

The reprogramming of cancer cell metabolism toward aerobic glycolysis, i.e. the Warburg effect, is a hallmark of cancer; the rationale for selecting such energy-inefficient metabolism is the need to increase cellular biomass and sustain the high proliferation rates of cancer cells.1 Metabolic reprogramming is generally considered as a phenotypic endpoint that occurs as a consequence of tumor development, as also indicated by the finding that oncogenes such as Ras, Myc and HIF1 actively induce expression of glucose transporters and glycolytic enzymes. A somewhat specular and newly emerging paradigm is that transcriptional networks and oncogenic signaling can also be regulated downstream of metabolic pathways, which assume causative roles in controlling cell behavior above their core biochemical functions.2
Starting from this provocative idea, we explored what happens to gene transcription in breast cancer cells upon inhibition of glucose metabolism by 2-deoxy-glucose, and found by gene set enrichment analyses (GSEA) a striking correlation between genes regulated by glucose metabolism and those regulated by YAP/TAZ, transcriptional cofactors downstream of the Hippo pathway.3 Prompted by this observation we then found that glucose uptake and a sustained flux of glucose through aerobic glycolysis directly support YAP/TAZ, because treatments that reprogram cellular metabolism away from aerobic glycolysis induce corresponding reductions in YAP/TAZ transcriptional activity. Accordingly, inhibition of glucose uptake, or knockdown of key enzymes of glycolysis, hampered YAP/TAZ-induced in vitro tumorigenic phenotypes in breast cancer cells, such as proliferation, clonogenic activity and self-renewal properties. This regulative axis is strikingly conserved in Drosophila, as we found that inhibiting glycolysis blunts clonal expansion induced by Yorkie, the YAP/TAZ homolog, and downregulates Yorkie target genes. Finally, and in keeping with the idea of oncogenic signaling at the service of a metabolic pathway, growth/survival promoted by glucose incorporates YAP/TAZ activity in human breast cancer cells.
We thus speculated that this mechanism would help tie together a metabolism suited for cell growth and the activity of pro-malignant factors such as YAP/TAZ. For this we performed bioinformatics analyses in an unprecedented metadataset collecting data from more than 3600 human primary breast cancers, and found that a gene expression signature experimentally associated with glucose uptake strongly correlates with YAP/TAZ activity and YAP/TAZ-dependent cancer features (tumor grade, disease-free survival). Given the recently identified links between glycolysis and breast tumor-initiating cells, and between YAP/TAZ and breast cancer stem cell properties, it is tempting to speculate that selection of a glycolytic phenotype in tumor cells carries the extra advantage of sustaining YAP/TAZ activity, which would then unleash tumor seeding ability and malignancy.
Mechanistically, we found that YAP/TAZ activity is linked to glycolysis by the enzyme phosphofructokinase (PFK1): we isolated PFK1 by proteomics as YAP partner, but then found that PFK1 directly interacts with the YAP/TAZ DNA-binding platforms TEADs. This interaction depends on the key allosteric regulation of PFK1 by fructose-2,6-bisphosphate, and overexpression of PFKFB3, the enzyme producing fructose-2,6-bisphosphate, coherently enhances YAP/TAZ activity and their cooperation with TEAD factors; this suggests that PFK1 metabolic activity, rather than its levels, are key to promote YAP/TAZ function. Strikingly, we then found that glucose metabolism and PFK1 are key determinants of the interaction between YAP/TAZ and TEAD proteins, and regulate the stability of YAP/TEAD transcriptional complexes at target gene promoters. These findings identify the first regulatory mechanism promoting YAP/TAZ activity at the level of TEADs, and build on the increasing emphasis on TEADs as key factors required for YAP/TAZ activity3. Finally, this mechanism appears distinct from the Hippo/LATS kinase cascade, from the AMPK energy-sensing system and from the mevalonate/RHO axis. We thus propose that the enzyme mediating the first committed step of glycolysis, at the heart of several allosteric and signaling mechanisms, also informs nuclear gene transcription through YAP/TAZ.
This study highlights the notion that YAP/TAZ are atypical oncogenic molecules that, rather than being activated by genetic events, lay downstream of multiple cancer hallmarks embedded in the tumor microenvironment3. Our data, together with the recent observation that cholesterol biosynthesis and energy stress also regulate YAP/TAZ,4-6 indicates that YAP/TAZ are activated downstream of metabolic pathways, possibly triggered by other oncogenic insults; in this view, YAP/TAZ may thus represent a general “bottleneck” for tumorigenesis, bridging information from the glucose and lipid metabolic state into cell proliferation and aggressive behaviors of cancer cells. More in general, this might open new paths to understand the crosstalk between tumor metabolism and cell signaling, to identify novel anti-YAP/TAZ compounds and to design combinatorial approaches to disable the powerful oncogenic activity of YAP/TAZ.
Figure 1.
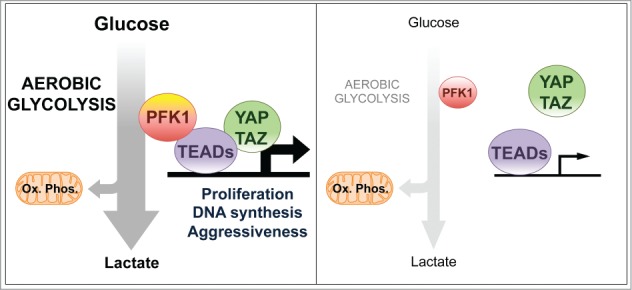
In cancer cells actively incorporating glucose and routing it through glycosysis, PFK1 (phosphofructokinase) activity is sustained and promotes the interaction of YAP/TAZ cofactors with their DNA-binding platform TEADs (left panel); when glucose metabolism is inhibited, also YAP/TAZ activity is weakened (right panel). This represents a mechanism by which the pro-tumorigenic functions of YAP/TAZ are incorporated and enhanced in cells displaying metabolic reprogramming toward aerobic glycolysis.
References
- 1.Lunt SY, Vander Heiden MG. Annu Rev Cell Dev Biol 2011; 27:441-64; PMID:21985671; http://dx.doi.org/ 10.1146/annurev-cellbio-092910-154237 [DOI] [PubMed] [Google Scholar]
- 2.Wellen KE, Thompson CB. Nat Rev Mol Cell Biol 2012; 13:270-6; PMID:22395772; http://dx.doi.org/ 10.1038/nrm3305 [DOI] [PubMed] [Google Scholar]
- 3.Piccolo S, et al.. Physiol Rev 2014; 94:1287-312; PMID:25287865; http://dx.doi.org/ 10.1152/physrev.00005.2014 [DOI] [PubMed] [Google Scholar]
- 4.Hariharan IK. Nat Cell Biol 2015; 17:362-3; PMID:25812520; http://dx.doi.org/ 10.1038/ncb3141 [DOI] [PubMed] [Google Scholar]
- 5.Sorrentino G, et al.. Nat Cell Biol 2014; 16:357-66; PMID:24658687; http://dx.doi.org/ 10.1038/ncb2936 [DOI] [PubMed] [Google Scholar]
- 6.Wang Z, et al.. Proc Natl Acad Sci USA 2014; 111(1):E89-98; PMID: 24367099; http://dx.doi.org/ 10.1073/pnas.1319190110 [DOI] [PMC free article] [PubMed] [Google Scholar]