

Abstract
Rhadomyosarcoma (RMS) is the most common soft tissue sarcoma of childhood. RMS cells resemble fetal myoblasts but are unable to complete myogenic differentiation. In previous work we showed that miR-206, which is low in RMS, when induced in RMS cells promotes the resumption of differentiation by modulating more than 700 genes. To better define the pathways involved in the conversion of RMS cells into their differentiated counterpart, we focused on 2 miR-206 effectors emerged from the microarray analysis, SMYD1 and G6PD. SMYD1, one of the most highly upregulated genes, is a H3K4 histone methyltransferase. Here we show that SMYD1 silencing does not interfere with the proliferative block or with the loss anchorage independence imposed by miR-206, but severely impairs differentiation of ERMS, ARMS, and myogenic cells. Thus SMYD1 is essential for the activation of muscle genes. Conversely, among the downregulated genes, we found G6PD, the enzyme catalyzing the rate-limiting step of the pentose phosphate shunt. In this work, we confirmed that G6PD is a direct target of miR-206. Moreover, we showed that G6PD silencing in ERMS cells impairs proliferation and soft agar growth. However, G6PD overexpression does not interfere with the pro-differentiating effect of miR-206, suggesting that G6PD downmodulation contributes to - but is not an absolute requirement for - the tumor suppressive potential of miR-206. Targeting cancer metabolism may enhance differentiation. However, therapeutic inhibition of G6PD is encumbered by side effects. As an alternative, we used DCA in combination with miR-206 to increase the flux of pyruvate into the mitochondrion by reactivating PDH. DCA enhanced the inhibition of RMS cell growth induced by miR-206, and sustained it upon miR-206 de-induction. Altogether these results link miR-206 to epigenetic and metabolic reprogramming, and suggest that it may be worth combining differentiation-inducing with metabolism-directed approaches.
Keywords: differentiation therapy, metabolism and cancer, miR-206, Rhabdomyosarcoma
Abbreviations
- DCA
Dichloroacetate
- DHEA
Dehydroepiandrosterone
- G6PD
Glucose 6 Phosphate Dehydrogenase
- HMT
Histone MethylTransferase
- MREs
MicroRNA Responsive Elements
- myomiRs
muscle-specific microRNAs
- MRFs
Myogenic Regulatory Factors
- PDH
Pyruvate Dehydrogenase
- PDK
Pyruvate Dehydrogenase Kinase
- PPP
Pentose Phosphate Pathway
- RMS
Rhabdomyosarcoma
- SMYD1
SET and MYND domain-containing protein 1
- TCA cycle
TriCarboxylic Acid cycle
Introduction
Rhabdomyosarcoma (RMS), the most common soft tissue sarcoma of childhood, consists of 2 major subtypes, alveolar (ARMS) and embryonal (ERMS), different in terms of presentation, clinical outcome and genetic lesions. Both subtypes are characterized by expression of skeletal muscle markers, such as Alpha-Actin, Myosin Heavy Chain, Desmin, Myoglobin, Z-band protein and MyoD.1,2 RMS cells resemble myogenic precursors arrested in their path to differentiation. The muscle regulatory factors (MRFs) MyoD and Myogenin are present, but largely non-functional.3 Among the genes which fail to be activated in RMS, are those for 2 closely related muscle-enriched microRNAs, miR-206 and miR-1,4 which collaborate with the MRFs in implementing the myogenic program.5,6 Since they share the seed sequence they are likely to downregulate common targets.7 However their transcription is temporally out of phase, miR-206 being more precocious and highest in newly formed myotubes, and miR-1 coming up later and remaining high in mature muscle, thus they may have at least some distinct functional roles.8
In previous work we reported that forced expression of miR-206 reprograms the global expression profile of both RMS subtypes toward that of normal muscle.9 During myogenesis there is a shift of MyoD from being in complex with transcriptional repressors to binding co-activators. It has been proposed that RMS cells are trapped in an undifferentiated state by the presence of inhibitors, which tip the balance of MyoD-containing complexes toward the inactive rather than the active status.10,11 MiR-206 acts as a genetic switch turning on myogenesis by repressing the MyoD inhibitor Musculin.12 This tilts the balance toward active MyoD, which further enhances miR-206 transcription. Besides freeing MyoD from its inhibitory partners, miR-206 cooperates with the MRFs by contributing to the block of proliferation via direct downregulation of targets such as Pola1,5 Cyclin D113,14 and Cyclin D2,15 Met,9,16 Pax3,15,17 Pax7,18 YY1,19 HDAC4,20,21 Snail,22 Notch3,23 and BAF53a,24 and by indirectly reactivating differentiation genes. In this work, in the effort of identifying new effectors of miR-206 critical for its differentiation-promoting activity, we focused on 2 genes emerged from the original global expression analysis of miR-206-converted RMS cells.9 The first one – the histone methyltransferase SMYD1- was upregulated, likely as a consequence of MyoD activation. The second one – glucose 6 phosphate dehydrogenase (G6PD) - was strongly downregulated, behaving as a potential direct target.9 These molecules link miR-206 to the modulation of the epigenetic machinery and metabolism, 2 aspects of cell physiology that are presently the focus of great interest in cancer research.
SMYD1 is one of the members of the SET-MYND histone methyltransferases subfamily (SMYD1-5).25 SMYD1 and 3 specifically methylate lysine 4 of H3, suggesting a function as transcriptional activators.26 In SMYD1 the MYND domain is required for interaction with skNAC, a transcriptional activator specific to heart and skeletal muscle.27 Targeted deletion of SMYD1 or skNAC in mice results in both cases in right ventricular hypoplasia.28,29 However, in SMYD1 null mice the defect is more severe, and the null embryos die in mid gestation.28 Conversely, some skNAC mutants survive. They show reduced muscle mass and impaired regeneration after injury,29 suggesting that the SMYD1-skNAC complex has a role also in skeletal muscle. SMYD1 is a transcriptional target of SRF and Myogenin, and it has been reported that its forced expression accelerates C2C12 myoblast differentiation.30 Studies in Zebrafish have unveiled a cytoplasmic function for SMYD1, which is independent from its methyltransferase activity31 and mediated by nuclear to cytoplasmic shuttling via sumoylation.32 In the cytoplasm SMYD1 binds myosin and works together with chaperones to control myosin folding, degradation, and assembly into sarcomeres during myofibrillogenesis.26,31,33,34 In RMS cells SMYD1 is strongly upregulated after miR-206 induction.9 In this work we show that this upregulation is essential for differentiation of RMS and myogenic cells. In fact, SMYD1 silencing, although without consequences on the block of proliferation imposed by miR-206, significantly impairs expression of muscle genes.
G6PD is the first enzyme of the pentose phosphate pathway (PPP), which in the oxidative phase yields NADPH (necessary for reductive biosynthesis) and ribose-5-phosphate, an essential precursor for nucleotides biosynthesis. In the anoxydative phase excess pentose-phosphates are converted into intermediates which may serve as biosynthetic precursors or fall back in the glycolytic route.35 In many tumors G6PD expression is altered, resulting in significant increase of activity.36 G6PD can be functionally defined as an oncogene since it transforms NIH3T3 fibroblasts and induces tumors in nude mice.37 It has been recently shown that transcriptional activation of G6PD plays a critical role in cell proliferation mediated by TAp73, a p53 homolog frequently overexpressed in human tumors.38 On the other hand, inhibition of G6PD activity by direct binding with the cytoplasmic form of p53 has been shown to contribute to its tumor suppressive function.39 G6PD is among the most strongly dowregulated genes in miR-206 induced RMS cells.9 In this work we show that miR-206 directly targets G6PD and that this occurs also during normal myogenesis. While sustained expression of G6PD did not interfere with the pro-differentiating action of miR-206, downregulation of G6PD caused a proliferative block in RMS cells. There is pre-clinical evidence that the adrenocortical steroid dehydroepiandrosterone (DHEA), a powerful inhibitor of G6PD, inhibits development of experimental tumors.40 However the therapeutic use of DHEA in humans is limited by its hormonal side effects. Reactivation of the Krebs cycle has been proposed as an alternative strategy to reprogram tumor cell metabolism.36 Dichloroacetate (DCA) is a well-tolerated and orally available inhibitor of pyruvate dehydrogenase kinase (PDK), which leads to pyruvate dehydrogenase (PDH) reactivation and ultimately causes metabolic remodeling from glycolysis to mitochondrial respiration.41 Our results show that DCA treatment is effective in inhibiting proliferation of ERMS and ARMS cells, both alone and in combination with miR-206. This suggests that the combination of differentiation strategies with metabolic targeting could be appropriate for RMS treatment.
Results
SMYD1 and G6PD are among the top modulated genes upon miR-206-induced ERMS cell differentiation and each of them is differentially expressed in primary tumors compared to normal muscle
To identify novel genes involved in RMS differentiation we took advantage of an expression profile previously obtained from RD18 ERMS cells forced to differentiate by miR-206 induction.9 We focused on SMYD1, one of the 30 genes upregulated more than fivefold, and on G6PD, one of the 70 most highly downregulated genes (Fig. 1A and Table S1). To assess the relevance of SMYD1 and G6PD modulation in RMS pathogenesis we made use of public data sets42,43 to compare the level of their transcripts in 101 primary tumors (including both ARMS and ERMS) and 30 normal muscles. SMYD1 and G6PD were, respectively, down and upregulated in RMS compared to normal muscles (Fig. 1B and C). Thereby, both genes were taken into consideration for further study.
Figure 1.

SMYD1 and G6PD are, respectively, among the top upregulated and downregulated genes following miR-206-induced ERMS cell differentiation and are differentially expressed in primary tumors compared to normal muscle. (A) Top 30 up-regulated genes and top 70 down-regulated genes in RD18 ERMS cells conditionally expressing miR-206 (RD18 NpBI-206 IND).9 SMYD1 is indicated with a red bar, G6PD is indicated with a blue bar. (B and C) Levels of expression of SMYD1 and G6PD mRNAs in human skeletal muscle (SM) and primary RMS tumors. ****P <0.0001 and **P<0.01. The publicly available datasets used for this analysis are specified in the Material and Methods section.
SMYD1 is strongly upregulated during myogenic differentiation
We verified by Western blot analysis whether SMYD1 was upregulated also at the protein level, together with various skeletal muscle markers, during miR-206-induced embryonal (RD18) and alveolar (RH4) RMS cells differentiation. SMYD1 was very low or absent in proliferating cells, but its level progressively increased throughout miR-206-induced differentiation, which occurred in high serum (Fig. 2A and B). In both RMS subtypes, strong SMYD1 upregulation was concomitant with the induction of muscle markers, such as Myogenin, Muscle Creatine Kinase (MCK), Desmin and Myosin Heavy Chain (MHC). C2C12 myoblasts are a widely used cellular model that recapitulates physiological myogenesis upon switching to low serum. In agreement with previous reports,27,30 we observed SMYD1 upregulation in differentiating C2C12 cells (Fig. 2C). Also in these cells, SMYD1 levels increased in concomitance with Myogenin, MCK, Desmin and MHC expression, consistent with our RMS differentiation model. NIH10T1/2 fibroblasts can be converted to myogenic cells upon expression of exogenous MyoD.44 SMYD1 upregulation was indeed observed also in this system, in concomitance with differentiation markers (Fig. 2D). These results altogether indicate that upregulation of SMYD1 is part of the genetic program controlling the myogenic differentiation process.
Figure 2.
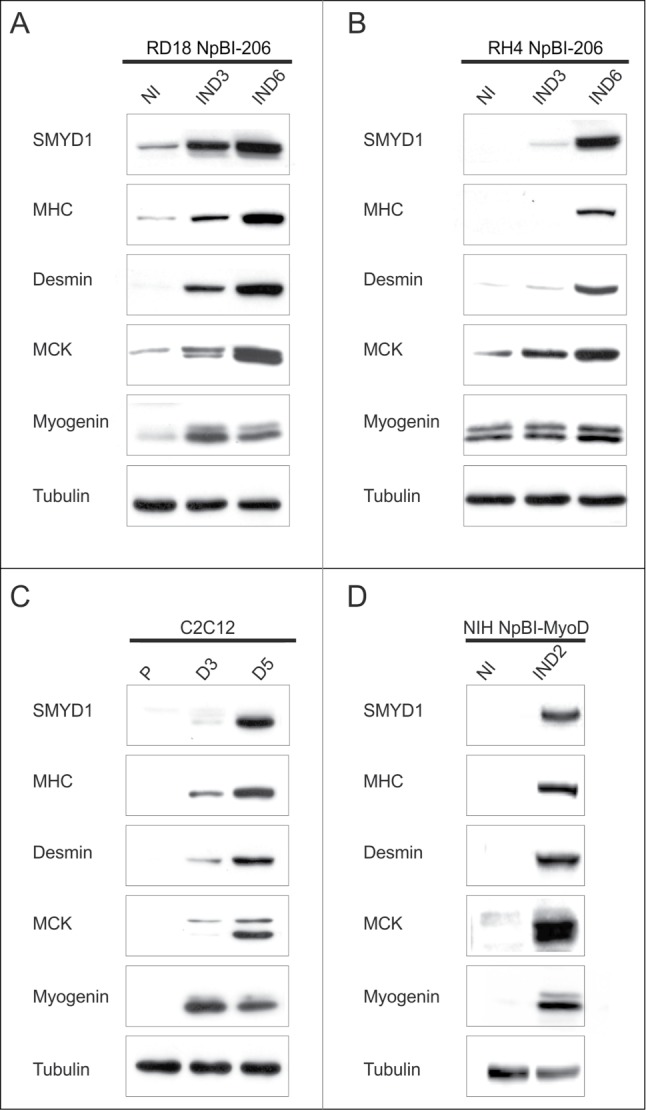
SMYD1 is upregulated during myogenic differentiation. (A and B) Western blot analysis of the indicated proteins in RD18 NpBI-206 and RH4 NpBI-206 cells, treated or not with doxycycline for the indicated days (miR-206 not induced, NI; miR-206 induced, IND). Cells were always kept in high serum (10%). (C) Western blot analysis of the indicated proteins in C2C12 cells grown in proliferation medium (P) and after 3 or 5 days in differentiation medium (D3, D5). (D) Western blot analysis of the indicated proteins in NIH10T1/2 fibroblasts infected with a conditional MyoD-expressing lentiviral vector (NIH NpBI-MyoD), treated or not with doxycycline for 2 days (MyoD not induced, NI; MyoD induced, IND).
SMYD1 is not involved in the inhibition of ERMS cells proliferation and soft agar growth imposed by miR-206
To better define the functional role of SMYD1, we tested the effect of SMYD1 silencing in ERMS cells, using a vector expressing a short hairpin RNA (shRNA) selected for silencing efficiency (Fig. S1A and B). The growth rate of proliferating RD18 cells (where SMYD1 is undetectable) was unaffected by SMYD1-downregulation, confirming the lack of off target effects of the selected shRNA (Fig. 3A). MiR-206-mediated reactivation of myogenesis in ERMS cells implicates, in concomitance with SMYD1 upregulation, cell cycle arrest and loss of anchorage-independent growth.9 Neither of them was affected by SMYD1 silencing (Fig. 3A and B). Thus, SMYD1 is not involved in the growth suppressive effects of miR-206.
Figure 3.
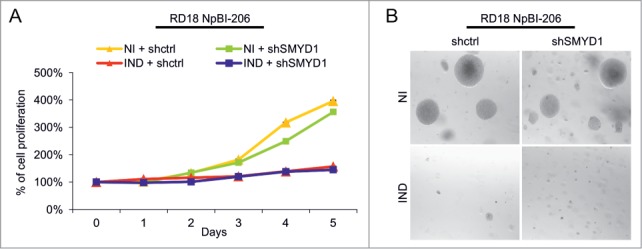
SMYD1 is not involved in the miR-206-mediated inhibition of proliferation and soft agar growth in ERMS cells. (A) MTT analysis of RD18 NpBI-206 cells infected with a constitutive control (shctrl) or SMYD1-directed (shSMYD1) shRNA, grown in the absence of doxycycline (miR-206 not induced, NI) or after doxycycline administration (miR-206 induced, IND) for the indicated days. The number of cells at day 0 was set at 100%. (B) Representative image of a soft agar growth assay of the cells described in (A).
SMYD1 is essential to implement the myogenic differentiation program
It was previously reported that in C2C12 myoblasts ectopic SMYD1 promotes differentiation.30 In our hands, overexpression of SMYD1 in C2C12 cells only slightly advanced differentiation after the switch to low serum, but in growth medium was without effect (Fig. S2A). In RD18 ERMS cells SMYD1 overexpression, both in high and low serum, was able to upregulate Myogenin but not MHC, which required miR-206 induction (Fig. S2B–D). On the other hand, overexpression of SMYD1 did not further enhance miR-206-induced differentiation (Fig. S2B and C). To verify whether SMYD1 is necessary for differentiation, we downregulated SMYD1 in miR-206-expressing ERMS and ARMS cells, and in 2 additional myogenic models. SMYD1 downregulation strongly impaired the induction of muscle markers in miR-206-expressing RD18 ERMS and RH4 ARMS cells (Fig. 4A and C). This was confirmed by immunofluorescence with an anti-MHC antibody (Fig. 4B and D). The effect of SMYD1 silencing on the expression of the muscle markers (global downregulation) and on the cell phenotype (loss of elongated or multinucleated MHC-positive myotubes) was even more severe in differentiating C2C12 cells (Fig. 4E and F). Moreover, SMYD1 silencing impaired MyoD-induced myogenic conversion of NIH 10T1/2 cells (Fig. 4G and H). Taken together, these results indicate that SMYD1, although not sufficient to promote differentiation by itself, is essential to complete the myogenic program.
Figure 4.
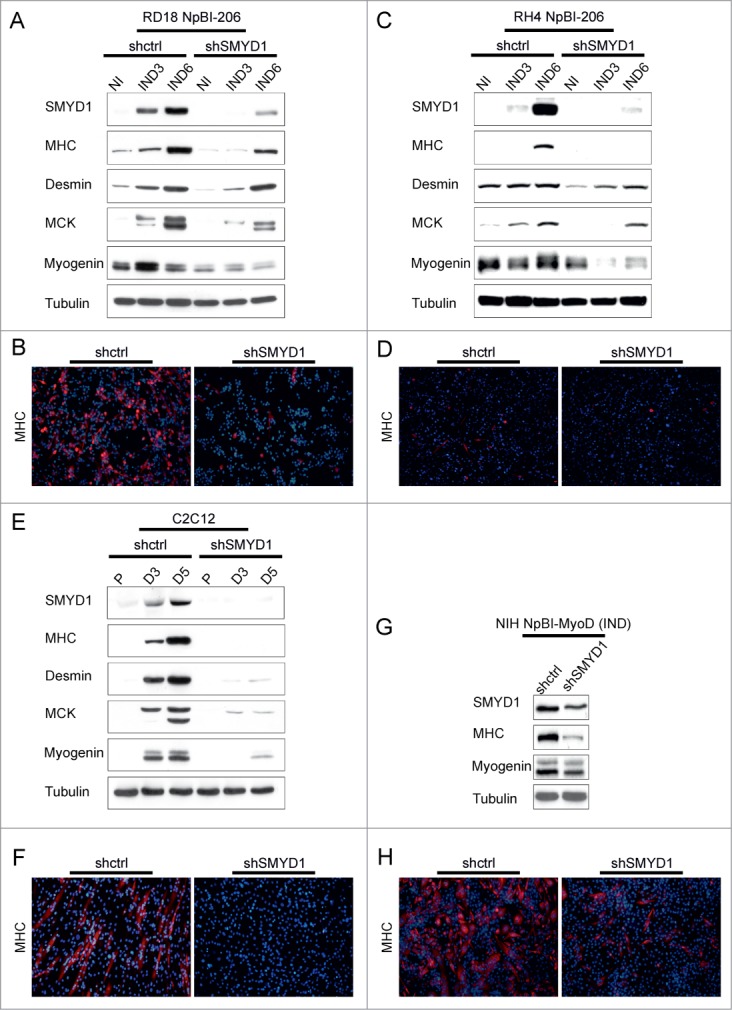
SMYD1 is essential to implement the myogenic differentiation program. (A) Western blot analysis of the indicated proteins in RD18 NpBI-206 cells infected with a constitutive control (shctrl) or SMYD1-directed (shSMYD1) shRNA, treated or not with doxycycline for the indicated days (miR-206 not induced, NI; miR-206 induced, IND). (B) Representative MHC immunostaining of the cells indicated in (A), treated with doxycycline for 6 days (miR-206 induced). Images were obtained at 10X magnification. (C) Western blot analysis of the indicated proteins in RH4 NpBI-206 cells infected with a constitutive control (shctrl) or SMYD1-directed (shSMYD1) shRNA, treated or not with doxycycline for the indicated days (miR-206 not induced, NI; miR-206 induced, IND). (D) Representative MHC immunostaining of the cells indicated in (C), treated with doxycycline for 6 days (miR-206 induced). Images were obtained at 10X magnification. (E) Western blot analysis of the indicated proteins in C2C12 cells infected with a constitutive control (shctrl) or SMYD1-directed (shSMYD1) shRNA, grown in proliferation medium (P) and after 3 or 5 days in differentiation medium (D3, D5). (F) Representative MHC immunostaining of C2C12 cells indicated in (E) after 5 days in differentiation medium. Images were obtained at 10X magnification. (G) Western blot analysis of the indicated proteins in NIH NpBI-MyoD cells infected with a constitutive control (shctrl) or SMYD1-directed (shSMYD1) shRNA, treated with doxycycline for 2 days (MyoD induced, IND). (H) Representative MHC immunostaining of the NIH NpBI-MyoD cells indicated in (G). Images were obtained at 10X magnification.
SMYD1 modulates transcription of myogenic genes
Initial work26,28 indicated a transcriptional activity for SMYD1, but more recent studies in Zebrafish have shown that during myogenesis SMYD1 is translocated to the cytoplasm, where it controls myosin folding and sarcomere assembly.31,33,34 To study the transcriptional function of SMYD1 we first verified its distribution in the nuclear and cytoplasmic fractions of differentiating RD18 ERMS and C2C12 cells. Comparable amounts of SMYD1 were found in the 2 cellular compartments (Fig. 5A and B), thus a substantial fraction of the SMYD1 protein is retained in the nucleus during differentiation. Next, we explored the effect of SMYD1 silencing on the transcription of myogenic genes. In RD18 cells upon miR-206-induced differentiation SMYD1 silencing severely reduced the transcript level of the MyoD, Myogenin, Mef2C, Mef2D transcription factors, and of the muscle markers MCK and MHC (Fig. 5C). The same set of transcripts was strongly reduced upon SMYD1 silencing also in differentiating C2C12 cells (Fig. 5D and E). These results confirm that SMYD1 is necessary for the transcriptional upregulation of myogenic genes during differentiation.
Figure 5.
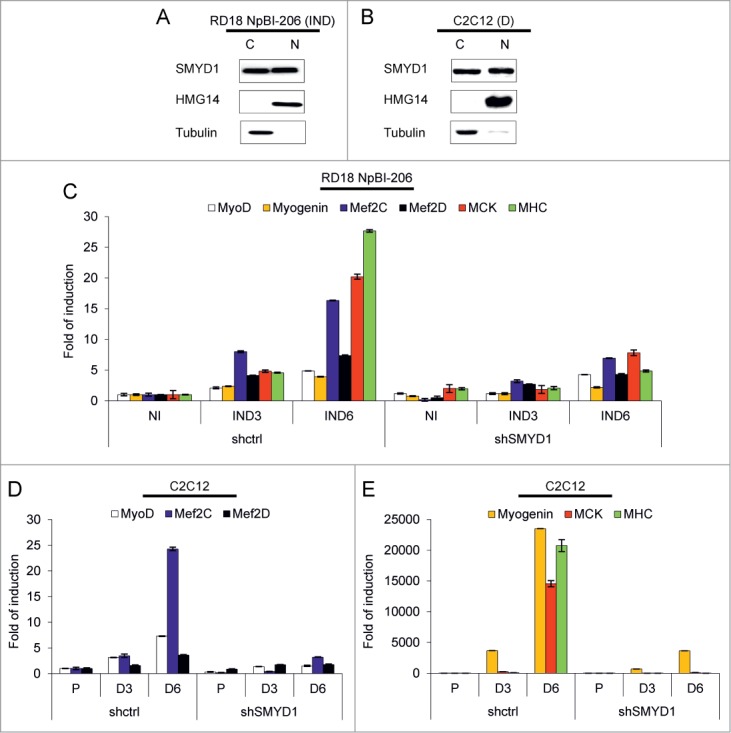
SMYD1 is present in both the cytoplasm and the nucleus of differentiating cells and its downmodulation impairs transcription of myogenic genes. (A) Western blot analysis of the indicated proteins in cytoplasmic (C) and nuclear (N) extracts of RD18 NpBI-206 cells treated with doxycycline for 3 days (miR-206 induced, IND). (B) Western blot analysis of the indicated proteins in cytoplasmic (C) and nuclear (N) extracts of C2C12 cells after 5 days in differentiation medium (D). (C) Real-time PCR analysis of the indicated transcripts in total RNA extracted from RD18 NpBI-206 cells infected with a constitutive control (shctrl) or SMYD1-directed (shSMYD1) shRNA, treated or not with doxycycline for the indicated days (miR-206 not induced, NI; miR-206 induced, IND). (D and E) Real-time PCR analysis of the indicated transcripts in total RNA extracted from C2C12 cells infected with a constitutive control (shctrl) or SMYD1-directed (shSMYD1) shRNA, grown in proliferation medium (P) and after 3 or 6 days in differentiation medium (D3, D6).
G6PD is downregulated during myogenic differentiation and is a direct target of miR-206
Glucose-6-phosphate dehydrogenase (G6PD) is one of the most significantly downregulated transcripts upon miR-206-induced ERMS cells differentiation (Fig. 1A) and its level of expression is higher in primary RMS tumors relative to skeletal muscle (Fig. 1C). Accordingly, the G6PD protein is almost completely downregulated in both RD18 ERMS and RH4 ARMS cells within 6 days of miR-206 induction (Fig. 6A and B). A gradual G6PD reduction also occurs in differentiating C2C12 and MyoD-converted NIH10T1/2 cells (Fig. 6C and D). The G6PD 3’UTR contains 3 miR-206 MREs, indicating that the G6PD transcript is a potential direct target of miR-206 (Fig. 6E). To verify this, we co-transfected a GFP sensor construct (wild type or mutated in the MREs) in 293T cells together with either miR-206 or an unrelated microRNA. FACS analysis performed 48 hours after transfection revealed a strong reduction of GFP fluorescence only in cells co-transfected with miR-206 and the wild-type sensor (Fig. 6F). We gained further evidence that this mechanism of G6PD downmodulation occurs physiologically, by transfecting C2C12 cells with a corresponding luciferase sensor construct. A progressive downregulation of the luciferase signal was observed during C2C12 differentiation, while that of the mutant construct remained unchanged (Fig. 6G).
Figure 6.
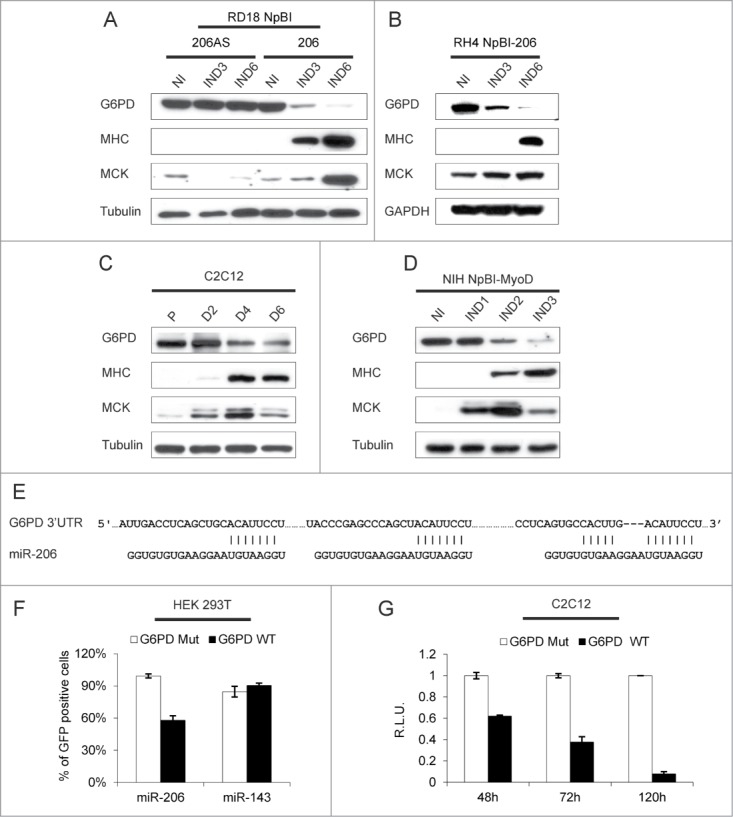
G6PD is downregulated during myogenic differentiation and is a direct target of miR-206. (A) Western blot analysis of the indicated proteins in RD18 cells infected with either a control (NpBI-206AS, antisense) or a miR-206-expressing (NpBI-206) lentiviral vector, treated or not with doxycycline for the indicated days (not induced, NI; induced, IND). (B) Western blot analysis of the indicated proteins in RH4 NpBI-206 cells, treated or not with doxycycline for the indicated days (not induced, NI; induced, IND). (C) Western blot analysis of the indicated proteins in C2C12 cells grown in proliferation medium (P) and after 2, 4 and 6 days in differentiation medium (D2, D4 and D6). (D) Western blot analysis of the indicated proteins in NIH NpBI-MyoD cells treated or not with doxycycline for the indicated days (MyoD not induced, NI; MyoD induced, IND). (E) Schematic representation of the 3 MREs of the human G6PD 3’ UTR aligned with the miR-206 sequence. The complementary of the miR-206 sequence with the MREs is indicated. (F) Flow cytometry quantification of GFP expression in HEK 293T cells co-transfected with wild type or mutant G6PD sensor construct along with miR-206 or miR-143 as a control. (G) Relative luciferase expression of C2C12 cells transfected with either wild type or mutant G6PD luciferase sensor construct, after 48, 72 and 120 hours in differentiation medium. For each time point, luciferase counts obtained with the control plasmid were set at 1.
G6PD is essential for the transformed phenotype of ERMS cells
To determine the role of G6PD in the transformed phenotype of RD18 ERMS cells, we generated a cell line where G6PD could be silenced in a doxycycline-dependent manner (Fig. 7A). Doxycycline treatment recapitulated the pattern of G6PD downregulation that we observed during miR-206-mediated differentiation (Fig. 6A). G6PD silencing resulted in strong reduction of RD18 ERMS cells proliferation (Fig. 7B) and soft agar growth (Fig. 7C and D), indicating that G6PD is essential for the transformed phenotype of these cells. We then tested the effect of overexpressing a G6PD cDNA devoid of 3’UTR. G6PD-overexpressing RD18 ERMS cells proliferated faster than controls (Fig. 7E). However, G6PD overexpression did not interfere with the proliferative block or with the induction of myogenic differentiation induced by miR-206 (Figs. 7E and F). Thus, while upregulating G6PD does not impair forced ERMS differentiation, the requirement for an anabolic metabolism sensitizes proliferating ERMS cells to variations of G6PD levels.
Figure 7.
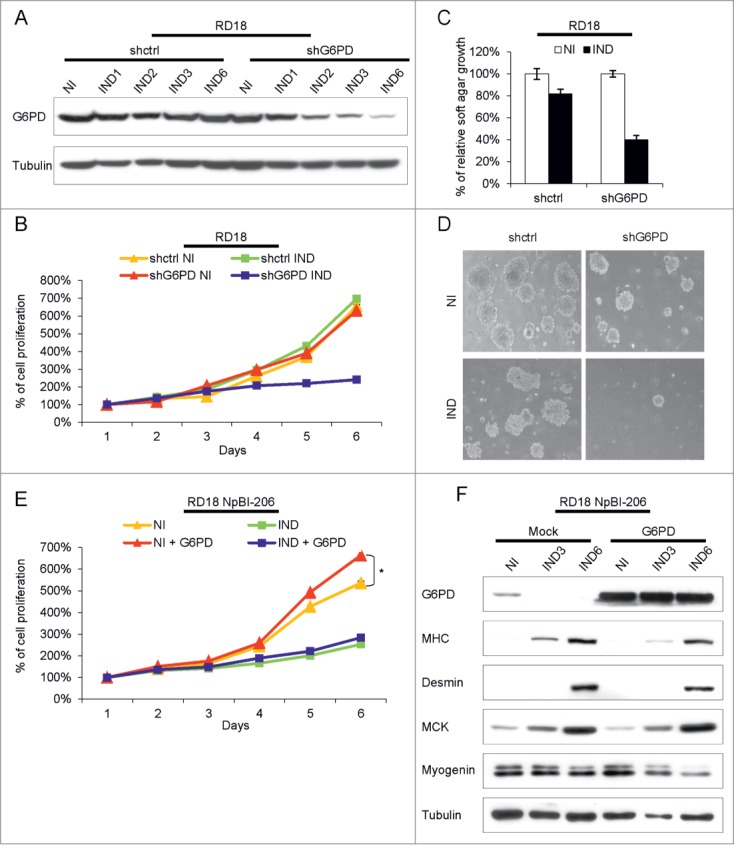
G6PD downregulation interferes with the transformed phenotype of ERMS cells but its overexpression does not affect miR-206-mediated ERMS cells differentiation. (A) Western Blot analysis of G6PD in RD18 cells conditionally expressing a control (shctrl) or a G6PD-directed (shG6PD) shRNA, treated or not with doxycycline for the indicated days (shRNA not induced, NI; shRNA induced, IND). (B) MTT analysis of the cells indicated in (A). Cells were analyzed for the indicated days in absence of doxycycline (shRNA not induced, NI) or after doxycycline administration (shRNA induced, IND). The number of cells at day 0 was set at 100%. (C) Quantification of soft agar growth assays of the cells indicated in (A). The number of colonies obtained from cells maintained in absence of doxycycline (shRNA not induced, NI) was set at 100%. (D) Representative images of the experiment indicated in (C). (E) MTT analysis of RD18 NpBI-206 cells overexpressing or not G6PD lacking the 3’UTR. Cells were analyzed for the indicated days in the absence of doxycycline (miR-206 not induced, NI) or after doxycycline administration (miR-206 induced, IND). The number of cells at day 0 was set at 100%. Student's t-test was used to evaluate statistical significance: *P <0.05. (F) Western blot analysis of the indicated proteins in the cells indicated in (E), treated or not with doxycycline for the indicated days (miR-206 not induced, NI; miR-206 induced, IND).
DCA inhibits ERMS and ARMS cell growth and enhances the tumor suppressive effects of miR-206
While G6PD inhibitors have unwanted side effects40 DCA, which negatively impacts on anabolic metabolism by increasing the flux of pyruvate to the TCA cycle, is well tolerated also after prolonged administration.41,45,46 Thus we tested the effects of DCA on ERMS and ARMS cells, either alone or in combination with miR-206 induction. When tested alone, DCA decreased proliferation of both ERMS and ARMS cells, with ARMS cells showing a much greater sensitivity (Fig. 8A and B). When combined, DCA treatment enhanced the effect of miR-206 induction. Furthermore, this synergy was particularly evident in both cell types upon miR-206 withdrawal (Fig. 8C and D). The cells were grown in doxycycline (miR-206 IND) with or without DCA, and then switched to doxycycline-free medium (DEIND). DCA prevented the resumption of cell proliferation occurring after miR-206 de-induction (Fig. 8C and D). In agreement with their higher sensitivity to DCA alone, the effect of DCA was particularly striking on de-induced ARMS cells, where very few cells survived. In ERMS cells, which are more prone to miR-206-induced differentiation,9 DCA treatment increased MHC expression in the surviving cells (Fig. 8E).
Figure 8.
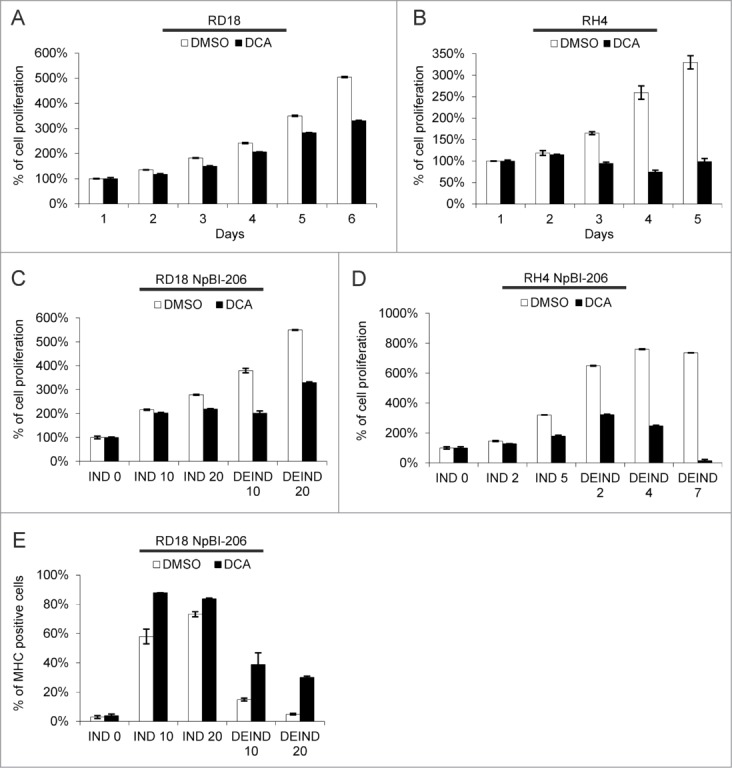
DCA inhibits ERMS and ARMS cell proliferation and sustains myogenic differentiation upon miR-206 withdrawal in ERMS cells. (A) MTT analysis of RD18 cells treated or not with 10 mM DCA for the indicated days. The number of cells at day 0 was set at 100%. (B) MTT analysis of RH4 cells treated or not with 10 mM DCA for the indicated days. The number of cells at day 0 was set at 100%. (C) MTT analysis of RD18 NpBI-206 cells treated or not with 10 mM DCA, in presence of doxycycline (miR-206 induced, IND) or after doxycycline withdrawal (miR-206 de-induced, DEIND) for the indicated days. The number of cells at day 0 was set at 100%. (D) MTT analysis of RH4 NpBI-206 cells treated or not with 10 mM DCA, in presence of doxycycline (miR-206 induced, IND) or after doxycycline withdrawal (miR-206 de-induced, DEIND) for the indicated days. The number of cells at day 0 was set at 100%. (E) Flow cytometry quantification of MHC expression in RD18 NpBI-206 cells treated or not with 10 mM DCA, in presence of doxycycline (miR-206 induced, IND) or after doxycycline withdrawal (miR-206 de-induced, DEIND) for the indicated days.
Discussion
We and others have previously shown that miR-206, a muscle-specific microRNA with a known pro-myogenic effect, can force RMS cells to resume differentiation.9,16 A series of direct targets have been described linking miR-206 to the proliferative block occurring at the onset of differentiation and to the epigenetic machinery involved in the activation of muscle genes.47–49 In this work we selected for further study 2 miR-206 effectors emerged from our original expression profile of miR-206-converted RMS cells, the histone methyltransferase SMYD1 and the pentose phosphate enzyme G6PD. Histone methylation is one of the mechanisms that enable transcription factors to implement the transition toward lineage-specific transcriptional profiles. H3K9 and H3K27 methylations are associated with gene silencing.50–52 In ARMS high levels of the H3K9 histone methyl-transferase Suv39h1 impair MyoD function, arresting myogenesis.10 Conversely, H3K27 methylation of late muscle genes is carried out by EZH2,53 a member of the Polycomb Repressive Complex 2, which is also associated with the adaptor JARID2.54 JARID2 and EZH2 have been found overexpressed, respectively, in ARMS and in all RMS.43,55
Replacement of repressive histone marks with their activating counterpart is necessary to initiate transcription of differentiation genes.56 In myogenesis upregulation of the methyltransferase Set7/9 activates muscle gene expression by precluding Suv39h1-mediated H3K9 methylation and by increasing H3K4 methylation at the promoter of myogenic genes.57 A second H3K4 histone methyltransferase, SMYD1, has been described as a regulator of myogenic differentiation.30 Contrary to Set7/9, which is ubiquitously expressed, SMYD1 expression is restricted to heart and skeletal muscle.28 Early work on SMYD1 established its activity as a H3K4 methyltransferase26 and its transcriptional role in myogenesis.27,30 Several papers describing work done in Zebrafish, have more recently revealed a role for SMYD1 in sarcomere assembly. A fraction of the SMYD1 protein localizes at the M-line where it physically associates with myosin.31,33,34 The cytoplasmic role of SMYD1 does not require the histone-methyltransferase activity which is responsible for its nuclear function.31
In the present work, by silencing SMYD1 in 3 different cell models (low-serum induced differentiation in C2C12 cells, MyoD-induced differentiation of NIH10T1/2 fibroblasts, and miR-206 induced differentiation of RMS cells), we confirmed that its upregulation is necessary for the expression of myogenic transcription factors and muscle proteins. Furthermore, we verified that upon SMYD1 silencing also the corresponding transcripts are dramatically downregulated, both in differentiating C2C12 myoblasts and RD18 ERMS cells. Thus SMYD1 is likely to regulate myogenic genes at the level of transcription. Considering that both Set7/9 and SMYD1 are upregulated during myogenesis it may be possible to envision a cooperation between the 2, related to the fact that Set7/9 activity is limited to converting unmodified H3K4 into monomethylated H3K4. For SMYD1, the preferred methylation state of the substrate is unclear.26 However, structural data indicate that the lysine-access channel in SMYD1 is more spacious than that of Set7/9,58 suggesting that it may accommodate mono and dimethylated lysine residues.
SMYD1 does not have a DNA binding motif and is thought to be recruited to muscle-specific target genes by its DNA-binding partner skNAC.27,29 Very recent work shows that skNAC expression can be induced by differentiation medium in RD18 ERMS cells.59 In the same conditions we observed only a very modest increase in SMYD1 (not shown). In our hands ectopic expression of SMYD1 in ERMS cells increased Myogenin levels but was without effect on MHC expression, which occurred only upon miR-206 upregulation. Thus, in ERMS, terminal differentiation may require removal of an additional block. According to Berkoltz et al., ARMS cell lines are unable to upregulate skNAC expression in low serum. However, forced skNAC expression was sufficient to induce differentiation and to inhibit their metastatic potential.59 This suggests that in combination with ectopic skNAC, SMYD1 may be upregulated by low serum at a level sufficient to trigger differentiation. On the other hand, the inability of ARMS cells to upregulate endogenous skNAC may explain why ARMS are less prone than ERMS cells to miR-206 induced differentiation.9
Cancer cells consume large quantities of glucose, producing lactate even in the presence of adequate oxygen (Warburg effect). The prevalence in cancer cells of the pyruvate kinase isoform PKM2 slows down the pyruvate flux to the mitochondrion, diverting glycolytic intermediates toward biosynthetic processes necessary for rapid growth and proliferation.60 An absolute requirement for biosynthesis is the availability of reducing power in the form of NADPH. The major source of NADPH is the pentose phosphate pathway (PPP), a glucose catabolic route alternative to glycolysis. G6PD catalyzes the rate-limiting step of the PPP and thus it is not surprising that it was found to be increased in many cancers. It is also to be expected that differentiating cells, which are not as biosynthetically active as proliferating cells, would downregulate G6PD. In this work we showed that G6PD is strongly downregulated in RMS cells upon miR-206-induced differentiation. We confirmed that G6PD is a direct miR-206 target, a finding previously described in a paper linking increased G6PD activity and dysregulation of the redox state of muscle cells in Duchenne Muscular Distrophy.61 Our results, showing that G6PD silencing inhibited RMS cells proliferation while exogenous G6PD increased it, confirm the link between G6PD level and cell growth.
The adrenocorticoid steroid DHEA is a potent uncompetitive inhibitor of G6PD. The supposed cancer-preventive and longevity-promoting effects of DHEA have been ascribed to its ability to lower NADPH levels and to reduce NADPH-dependent oxygen-free radical production.62 However clinical trials with DHEA were hampered by the high oral doses required, as well as by the conversion of DHEA into active androgens.40 An alternative strategy to normalize tumor metabolism is based on the idea of potentiating the flux of pyruvate toward the TCA cycle through the reactivation of the PDH complex via inhibition of its negative regulator PDK1 with DCA.41 DCA can be given orally and has been used for more than 30 years to treat lactic acidosis arising from a number of conditions. Initial results on a limited number of glioblastoma patients treated for up to 15 months suggested that this drug can be administer safely, it increases PDH activity in the tumors, and showed clinical efficacy in 3 out of 5 of them.45 Recently, DCA has been approved for phase 1 clinical trial in adults with recurrent malignant brain tumors.46 When tested alone DCA significantly impaired cell growth in both RMS subtypes, but ARMS showed much higher sensitivity. The effect of the combination with miR-206 was modest, both in terms of inhibition of proliferation and promotion of differentiation. However, DCA made a substantial difference upon miR-206 withdrawal, sustaining the differentiation of ERMS cells and inhibiting the emergence of resistant clones. The reason for the greater response of RH4 ARMS cells to DCA is unclear. The metabolic switch imposed by DCA treatment should have a higher impact on proliferating than on differentiating cells. Thus the resistance of ARMS cells to differentiation may explain their higher sensitivity to DCA. If the efficacy of DCA could be proven also in additional ARMS cell lines these observations could have a significant translational value.
The myomiRs miR-206 and miR-1 have been previously shown to promote the transition from proliferating myoblasts to differentiating myotubes by targeting, among other genes, the negative epigenetic regulators YY119 and HDAC4.20,21 In this work by showing that SMYD1 upregulation following miR-206 induction, is essential to myogenesis, we further linked the effects of this microRNA with epigenetic reprogramming. Furthermore, by demonstrating that miR-206 is directly responsible for downregulating G6PD, we also linked this microRNA to metabolic reprogramming, in a direction opposite to the requirement of cancer cells. MiR-206 is low in RMS. These new data further substantiate the idea that miR-206 or miR-206 mimetics could have value as an anticancer drug in this malignancy and suggest that combination with DCA may potentiate its effect.
Materials and Methods
Reagents
All reagents, unless otherwise specified, were from Sigma-Aldrich.
Cell cultures
RD18 NpBI-206 cells, RD18 NpBI-206AS cells, RH4 NpBI-206 cells and NIH NpBI-MyoD have been previously described.9,24 Cells were grown in DMEM (Euroclone) supplemented with 10% FBS (Euroclone). To obtain differentiation of C2C12 myoblast into myotubes, cells were plated at subconfluence, kept in growth medium for 24 hours, and then switched to differentiation medium (DMEM containing 2% HS). A similar procedure was used to improve differentiation of RD18 cells, switched to 2% HS after plating. All cells were incubated at 37°C in a 5% CO2-water–saturated atmosphere, and media were supplemented with 2 mM l-glutamine, 100 U/ml penicillin, and 0.1 mg/ml streptomycin.
Western blot
Cells were washed with ice-cold PBS, lysed, and scraped in RIPA buffer (50mM TrisHCl pH 8, 150mM NaCl, 0.1% SDS, 0.5% sodium deoxycholate, 1% NP40) with 1 mM phenylmethylsulfonyl fluoride, 10 mM NaF, 1 mM Na3VO4, and protease inhibitor cocktail. Protein lysates were cleared of cellular debris by centrifugation at 4°C for 10 minutes at 12,000 g, quantified using Bio-Rad protein assay, resolved in 10% SDS-PAGE gels, and transferred to Hybond ECL Nitrocellulose Membranes (Amersham Biosciences). Proteins were visualized with horseradish peroxidise-conjugated secondary antibodies and SuperSignal West Pico Chemiluminescent Substrate (Pierce). Tubulin, Actin and GAPDH were used as loading controls.
Cytoplasmic and nuclear extraction
Cytoplasmic and nuclear extracts were obtained from plated cells using the Nuclear Extract Kit (Active Motif), according to the manufacturer's protocol. Protein lysates were analyzed by Western blot using the aforementioned protocol. HMG14 was used as a control for nuclear loading, Tubulin was used as a control for cytoplasmic loading.
Antibodies
Anti-MHC and anti-Myogenin were from Santa Cruz Biotechnology Inc..; anti-α-Tubulin and anti-Actin were from Sigma-Aldrich; anti-GAPDH was from Cell Signaling Technology; anti-Desmin was from DAKO; anti-MCK was from Hybridoma Bank; anti-G6PD, anti-HMG14 was from Abcam; anti-SMYD1 was from Abcam or Gene Tex.
Immunofluorescence
For MHC detection, cells seeded on 24-wells, were fixed for 15 minutes with 4% paraformaldehyde, washed in PBS, and saturated in blocking solution (3% BSA in PBS) for 1 hour. Once permeabilized with 0.3% Triton X-100 for 5 minutes, cells were incubated with 1:50 primary antibody (MHC, Hybridoma Bank) for 1 hour and then with secondary antibody (Alexa-555 Invitrogen) for 30 minutes. Nuclei were stained with DAPI. The photos were taken through the fluorescent-microscope.
Intracytoplasmic staining of MHC for FACS analysis
In order to measure by flow cytometry the positivity for MHC, cells were fixed for 15 minutes with 2% paraformaldehyde, washed in PBS and saturated by washing twice in blocking solution (3% BSA in PBS). Cells were then incubated with 1 ug of primary antibody (MHC, Hybridoma Bank) resuspended in a saponin-PBS-3% BSA solution for 30 minutes and, after that, with secondary antibody (Alexa-555 Invitrogen) resuspended in the aforementioned solution for other 30 minutes. Cells were analyzed by FACS scan using CellQuest Software. Mean values (± SD) are from 3 independent experiments.
Cell proliferation assay
Cells were seeded in 96-well plates at a density of 2 × 103 cells/well. Proliferation was evaluated by MTT labeling reagent (Roche). Mean values (±SD) are from 3 independent experiments.
Anchorage-independent cell-growth assay
Cells were suspended in 0.45% type VII low-melting agarose in 10% DMEM at 2 × 104 per well and plated on a layer of 0.9% type VII low-melting agarose in 10% DMEM in 6-well plates and cultured at 37°C with 5% CO2. After 2 weeks, colonies of more than 100 μm in diameter were counted. Mean values (± SD) are from 3 independent experiments.
Real-time PCR and gene expression profile analysis
RNA was extracted from cells using TRIzol (Invitrogen). 1 μg of total RNA was used for reverse transcription with iScript cDNA Synthesis Kit (Bio-Rad) according to the manufacturer's protocol. Real-time PCR was performed with iQ SYBR Green (Bio-Rad) using the following primers:
hMCK forward 5′-TGGAGAAGCTCTCTGTGGAAGCTC-3′
h/mMCK reverse 5′-TCCGTCATGCTCTTCAGAGGGTAGT-3′
mMCK forward 5′-TGGAGAAGCTGTCCGTGGAAGCTC-3′
h/mMCK reverse 5′-TCCGTCATGCTCTTCAGAGGGTAGT-3′
hMHC forward 5′-CTGAGGTGTAACGGTGTGCT-3′
hMHC reverse 5′-AAGACCTTGGTGTGCCCAAA-3′
mMHC forward 5′-ACAGACATTTCCCAAATCCA-3′
mMHC reverse 5′-ATGTTCTTCTTCATCCGCTC-3′
hMyogenin forward 5′-TCAGCTCCCTCAACCAGGAG-3′
hMyogenin reverse 5′-CCGTGAGCAGATGATCCCC-3′
mMyogenin forward 5′- AATGCAACTCCCACAGCGCCTC-3′
mMyogenin reverse 5′- TCAGCCGCGAGCAAATGATCT-3′
hMyoD forward 5′-CGGCATGATGGACTACAGCG-3′
hMyoD reverse 5′-AGGCAGTCTAGGCTCGACAC-3′
mMyoD forward 5′-GCTGCCTTCTACGCACCTG-3′
mMyoD reverse 5′-GCCGCTGTAATCCATCATGC-3′
h/mMef2C forward 5′-CTGCTGGTCTCACCTGGTAAC-3′
h/mMef2C reverse 5′-TAGCCAATGACTGAGCCGAC-3′
h/mMef2D forward 5′-GGAAAAAGATTCAGATCCAGCGA-3′
h/mMef2D reverse 5′-TTGAGCAGCACCTTGTCCAT-3′
mSmyd1 forward 5′-CAGAGCCAGCAGTTCAGCAT-3′
mSmyd1 reverse total 5′-ATATGACAGTGCAGTTTGGC-3′
mSmyd1 forward 5′-CAGAGCCAGCAGTTCAGCAT-3′
mSmyd1 reverse transcriptional variant 1 (TV1) 5′-TGGATTTCACTGCCTCATGA-3′
mSmyd1 forward 5′-CAGAGCCAGCAGTTCAGCAT-3′
mSmyd1 reverse transcriptional variant 2 (TV2) 5′-AGCTCAATCTTGCCATTGTT-3′
mHPRT forward 5′-TGACACTGGTAAAACAATGCA-3′
mHPRT reverse 5′-GGTCCTTTTCACCAGCAAGCT-3′
hHuPO forward 5′-GCTTCCTGGAGGGTGTCC-3′
hHuPO reverse 5′-GGACTCGTTTGTACCCGTTG-3′
Real-time PCR parameters were as follows: cycle 1, 95°C for 3 minutes; cycle 2, 95°C for 15 seconds, 60°C 30 seconds for 40 cycles. The 2-ΔΔCT method was used to analyze the data.
Gene expression profile of RD18 NpBI-206 cells has been previously described.9 Briefly, The array data were analyzed with the Partek Genomics Suite version 6.3 software (Partek Inc.). Genes showing differential expression between the 2 experimental conditions in RD18 cells found to be significant by ANOVA (fold change compared to the mean across the whole panel was greater than 2 and the Student's t test p-value was lower than 0.05). Gene expression analysis of SMYD1 and G6PD was assessed in a panel of 101 RMS patient samples relative to 30 skeletal muscle samples as previously described.24 Briefly, all samples, assayed on the Affymetrix Human Genome U133 Plus 2.0 Array platform (Santa Clara, CA, USA) have been normalized by the RMA algorithm as implemented in R free software environment, and annoted with a custom CDF.63 Differential expression has been evaluated by limma package.
Lentiviral and retroviral vectors construction and trasduction
PLKO.1 lentiviral vectors (SMYD1: code TRCN0000129092, G6PD: TRCN0000025874 and control code SHC002) were purchased from Sigma-Aldrich. The conditional G6PD shRNA and shctrl lentiviral vectors were generated as previously described.24 Murine SMYD1 cDNA was PCR amplified from genomic DNA using the following primers: forward 5′-GCGGATCCATGACAATAGGCAGCATGGAG-3′, reverse 5′-GCGTCGACTCACTGCTT
CTTATGGAACAG-3′ and subcloned into the BamHI/SalI restriction sites of the pCCL.sin.PPT.hPGK.GFPWpre vector provided by Luigi Naldini (San Raffaele-Telethon Institute for Gene Therapy, Milano, Italy). The inducible SMYD1 lentiviral vector was generated by subcloning the human SMYD1 cDNA (imaGenes GmbH) into the blunted-NheI NpBI-206AS vector previously described.9,24 G6PD Δ-3′UTR retroviral vector, gently provided by Dr. Claudia Voena (CeRMS, University of Turin) was previously described.64 High titer lentiviral and retroviral vector stock was produced in HEK 293T cells as previously described.9 To induce the expression of the inducible vectors cells were treated with 1 ug/ml of doxycycline for the indicated times.
Sensor vectors generation and assessment of miRNA activity
The EIMMo miRNA target prediction server (http://www.mirz.unibas.ch/ElMMo2/) was used to identify putative miR-206 targets among the downregulated transcripts RD18 NpBI-206 cells. The GFP-G6PD 4x sensor vector (containing 2 repetition of the first 2 miR-206 MREs) was obtained by annealing the following oligonucleotides: forward 1, 5′-GGCAGCTGCACATTCCTGGCCCCGGCGATCAGCTGCACATTCCTGG-CCC CGGA-3′; forward 2, 5′-AGCTTCCCAGCTACATTCCTC AGCTGCCCGATCCCAGCTACATTCCTCAGCTGCCGGTA-3′; reverse 1, 5′-AGCTTCCGGGGCCAGGAATGTGCAGCTGATCGCCG GGGCCAGGAATGTGCAGCTGCCG-3′, reverse 2, 5′-CGGCAGCTGAGGAATGTAGCTG
GGATCGGGCAGCTGAGGAATGTAGCTGGGA-3′. We then subcloned the annealed oligonucleotides into the SacII/KpnI of the pCCL.sin.PPT.hPGK.GFP.Wpre vector as previously described. The same procedure was used to generate the 4X mutated sensor using the following oligonucleotides: forward 1, 5′-GGCTTCGACGACGCTTGGGATACAG
CGATCTTCGACGACGCTTGGGATACAGA-3′; forward 2, 5′-AGCTTACGTGATGACGCTT
GCTTCTACGCGATACGTGATGACGCTTGCTTCTAC GGGTAC-3′; reverse 1, 5′-AGCTTCTGTATCCCAAGCGT CGTCGAAGATCGCTGTATCCCAAGCGTCGTCGAAGC CGC-3′; reverse 2, 5′-CCGTAGAAGCAAGCGTCATCACGTATCGCGTAGAAGCAAGCGTC
ATCACGTA-3′. The GFP sensors vectors and synthetic mature miRNAs (Invitrogen) were cotransfected in HEK 293T cells with Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. The cells were harvest after 48 hours and green fluorescence was measured by flow cytometry using CellQuest Software. The GFP level of control cells was set at 100%. Mean values (± SD) are from 3 independent experiments.
The luciferase-G6PD 4x sensor vector was obtained by substituting the GFP with the luciferase. The luciferase sensors vectors were transfected in C2C12 cells with the indicated miRNAs and control luciferase (for normalization) by using FuGENE (Promega) according to the manufacturer's instructions. Cells were kept in growth medium for 24 hours, switched to differentiation medium and then harvest at 48, 72 and 120 hours. Luciferase assay was performed by using the Dual-Luciferase Reporter Assay System (Promega) according to the manufacturer's instructions. The luminescence was measured with the “Dual Glow” protocol of the Glowmax MULTI + Detection System (Promega).The values obtained for the mutated sensor were set as 1 at each time point. Mean values (± SD) are from 3 independent experiments.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Sharon Mazzero and Nicola Maestro for technical help. The MCK antibody was obtained from the Developmental Studies Hybridoma Bank, created by the NICHD of the NIH and maintained at The University of Iowa, Department of Biology, Iowa City, IA.
Funding
This work was supported by funding from the Italian Association for Cancer Research (AIRC Project number IG12089) and the Regione Piemonte (IMMONC Project). The support of the Fondazione Ricerca Molinette Onlus is gratefully acknowledged.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- 1. Merlino G, Helman LJ. Rhabdomyosarcoma–working out the pathways. Oncogene 1999; 18:5340-8; PMID:10498887; http://dx.doi.org/ 10.1038/sj.onc.1203038 [DOI] [PubMed] [Google Scholar]
- 2. Belyea B, Kephart JG, Blum J, Kirsch DG, Linardic CM. Embryonic signaling pathways and rhabdomyosarcoma: contributions to cancer development and opportunities for therapeutic targeting. Sarcoma 2012; 2012:406239; PMID:22619564; http://dx.doi.org/ 10.1155/2012/406239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Keller C, Guttridge DC. Mechanisms of impaired differentiation in rhabdomyosarcoma. FEBS J 2013; 280:4323-34; PMID:23822136; http://dx.doi.org/ 10.1111/febs.12421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. McCarthy JJ. MicroRNA-206: the skeletal muscle-specific myomiR. Biochim Biophys Acta 2008; 1779:682-91; PMID:18381085; http://dx.doi.org/ 10.1016/j.bbagrm.2008.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kim HK, Lee YS, Sivaprasad U, Malhotra A, Dutta A. Muscle-specific microRNA miR-206 promotes muscle differentiation. J Cell Biol 2006; 174:677-87; PMID:16923828; http://dx.doi.org/ 10.1083/jcb.200603008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Goljanek-Whysall K, Sweetman D, Münsterberg AE. microRNAs in skeletal muscle differentiation and disease. Clin Sci 2012; 123:611-25; PMID:22888971; http://dx.doi.org/ 10.1042/CS20110634 [DOI] [PubMed] [Google Scholar]
- 7. Goljanek-Whysall K, Pais H, Rathjen T, Sweetman D, Dalmay T, Münsterberg A. Regulation of multiple target genes by miR-1 and miR-206 is pivotal for C2C12 myoblast differentiation. J Cell Sci 2012; 125:3590-600; PMID:22595520; http://dx.doi.org/ 10.1242/jcs.101758 [DOI] [PubMed] [Google Scholar]
- 8. Lin C-Y, Lee H-C, Fu C-Y, Ding Y-Y, Chen J-S, Lee M-H, Huang W-J, Tsai H-J. MiR-1 and miR-206 target different genes to have opposing roles during angiogenesis in zebrafish embryos. Nat Commun 2013; 4:2829; PMID:24264597 [DOI] [PubMed] [Google Scholar]
- 9. Taulli R, Bersani F, Foglizzo V, Linari A, Vigna E, Ladanyi M, Tuschl T, Ponzetto C. The muscle-specific microRNA miR-206 blocks human rhabdomyosarcoma growth in xenotransplanted mice by promoting myogenic differentiation. J Clin Invest 2009; 119:2366-78; PMID:19620785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lee M-H, Jothi M, Gudkov AV, Mal AK. Histone methyltransferase KMT1A restrains entry of alveolar rhabdomyosarcoma cells into a myogenic differentiated state. Cancer Res 2011; 71:3921-31; PMID:21493592; http://dx.doi.org/ 10.1158/0008-5472.CAN-10-3358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. MacQuarrie KL, Tapscott SJ. Stuck in a balancing act: histone methyltransferase activity of KMT1A traps alveolar rhabdomyosarcomas in an undifferentiated state. Cell Cycle 2011; 10:3225-6; PMID:21941084; http://dx.doi.org/ 10.4161/cc.10.19.17451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Macquarrie KL, Yao Z, Young JM, Cao Y, Tapscott SJ. miR-206 integrates multiple components of differentiation pathways to control the transition from growth to differentiation in rhabdomyosarcoma cells. Skelet Muscle 2012; 2:7; PMID:22541669; http://dx.doi.org/ 10.1186/2044-5040-2-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Alteri A, De Vito F, Messina G, Pompili M, Calconi A, Visca P, Mottolese M, Presutti C, Grossi M. Cyclin D1 is a major target of miR-206 in cell differentiation and transformation. Cell Cycle 2013; 12:3781-90; PMID:24107628; http://dx.doi.org/ 10.4161/cc.26674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Elliman SJ, Howley BV, Mehta DS, Fearnhead HO, Kemp DM, Barkley LR. Selective repression of the oncogene cyclin D1 by the tumor suppressor miR-206 in cancers. Oncogenesis 2014; 3:e113; PMID:25111862; http://dx.doi.org/ 10.1038/oncsis.2014.26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Li L, Sarver AL, Alamgir S, Subramanian S. Downregulation of microRNAs miR-1, -206 and -29 stabilizes PAX3 and CCND2 expression in rhabdomyosarcoma. Lab Investig 2012; 92:571-83; PMID:22330340; http://dx.doi.org/ 10.1038/labinvest.2012.10 [DOI] [PubMed] [Google Scholar]
- 16. Yan D, Dong XDE, Chen X, Wang L, Lu C, Wang J, Qu J, Tu L. MicroRNA-1/206 targets c-Met and inhibits rhabdomyosarcoma development. J Biol Chem 2009; 284:29596-604; PMID:19710019; http://dx.doi.org/ 10.1074/jbc.M109.020511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Boutet SC, Cheung TH, Quach NL, Liu L, Prescott SL, Edalati A, Iori K, Rando TA. Alternative polyadenylation mediates microRNA regulation of muscle stem cell function. Cell Stem Cell 2012; 10:327-36; PMID:22385659; http://dx.doi.org/ 10.1016/j.stem.2012.01.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dey BK, Gagan J, Dutta A. miR-206 and -486 induce myoblast differentiation by downregulating Pax7. Mol Cell Biol 2011; 31:203-14; PMID:21041476; http://dx.doi.org/ 10.1128/MCB.01009-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lu L, Zhou L, Chen EZ, Sun K, Jiang P, Wang L, Su X, Sun H, Wang H. A Novel YY1-miR-1 regulatory circuit in skeletal myogenesis revealed by genome-wide prediction of YY1-miRNA network. PloS One 2012; 7:e27596; PMID:22319554; http://dx.doi.org/ 10.1371/journal.pone.0027596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chen J-F, Mandel EM, Thomson JM, Wu Q, Callis TE, Hammond SM, Conlon FL, Wang D-Z. The role of microRNA-1 and microRNA-133 in skeletal muscle proliferation and differentiation. Nat Genet 2006; 38:228-33; PMID:16380711; http://dx.doi.org/ 10.1038/ng1725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Winbanks CE, Wang B, Beyer C, Koh P, White L, Kantharidis P, Gregorevic P. TGF-beta regulates miR-206 and miR-29 to control myogenic differentiation through regulation of HDAC4. J Biol Chem 2011; 286:13805-14; PMID:21324893; http://dx.doi.org/ 10.1074/jbc.M110.192625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Soleimani VD, Yin H, Jahani-Asl A, Ming H, Kockx CEM, van Ijcken WFJ, Grosveld F, Rudnicki MA. Snail regulates MyoD binding-site occupancy to direct enhancer switching and differentiation-specific transcription in myogenesis. Mol Cell 2012; 47:457-68; PMID:22771117; http://dx.doi.org/ 10.1016/j.molcel.2012.05.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Song G, Zhang Y, Wang L. MicroRNA-206 targets notch3, activates apoptosis, and inhibits tumor cell migration and focus formation. J Biol Chem 2009; 284:31921-7; PMID:19723635; http://dx.doi.org/ 10.1074/jbc.M109.046862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Taulli R, Foglizzo V, Morena D, Coda DM, Ala U, Bersani F, Maestro N, Ponzetto C. Failure to downregulate the BAF53a subunit of the SWI/SNF chromatin remodeling complex contributes to the differentiation block in rhabdomyosarcoma. Oncogene 2014; 33:2354-62; PMID:23728344; http://dx.doi.org/ 10.1038/onc.2013.188 [DOI] [PubMed] [Google Scholar]
- 25. Dillon SC, Zhang X, Trievel RC, Cheng X. The SET-domain protein superfamily: protein lysine methyltransferases. Genome Biol 2005; 6:227; PMID:16086857; http://dx.doi.org/ 10.1186/gb-2005-6-8-227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tan X, Rotllant J, Li H, De Deyne P, DeDeyne P, Du SJ. SmyD1, a histone methyltransferase, is required for myofibril organization and muscle contraction in zebrafish embryos. Proc Natl Acad Sci 2006; 103:2713-8; PMID:16477022; http://dx.doi.org/ 10.1073/pnas.0509503103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sims RJ, Weihe EK, Zhu L, O’Malley S, Harriss JV, Gottlieb PD. m-Bop, a repressor protein essential for cardiogenesis, interacts with skNAC, a heart- and muscle-specific transcription factor. J Biol Chem 2002; 277:26524-9; PMID:12011100; http://dx.doi.org/ 10.1074/jbc.M204121200 [DOI] [PubMed] [Google Scholar]
- 28. Gottlieb PD, Pierce SA, Sims RJ, Yamagishi H, Weihe EK, Harriss JV, Maika SD, Kuziel WA, King HL, Olson EN, et al. Bop encodes a muscle-restricted protein containing MYND and SET domains and is essential for cardiac differentiation and morphogenesis. Nat Genet 2002; 31:25-32; PMID:11923873 [DOI] [PubMed] [Google Scholar]
- 29. Park CY, Pierce SA, von Drehle M, Ivey KN, Morgan JA, Blau HM, Srivastava D. skNAC, a Smyd1-interacting transcription factor, is involved in cardiac development and skeletal muscle growth and regeneration. Proc Natl Acad Sci 2010; 107:20750-5; PMID:21071677; http://dx.doi.org/ 10.1073/pnas.1013493107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Li D, Niu Z, Yu W, Qian Y, Wang Q, Li Q, Yi Z, Luo J, Wu X, Wang Y, et al. SMYD1, the myogenic activator, is a direct target of serum response factor and myogenin. Nucleic Acids Res 2009; 37:7059-71; PMID:19783823; http://dx.doi.org/ 10.1093/nar/gkp773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Just S, Meder B, Berger IM, Etard C, Trano N, Patzel E, Hassel D, Marquart S, Dahme T, Vogel B, et al. The myosin-interacting protein SMYD1 is essential for sarcomere organization. J Cell Sci 2011; 124:3127-36; PMID:21852424; http://dx.doi.org/ 10.1242/jcs.084772 [DOI] [PubMed] [Google Scholar]
- 32. Berkholz J, Michalick L, Munz B. The E3 SUMO ligase Nse2 regulates sumoylation and nuclear-to-cytoplasmic translocation of skNAC-Smyd1 in myogenesis. J Cell Sci 2014; 127:3794-804; PMID:25002400; http://dx.doi.org/ 10.1242/jcs.150334 [DOI] [PubMed] [Google Scholar]
- 33. Li H, Xu J, Bian Y-H, Rotllant P, Shen T, Chu W, Zhang J, Schneider M, Du SJ. Smyd1b_tv1, a key regulator of sarcomere assembly, is localized on the M-line of skeletal muscle fibers. PloS One 2011; 6:e28524; PMID:22174829; http://dx.doi.org/ 10.1371/journal.pone.0028524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Li H, Zhong Y, Wang Z, Gao J, Xu J, Chu W, Zhang J, Fang S, Du SJ. Smyd1b is required for skeletal and cardiac muscle function in zebrafish. Mol Biol Cell 2013; 24:3511-21; PMID:24068325; http://dx.doi.org/ 10.1091/mbc.E13-06-0352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Stanton RC. Glucose-6-phosphate dehydrogenase, NADPH, and cell survival. IUBMB Life 2012; 64:362-9; PMID:22431005; http://dx.doi.org/ 10.1002/iub.1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jones NP, Schulze A. Targeting cancer metabolism–aiming at a tumour's sweet-spot. Drug Discov Today 2012; 17:232-41; PMID:22207221; http://dx.doi.org/ 10.1016/j.drudis.2011.12.017 [DOI] [PubMed] [Google Scholar]
- 37. Kuo W, Lin J, Tang TK. Human glucose-6-phosphate dehydrogenase (G6PD) gene transforms NIH 3T3 cells and induces tumors in nude mice. Int J Cancer 2000; 85:857-64; PMID:10709108; http://dx.doi.org/ 10.1002/(SICI)1097-0215(20000315)85:6%3c857::AID-IJC20%3e3.0.CO;2-U [DOI] [PubMed] [Google Scholar]
- 38. Du W, Jiang P, Mancuso A, Stonestrom A, Brewer MD, Minn AJ, Mak TW, Wu M, Yang X. TAp73 enhances the pentose phosphate pathway and supports cell proliferation. Nat Cell Biol 2013; 15:991-1000; PMID:23811687; http://dx.doi.org/ 10.1038/ncb2789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jiang P, Du W, Wang X, Mancuso A, Gao X, Wu M, Yang X. p53 regulates biosynthesis through direct inactivation of glucose-6-phosphate dehydrogenase. Nat Cell Biol 2011; 13:310-6; PMID:21336310; http://dx.doi.org/ 10.1038/ncb2172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Schwartz AG, Pashko LL. Cancer prevention with dehydroepiandrosterone and non-androgenic structural analogs. J Cell Biochem Suppl 1995; 22:210-7; PMID:8538200; http://dx.doi.org/ 10.1002/jcb.240590826 [DOI] [PubMed] [Google Scholar]
- 41. Michelakis ED, Webster L, Mackey JR. Dichloroacetate (DCA) as a potential metabolic-targeting therapy for cancer. Br J Cancer 2008; 99:989-94; PMID:18766181; http://dx.doi.org/ 10.1038/sj.bjc.6604554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Williamson D, Missiaglia E, de Reyniès A, Pierron G, Thuille B, Palenzuela G, Thway K, Orbach D, Laé M, Fréneaux P, et al. Fusion gene-negative alveolar rhabdomyosarcoma is clinically and molecularly indistinguishable from embryonal rhabdomyosarcoma. J Clin Oncol 2010; 28:2151-8; PMID:20351326; http://dx.doi.org/ 10.1200/JCO.2009.26.3814 [DOI] [PubMed] [Google Scholar]
- 43. Walters ZS, Villarejo-Balcells B, Olmos D, Buist TWS, Missiaglia E, Allen R, Al-Lazikani B, Garrett MD, Blagg J, Shipley J. JARID2 is a direct target of the PAX3-FOXO1 fusion protein and inhibits myogenic differentiation of rhabdomyosarcoma cells. Oncogene 2013; 33:1148-57; PMID:23435416; http://dx.doi.org/ 10.1038/onc.2013.46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Weintraub H, Tapscott SJ, Davis RL, Thayer MJ, Adam MA, Lassar AB, Miller AD. Activation of muscle-specific genes in pigment, nerve, fat, liver, and fibroblast cell lines by forced expression of MyoD. Proc Natl Acad Sci 1989; 86:5434-8; PMID:2748593; http://dx.doi.org/ 10.1073/pnas.86.14.5434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Michelakis ED, Sutendra G, Dromparis P, Webster L, Haromy A, Niven E, Maguire C, Gammer T-L, Mackey JR, Fulton D, et al. Metabolic modulation of glioblastoma with dichloroacetate. Sci Transl Med 2010; 2:31ra34; PMID:20463368 [DOI] [PubMed] [Google Scholar]
- 46. Dunbar EM, Coats BS, Shroads AL, Langaee T, Lew A, Forder JR, Shuster JJ, Wagner DA, Stacpoole PW. Phase 1 trial of dichloroacetate (DCA) in adults with recurrent malignant brain tumors. Invest New Drugs 2014; 32:452-64; PMID:24297161; http://dx.doi.org/ 10.1007/s10637-013-0047-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Taulli R, Bersani F, Ponzetto C. Micro-orchestrating differentiation in cancer. Cell Cycle 2010; 9:918-22; PMID:20160508; http://dx.doi.org/ 10.4161/cc.9.5.10864 [DOI] [PubMed] [Google Scholar]
- 48. Rota R, Ciarapica R, Giordano A, Miele L, Locatelli F. MicroRNAs in rhabdomyosarcoma: pathogenetic implications and translational potentiality. Mol Cancer 2011; 10:120; PMID:21943149; http://dx.doi.org/ 10.1186/1476-4598-10-120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Cieśla M, Dulak J, Józkowicz A. MicroRNAs and epigenetic mechanisms of rhabdomyosarcoma development. Int J Biochem Cell Biol 2014; 53C:482-92; http://dx.doi.org/ 10.1016/j.biocel.2014.05.003 [DOI] [PubMed] [Google Scholar]
- 50. Kouzarides T. Histone methylation in transcriptional control. Curr Opin Genet Dev 2002; 12:198-209; PMID:11893494; http://dx.doi.org/ 10.1016/S0959-437X(02)00287-3 [DOI] [PubMed] [Google Scholar]
- 51. Mal AK. Histone methyltransferase Suv39h1 represses MyoD-stimulated myogenic differentiation. EMBO J 2006; 25:3323-34; PMID:16858404; http://dx.doi.org/ 10.1038/sj.emboj.7601229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Gillespie MA, Le Grand F, Scimè A, Kuang S, von Maltzahn J, Seale V, Cuenda A, Ranish JA, Rudnicki MA. p38-{gamma}-dependent gene silencing restricts entry into the myogenic differentiation program. J Cell Biol 2009; 187:991-1005; PMID:20026657; http://dx.doi.org/ 10.1083/jcb.200907037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Caretti G, Di Padova M, Micales B, Lyons GE, Sartorelli V. The Polycomb Ezh2 methyltransferase regulates muscle gene expression and skeletal muscle differentiation. Genes Dev 2004; 18:2627-38; PMID:15520282; http://dx.doi.org/ 10.1101/gad.1241904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Pasini D, Cloos PAC, Walfridsson J, Olsson L, Bukowski J-P, Johansen JV, Bak M, Tommerup N, Rappsilber J, Helin K. JARID2 regulates binding of the Polycomb repressive complex 2 to target genes in ES cells. Nature 2010; 464:306-10; PMID:20075857; http://dx.doi.org/ 10.1038/nature08788 [DOI] [PubMed] [Google Scholar]
- 55. Ciarapica R, Russo G, Verginelli F, Raimondi L, Donfrancesco A, Rota R, Giordano A. Deregulated expression of miR-26a and Ezh2 in rhabdomyosarcoma. Cell Cycle 2009; 8:172-5; PMID:19106613; http://dx.doi.org/ 10.4161/cc.8.1.7292 [DOI] [PubMed] [Google Scholar]
- 56. Barski A, Cuddapah S, Cui K, Roh T-Y, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K. High-Resolution Profiling of Histone Methylations in the Human Genome. Cell 2007; 129:823-37; PMID:17512414; http://dx.doi.org/ 10.1016/j.cell.2007.05.009 [DOI] [PubMed] [Google Scholar]
- 57. Tao Y, Neppl RL, Huang Z-P, Chen J, Tang R-H, Cao R, Zhang Y, Jin S-W, Wang D-Z. The histone methyltransferase Set7/9 promotes myoblast differentiation and myofibril assembly. J Cell Biol 2011; 194:551-65; PMID:21859860; http://dx.doi.org/ 10.1083/jcb.201010090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sirinupong N, Brunzelle J, Ye J, Pirzada A, Nico L, Yang Z. Crystal structure of cardiac-specific histone methyltransferase SmyD1 reveals unusual active site architecture. J Biol Chem 2010; 285:40635-44; PMID:20943667; http://dx.doi.org/ 10.1074/jbc.M110.168187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Berkholz J, Kuzyniak W, Hoepfner M, Munz B. Overexpression of the skNAC gene in human rhabdomyosarcoma cells enhances their differentiation potential and inhibits tumor cell growth and spreading. Clin Exp Metastasis 2014; 31:869-79; PMID:25209525; http://dx.doi.org/ 10.1007/s10585-014-9676-z [DOI] [PubMed] [Google Scholar]
- 60. Vander Heiden MG, Lunt SY, Dayton TL, Fiske BP, Israelsen WJ, Mattaini KR, Vokes NI, Stephanopoulos G, Cantley LC, Metallo CM, et al. Metabolic pathway alterations that support cell proliferation. Cold Spring Harb Symp Quant Biol 2011; 76:325-34; PMID:22262476; http://dx.doi.org/ 10.1101/sqb.2012.76.010900 [DOI] [PubMed] [Google Scholar]
- 61. Cacchiarelli D, Martone J, Girardi E, Cesana M, Incitti T, Morlando M, Nicoletti C, Santini T, Sthandier O, Barberi L, et al. MicroRNAs involved in molecular circuitries relevant for the duchenne muscular dystrophy pathogenesis are controlled by the dystrophin/nNOS pathway. Cell Metab 2010; 12:341-51; PMID:20727829; http://dx.doi.org/ 10.1016/j.cmet.2010.07.008 [DOI] [PubMed] [Google Scholar]
- 62. Schwartz AG, Pashko LL. Dehydroepiandrosterone, glucose-6-phosphate dehydrogenase, and longevity. Ageing Res Rev 2004; 3:171-87; PMID:15177053; http://dx.doi.org/ 10.1016/j.arr.2003.05.001 [DOI] [PubMed] [Google Scholar]
- 63. Lembo A, Di Cunto F, Provero P. Shortening of 3′UTRs correlates with poor prognosis in breast and lung cancer. PloS One 2012; 7:e31129; PMID:22347440; http://dx.doi.org/ 10.1371/journal.pone.0031129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Polimeni M, Voena C, Kopecka J, Riganti C, Pescarmona G, Bosia A, Ghigo D. Modulation of doxorubicin resistance by the glucose-6-phosphate dehydrogenase activity. Biochem J 2011; 439:141-9; PMID:21679161; http://dx.doi.org/ 10.1042/BJ20102016 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.