah PD, Hollstein M. p53 polymorphisms: cancer implications. Nat Rev Cancer 2009; 9:95-107; PMID:19165225; http://dx.doi.org/ 10.1038/nrc2584 [DOI] [PubMed] [Google Scholar]
- 30.Contente A, Zischler H, Einspanier A, Dobbelstein M. A promoter that acquired p53 responsiveness during primate evolution. Cancer Res 2003; 63:1756-8; PMID:12702557 [PubMed] [Google Scholar]
- 31.Sohr S, Engeland K. The tumor suppressor p53 induces expression of the pregnancy-supporting human chorionic gonadotropin (hCG) CGB7 gene. Cell Cycle 2011; 10:3758-67; PMID:22032922; http://dx.doi.org/ 10.4161/cc.10.21.17946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wade M, Wang YV, Wahl GM. The p53 orchestra: Mdm2 and Mdmx set the tone. Trends Cell Biol 2010; 20:299-309; PMID:20172729; http://dx.doi.org/ 10.1016/j.tcb.2010.01.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bond GL, Levine AJ. A single nucleotide polymorphism in the p53 pathway interacts with gender, environmental stresses and tumor genetics to influence cancer in humans. Oncogene 2007; 26:1317-23; PMID:17322917; http://dx.doi.org/ 10.1038/sj.onc.1210199 [DOI] [PubMed] [Google Scholar]
- 34.Lower R, Boller K, Hasenmaier B, Korbmacher C, Muller-Lantzsch N, Lower J, Kurth R. Identification of human endogenous retroviruses with complex mRNA expression and particle formation. Proc Natl Acad Sci U S A 1993; 90:4480-4; PMID:8506289; http://dx.doi.org/ 10.1073/pnas.90.10.4480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Armbruester V, Sauter M, Roemer K, Best B, Hahn S, Nty A, Schmid A, Philipp S, Mueller A, Mueller-Lantzsch N. Np9 protein of human endogenous retrovirus K interacts with ligand of numb protein X. J Virol 2004; 78:10310-9; PMID:15367597; http://dx.doi.org/ 10.1128/JVI.78.19.10310-10319.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen T, Meng Z, Gan Y, Wang X, Xu F, Gu Y, Xu X, Tang J, Zhou H, Zhang X, et al.. The viral oncogene Np9 acts as a critical molecular switch for co-activating beta-catenin, ERK, Akt and Notch1 and promoting the growth of human leukemia stem/progenitor cells. Leukemia 2013; 27:1469-78; PMID:23307033; http://dx.doi.org/ 10.1038/leu.2013.8 [DOI] [PubMed] [Google Scholar]
- 37.Gross H, Barth S, Pfuhl T, Willnecker V, Spurk A, Gurtsevitch V, Sauter M, Hu B, Noessner E, Mueller-Lantzsch N, et al.. The NP9 protein encoded by the human endogenous retrovirus HERV-K(HML-2) negatively regulates gene activation of the Epstein-Barr virus nuclear antigen 2 (EBNA2). Int J Cancer 2011; 129:1105-15; PMID:21710493; http://dx.doi.org/ 10.1002/ijc.25760 [DOI] [PubMed] [Google Scholar]
- 38.Armbruester V, Sauter M, Roemer K, Best B, Hahn S, Nty A, Schmid A, Philipp S, Mueller A, Mueller-Lantzsch N. Np9 protein of human endogenous retrovirus K interacts with ligand of numb protein X A novel gene from the human endogenous retrovirus K expressed in transformed cells. J Virol 2004; 78:10310-9; PMID:15367597; http://dx.doi.org/ 10.1128/JVI.78.19.10310-10319.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kisselev AF, van der Linden WA, Overkleeft HS. Proteasome inhibitors: an expanding army attacking a unique target. Chem Biol 2012; 19:99-115; PMID:22284358; http://dx.doi.org/ 10.1016/j.chembiol.2012.01.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Denne M, Sauter M, Armbruester V, Licht JD, Roemer K, Mueller-Lantzsch N. Physical and functional interactions of human endogenous retrovirus proteins Np9 and rec with the promyelocytic leukemia zinc finger protein. J Virol 2007; 81:5607-16; PMID:17360752; http://dx.doi.org/ 10.1128/JVI.02771-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Buaas FW, Kirsh AL, Sharma M, McLean DJ, Morris JL, Griswold MD, de Rooij DG, Braun RE. Plzf is required in adult male germ cells for stem cell self-renewal. Nat Genet 2004; 36:647-52. Epub 2004 May 23; PMID:15156142; http://dx.doi.org/ 10.1038/ng1366 [DOI] [PubMed] [Google Scholar]
- 42.Nie J, McGill MA, Dermer M, Dho SE, Wolting CD, McGlade CJ. LNX functions as a RING type E3 ubiquitin ligase that targets the cell fate determinant Numb for ubiquitin-dependent degradation. EMBO J 2002; 21:93-102; PMID:11782429; http://dx.doi.org/ 10.1093/emboj/21.1.93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Armbruester V, Sauter M, Roemer K, Best B, Hahn S, Nty A, Schmid A, Philipp S, Mueller A, Mueller-Lantzsch N. Np9 protein of human endogenous retrovirus K interacts with ligand of numb protein X. J Virol 2004; 78:10310-9; PMID:15367597; http://dx.doi.org/ 10.1128/JVI.78.19.10310-10319.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Poyurovsky MV, Katz C, Laptenko O, Beckerman R, Lokshin M, Ahn J, Byeon IJ, Gabizon R, Mattia M, Zupnick A, et al.. The C terminus of p53 binds the N-terminal domain of MDM2. Nat Struct Mol Biol 2010; 17:982-9; PMID:20639885; http://dx.doi.org/ 10.1038/nsmb.1872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zeron-Medina J, Wang X, Repapi E, Campbell MR, Su D, Castro-Giner F, Davies B, Peterse EF, Sacilotto N, Walker GJ, et al.. A polymorphic p53 response element in KIT ligand influences cancer risk and has undergone natural selection. Cell 2013; 155:410-22; PMID:24120139; http://dx.doi.org/ 10.1016/j.cell.2013.09.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lane DP, Verma C. Mdm2 in evolution. Genes Cancer 2012; 3:320-4; PMID:23150765; http://dx.doi.org/ 10.1177/1947601912458285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Momand J, Villegas A, Belyi VA. The evolution of MDM2 family genes. Gene 2011; 486:23-30; PMID:21762762; http://dx.doi.org/ 10.1016/j.gene.2011.06.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Weber JD, Taylor LJ, Roussel MF, Sherr CJ, Bar-Sagi D. Nucleolar Arf sequesters Mdm2 and activates p53. Nat Cell Biol 1999; 1:20-6; PMID:10559859; http://dx.doi.org/ 10.1038/8991 [DOI] [PubMed] [Google Scholar]
- 49.Zhang Y, Lu H. Signaling to p53: ribosomal proteins find their way. Cancer Cell 2009; 16:369-77; PMID:19878869; http://dx.doi.org/ 10.1016/j.ccr.2009.09.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gokhale PJ, Andrews PW. The development of pluripotent stem cells. Curr Opin Genet Dev 2012; 22:403-8; PMID:22868175; http://dx.doi.org/ 10.1016/j.gde.2012.07.006 [DOI] [PubMed] [Google Scholar]
- 51.Almstrup K, Nielsen JE, Mlynarska O, Jansen MT, Jorgensen A, Skakkebaek NE, Rajpert-De Meyts E. Carcinoma in situ testis displays permissive chromatin modifications similar to immature foetal germ cells. Br J Cancer 2010; 103:1269-76; PMID:20823885; http://dx.doi.org/ 10.1038/sj.bjc.6605880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lavie L, Kitova M, Maldener E, Meese E, Mayer J. CpG methylation directly regulates transcriptional activity of the human endogenous retrovirus family HERV-K(HML-2). Journal of virology 2005; 79:876-83; PMID:15613316; http://dx.doi.org/ 10.1128/JVI.79.2.876-883.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shi G, Chen D, Zhai G, Chen MS, Cui QC, Zhou Q, He B, Dou QP, Jiang G. The proteasome is a molecular target of environmental toxic organotins. Environ Health Perspect 2009; 117:379-86; PMID:19337512; http://dx.doi.org/ 10.1289/ehp.11865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Si J, Li P, Xin Q, Li X, An L, Li J. Perinatal exposure to low doses of tributyltin chloride reduces sperm count and quality in mice. Environ Toxicol 2015; 30:44-52; PMID:23913619; http://dx.doi.org/ 10.1002/tox.21892 [DOI] [PubMed] [Google Scholar]
- 55.Kaufmann S, Sauter M, Schmitt M, Baumert B, Best B, Boese A, Roemer K, Mueller-Lantzsch N. Human endogenous retrovirus protein Rec interacts with the testicular zinc-finger protein and androgen receptor. J Gen Virol 2010; 91:1494-502; PMID:20147518; http://dx.doi.org/ 10.1099/vir.0.014241-0 [DOI] [PubMed] [Google Scholar]
- 56.Costoya JA, Hobbs RM, Barna M, Cattoretti G, Manova K, Sukhwani M, Orwig KE, Wolgemuth DJ, Pandolfi PP. Essential role of Plzf in maintenance of spermatogonial stem cells. Nat Genet 2004; 36:653-9. Epub 2004 May 23; PMID:15156143; http://dx.doi.org/ 10.1038/ng1367 [DOI] [PubMed] [Google Scholar]
- 57.Spike BT, Wahl GM. p53, Stem Cells, and Reprogramming: Tumor Suppression beyond Guarding the Genome. Genes Cancer 2011; 2:404-19; PMID:21779509; http://dx.doi.org/ 10.1177/1947601911410224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, et al.. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004; 303:844-8; PMID:14704432; http://dx.doi.org/ 10.1126/science.1092472 [DOI] [PubMed] [Google Scholar]
- 59.Kent WJ. BLAT–the BLAST-like alignment tool. Genome Res 2002; 12:656-64; PMID:11932250; http://dx.doi.org/ 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D. The human genome browser at UCSC. Genome Res 2002; 12:996-1006; PMID:12045153; http://dx.doi.org/ 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Karolchik D, Hinrichs AS, Furey TS, Roskin KM, Sugnet CW, Haussler D, Kent WJ. The UCSC Table Browser data retrieval tool. Nucleic acids Res 2004; 32:D493-6; PMID:14681465; http://dx.doi.org/ 10.1093/nar/gkh103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 2013; 30:772-80; PMID:23329690; http://dx.doi.org/ 10.1093/molbev/mst010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schmitt K, Reichrath J, Roesch A, Meese E, Mayer J. Transcriptional profiling of human endogenous retrovirus group HERV-K(HML-2) loci in melanoma. Genome Biol Evol 2013; 5:307-28; PMID:23338945; http://dx.doi.org/ 10.1093/gbe/evt010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Heyne K, Willnecker V, Schneider J, Conrad M, Raulf N, Schule R, Roemer K. NIR, an inhibitor of histone acetyltransferases, regulates transcription factor TAp63 and is controlled by the cell cycle. Nucleic Acids Res 2010; 38:3159-71; PMID:20123734; http://dx.doi.org/ 10.1093/nar/gkq016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Heyne K, Forster J, Schule R, Roemer K. Transcriptional repressor NIR interacts with the p53-inhibiting ubiquitin ligase MDM2. Nucleic Acids Res 2014; 42:3565-79; PMID:24413661; http://dx.doi.org/ 10.1093/nar/gkt1371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Martin RD. Primates. Curr Biol 2012; 22:R785-90; PMID:23017987; http://dx.doi.org/ 10.1016/j.cub.2012.07.015 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.