

The control of transcriptional pathways during the cell cycle is paramount to regulating cell growth and differentiation, and maintaining cells in a non-cancerous state. Human CGGBP1 (CGG triplet repeat-binding protein 1) was first identified as a protein that binds unmethylated CGG repeats,1 and has more recently been implicated in cell cycle control.2 Depletion of CGGBP1 causes cells to remain in G0/G1, thereby reducing entry of cells into S phase.2 Moreover, CGGBP1 was found to regulate the expression of genes important for controlling the cell cycle (e.g. GAS1 and CDKN1A).2 However, the mechanism by which CGGBP1 controls expression of select genes remained unknown.
Alu SINEs (short interspersed elements) are repeat elements that are abundantly dispersed throughout the human genome. Some Alu SINEs are transcribed by RNA polymerase III (Pol III) to give rise to ∼300 nucleotide, non-coding transcripts (Alu RNA).3 Transcription of Alu RNA is increased upon heat shock, where it acts as a potent repressor of RNA polymerase II (Pol II) transcription.4 How transcription of Alu SINEs is controlled and whether Alu RNA functions as a transcriptional repressor under conditions other than heat shock were unanswered questions.
In an article in this issue of Cell Cycle, Agarwal et al. provide evidence for a new role for CGGBP1 in inhibiting the production of Alu RNAs during serum activation, thereby indirectly controlling mRNA transcription (Fig. 1).5 Global microarray experiments revealed that depleting CGGBP1 from actively growing cells caused a significant shift in RNA patterns, and this effect was diminished in quiescent (serum starved) cells. Moreover, depletion of CGGBP1 resulted in a significant decrease in total mRNA in cells. These results suggest that during serum activation a general pattern of increased mRNA production occurs and that CGGBP1 is needed for this to happen. Curiously, a common motif in promoter sequences could not be found among the genes whose mRNA levels were altered by serum starvation; hence, the role of CGGBP1 appeared to be indirect.
Figure 1.
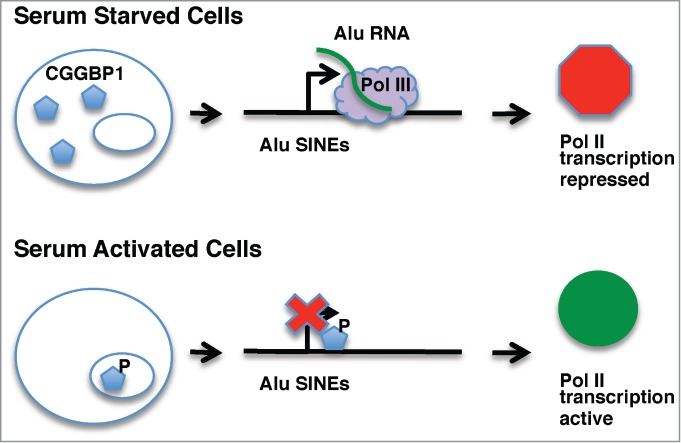
Model of the CGGBP1-Alu RNA pathway. In serum starved cells, CGGBP1 is pan-cellular, Alu SINEs are transcribed by Pol III, and Alu RNA represses Pol II transcription. Upon serum activation, CGGBP1 is phosphorylated, becomes nuclear, binds to Alu SINEs, and blocks Pol III transcription. The resulting decrease in Alu RNA levels results in active Pol II transcription.
The widespread transcriptional changes indirectly regulated by CGGBP1 upon serum activation were found to be mediated by a reduction in levels of Alu RNA, a known repressor of mRNA transcription.5 A series of experiments showed that serum activation caused phosphorylation of a specific tyrosine (Y20) on over-expressed CGGBP1, which resulted in its nuclear localization. ChIP experiments showed that CGGBP1 was enriched at a variety of genomic regions including Alu SINEs, where its occupancy selectively increased between serum starved and serum activated cells. Importantly, ChIP experiments also revealed that the depletion of CGGBP1 in cells significantly reduced Pol II occupancy on the bodies of 4 genes previously found to be transcriptionally repressed by Alu RNA.4 Moreover, treatment of CGGBP1 knockdown cells with an oligonucleotide antisense to Alu RNA rescued Pol II occupancy in the bodies of these 4 genes. These data indicate that serum activation causes CGGBP1 to bind Alu SINEs and inhibit Alu RNA production by Pol III, thereby increasing Pol II transcription of the 4 Alu-sensitive genes.
Taken together the results in the Agarwal et al. paper5 lead to the following model (Fig. 1): in the presence of serum, CGGBP1 is phosphorylated, localizes to the nucleus, and binds Alu SINEs where it inhibits Pol III transcription. This results in a decrease in the level of Alu RNA, thereby increasing Pol II transcription at genes sensitive to repression by Alu RNA. This model raises many new questions. What is the breadth of genes regulated by the CGGBP1-Alu RNA pathway in response to serum stimulation? What serum-responsive signaling pathways are involved in the phosphorylation and regulation of CGGBP1? CGGBP1 was also found to occupy LINEs and satellite regions, to a higher level in the absence of serum than the presence; what is the function of this CGGBP1 occupancy? It will be interesting to watch this story further unfold in coming years.
References
- 1. Deissler H, et al. . J Biol Chem 1997; 272:16761-8; PMID:9201980; http://dx.doi.org/ 10.1074/jbc.272.27.16761 [DOI] [PubMed] [Google Scholar]
- 2. Singh U, et al. . BMC Mol Biol 2011; 12:28; PMID:21733196; http://dx.doi.org/ 10.1186/1471-2199-12-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sinnett D, et al. . J Biol Chem 1991; 266 (14): 8675-8; PMID:1709156 [PubMed] [Google Scholar]
- 4. Mariner PD, et al. . Mol Cell 2008; 29: 499-509; PMID:18313387; http://dx.doi.org/ 10.1016/j.molcel.2007.12.013 [DOI] [PubMed] [Google Scholar]
- 5.Agarwal P, et al. Cell Cycle. 2014 [Google Scholar]