

Epileptic discharges are increased during non-REM sleep. By studying the sleep EEG in patients with focal epilepsies, Frauscher et al. show that the increase is specifically associated with high-amplitude slow waves. In contrast to physiological activity, it occurs at transitions from activation to deactivation states, a period of high synchronization.
Keywords: epilepsy, sleep, high frequency oscillations, slow wave, intracerebral electroencephalography
Epileptic discharges are increased during non-REM sleep. By studying the sleep EEG in patients with focal epilepsies, Frauscher et al. show that the increase is specifically associated with high-amplitude slow waves. In contrast to physiological activity, it occurs at transitions from activation to deactivation states, a period of high synchronization.
Abstract
Epileptic discharges in focal epilepsy are frequently activated during non-rapid eye movement sleep. Sleep slow waves are present during this stage and have been shown to include a deactivated (‘down’, hyperpolarized) and an activated state (‘up’, depolarized). The ‘up’ state enhances physiological rhythms, and we hypothesize that sleep slow waves and particularly the ‘up’ state are the specific components of non-rapid eye movement sleep that mediate the activation of epileptic activity. We investigated eight patients with pharmaco-resistant focal epilepsies who underwent combined scalp-intracerebral electroencephalography for diagnostic evaluation. We analysed 259 frontal electroencephalographic channels, and manually marked 442 epileptic spikes and 8487 high frequency oscillations during high amplitude widespread slow waves, and during matched control segments with low amplitude widespread slow waves, non-widespread slow waves or no slow waves selected during the same sleep stages (total duration of slow wave and control segments: 49 min each). During the slow waves, spikes and high frequency oscillations were more frequent than during control segments (79% of spikes during slow waves and 65% of high frequency oscillations, both P ∼ 0). The spike and high frequency oscillation density also increased for higher amplitude slow waves. We compared the density of spikes and high frequency oscillations between the ‘up’ and ‘down’ states. Spike and high frequency oscillation density was highest during the transition from the ‘up’ to the ‘down’ state. Interestingly, high frequency oscillations in channels with normal activity expressed a different peak at the transition from the ‘down’ to the ‘up’ state. These results show that the apparent activation of epileptic discharges by non-rapid eye movement sleep is not a state-dependent phenomenon but is predominantly associated with specific events, the high amplitude widespread slow waves that are frequent, but not continuous, during this state of sleep. Both epileptic spikes and high frequency oscillations do not predominate, like physiological activity, during the ‘up’ state but during the transition from the ‘up’ to the ‘down’ state of the slow wave, a period of high synchronization. Epileptic discharges appear therefore more associated with synchronization than with excitability. Furthermore, high frequency oscillations in channels devoid of epileptic activity peak differently during the slow wave cycle from those in channels with epileptic activity. This property may allow differentiating physiological from pathological high frequency oscillations, a problem that is unresolved until now.
Introduction
Sleep causes important modifications of the EEG characteristics of epilepsy, with increased and more widespread interictal spiking occurring during non-rapid eye movement (non-REM) sleep compared to wakefulness or REM sleep (Montplaisir et al., 1987; Sammaritano et al., 1991; Malow et al., 1998). High frequency oscillations (HFOs) are more frequent during non-REM sleep (Staba et al., 2004; Bagshaw et al., 2009) and may also be better biomarkers for epileptogenic tissue than spikes (Zijlmans et al., 2012). It has been suggested that the increased temporal and spatial synchronization during non-REM sleep plays an important role for the facilitation of epileptic discharges during sleep (Steriade et al., 1994). Here, we hypothesized that this facilitation is explained by the low frequency slow waves occurring during non-REM sleep.
Slow waves with frequencies below 1 Hz were first described in the rodent auditory cortex (Metherate et al., 1992), and later systematically investigated in the feline association cortex (Steriade et al., 1993a, b). Besides sleep spindles and delta activity between 1 and 4 Hz, slow waves between 0.5 and 1 Hz represent the most important feature of the EEG during non-REM sleep (Steriade, 2006). They are characterized by a rhythmic alternation between activated (‘up’, depolarized) and deactivated (‘down’, hyperpolarized) states, and are shown to influence physiological brain rhythms (Steriade, 2006; Haider and McCormick, 2009; Crunelli and Hughes, 2010). Several authors demonstrated that sleep spindles and EEG activities in the beta, gamma and ripple (>80 Hz) bands are linked to the slow wave ‘up’ state as known from in vivo local field potential recordings of the rodent and feline cortex, and in vitro recordings from brain slices (Steriade et al., 1993a, b; Sanchez-Vives and McCormick, 2000; Grenier et al., 2001; Mölle et al., 2002; Mukovski et al., 2007; Valderrama et al., 2012; Valencia et al., 2013). Moreover, the ‘up’ state was shown to coordinate the coupling between hippocampal HFOs and scalp spindles, which is of importance for memory consolidation (Sirota et al., 2003; Battaglia et al., 2004; Isomura et al., 2006; Mölle et al., 2006; Clemens et al., 2007, 2011). Evidence from thalamic lesional studies, in vitro studies, as well as decorticated hemisphere studies points to an intracortical generation of slow waves (Steriade et al., 1993a; Timofeev and Steriade, 1996; Sanchez-Vives and McCormick, 2000). This intracortical generation of slow waves was demonstrated to have its onset in layer five of the cerebral cortex (Sanchez-Vives and McCormick, 2000; Chauvette et al., 2010; Beltramo et al., 2013).
Based on the above considerations, we speculated that the activation of epileptic discharges (interictal spikes and HFOs) by non-REM sleep is in fact due to a specific element of this sleep stage, the sleep slow waves, which are frequent during non-REM sleep. If this hypothesis proves true, it would provide a mechanistic explanation for this activation and one might further speculate that epileptic discharges might predominate during the ‘up’ state in line with findings for physiological brain activity (Steriade, 2006; Haider and McCormick, 2009; Crunelli and Hughes, 2010). To answer these questions, we studied the non-REM sleep EEG activity in patients with pharmaco-resistant focal epilepsy who underwent combined scalp-intracerebral EEG recordings for diagnostic work-up.
Material and methods
Patient selection
We selected consecutive patients with pharmaco-resistant epilepsies who underwent continuous intracerebral (depth electrodes) EEG recordings at the Montreal Neurological Institute and Hospital between January 2010 and April 2014 for seizure foci identification and potential surgical treatment. Inclusion criteria were: first, only patients who underwent a combined intracerebral and scalp EEG investigation were included, as polarity of the sleep slow waves can easily be determined in the scalp EEG, but is difficult to assess in intracerebral channels (the polarity of slow waves depends on the location of the grey matter with respect to electrode contacts). Second, we selected only patients for whom we had at least one continuous whole night recording, obtained at least 72 h post-electrode implantation. Third, patients’ recordings needed to have at least one frontal intracerebral EEG channel with at least 10 unambiguous spike events during the first sleep cycle appearing over an EEG background without major abnormality, and at least one frontal intracerebral EEG channel with normal EEG activity (absence of spikes or non-epileptic anomaly). Finally, we only included subjects in whom epileptic activity or non-epileptic anomalies did not interfere with sleep scoring in the scalp EEG. Patients with a secondarily generalized seizure during the 12 h or a partial seizure during the 6 h before the evaluated sleep recording were excluded (Frauscher et al., 2015).
Eight of 58 patients with combined scalp-intracerebral EEG recordings met these selection criteria. Demographic and clinical data of these patients are provided in Supplementary Table 1. Reasons for exclusion were: recordings with fewer than 10 clear-cut isolated spike events in frontal lobe channels during the first sleep cycle N2 and N3 sleep (n = 32); continuous spiking in the frontal lobe (n = 7); no normal (non-epileptic) channels in the frontal lobe (n = 4); no electrodes in the frontal lobe (n = 3), continuous generalized and multifocal epileptic discharges or continuous slow wave anomaly in the scalp EEG that interfered with the scoring of sleep and sleep slow waves (n = 3); recording obtained <72 h after electrode implantation (n = 1). In patients with several full-night recordings, the first available night after 72 h post-implantation was analysed.
This study was approved by the Review Ethics Board at the Montreal Neurological Institute and Hospital. All patients signed an ethical board approved written informed consent.
Intracerebral and scalp EEG recordings
A mean of nine depth electrodes (range 6–11) was implanted stereotactically using an image-guided system (ROSA robotic neuronavigation system or Medtronic Stealth neuronavigation system), and of these, a mean of three (range 1–6) was implanted in one (n = 3) or both (n = 5) frontal lobes. For localizations of intracerebral electrodes see Supplementary Table 1.
In four patients, implantations consisted of electrodes manufactured on site (nine contacts, 0.5–1 mm in length and separated by 5 mm) and in four patients, commercially available electrodes (5–18 contacts, 2 mm in length, separated by 1.5 mm) were used (DIXI Medical). Electrode locations were determined by either post-implantation MRI (n = 4), post-implantation CT co-registered with a pre-implantation MRI using Statistical Parametric Mapping 8 software (n = 3), or the information from the reconstructed planned position of the electrodes from the ROSA robotic navigation system (n = 1).
Subdermal thin wire electrodes (Ives, 2005) were placed at the time of implantation at positions F3, F4, Fz, C3, C4, Cz, P3, P4, Pz allowing for identification of sleep staging and of sleep slow waves. Intracerebral electrode positions were tailored for each patient and depended on the clinical hypothesis made on the location of the seizure generator and propagation of ictal discharge.
The EEG signal was high pass-filtered at 0.1 Hz, low-pass filtered at 500 Hz, and sampled at 2000 Hz. EEGs were recorded using the Harmonie EEG system (Stellate). The signals were anti-causally filtered with a digital single-pole high-pass infinite impulse response filter with 0.1 Hz cut-off frequency to correct the phase distortion introduced by the analogue high-pass filter.
Scoring of sleep and selection of intracerebral EEG channels in the frontal lobe
Sleep was scored manually in 30-s epochs in line with the 2012 criteria of the American Academy of Sleep Medicine (Berry et al., 2012) by one board certified sleep expert. A flow chart of the principal steps of this study is provided in Fig. 1. After sleep scoring, negative sleep slow wave halves were detected in F3 and F4 referred to the average of all scalp electrodes. Positions F3 and F4 were chosen as slow waves predominantly originate from the prefrontal cortex (Massimini et al., 2004). We did not use a traditional mastoid reference for the scoring of sleep, as this was not feasible for all investigated patients due to the proximity of some depth electrodes and the risk of infection. Moreover, a temporal epileptic focus would certainly contaminate a mastoid reference. However, in three healthy subjects we obtained a conventional sleep recording with scalp electrodes at the same positions in both an average and a mastoid reference montage that revealed an 85% concordance in occurrence and polarity of high amplitude sleep slow waves.
Figure 1.

Flow chart illustrating the principal steps of the study analysis.
We used intracerebral channels in the frontal lobe only, as slow waves were shown to spread from the frontal to the occipital lobe (Massimini et al., 2004), and that could have influenced our findings. We evaluated intracerebral EEG channels in the frontal lobe in two groups: (i) channels with isolated epileptic spikes during the first sleep cycle N2 and N3 sleep; and (ii) channels with normal EEG activity (absence of seizure onset zone or interictal spikes and absence of non-epileptic anomaly) located in brain regions with no structural abnormalities, as revealed by high resolution MRI; referred to as ‘intracerebral channels with normal EEG activity’ below. Channels displaying non-epileptic abnormalities, artefacts interfering with the identification of HFOs, or channels outside the brain were excluded from the analysis. Suitable channels were selected independently by two of the authors after reviewing all channels of frontal depth electrodes of all patients in a bipolar montage. For all ambiguous channels, a consensus was found in a common scoring session.
Detection of sleep slow waves
We automatically detected widespread frontal physiological slow waves during the first cycle of non-REM sleep stages N2 and N3. We chose the first sleep cycle to increase the probability of having high amplitude slow waves, as the literature suggests that earlier sleep cycles are associated with slow waves with a steeper slope and higher amplitude compared to later sleep cycles due to changes in homeostatic sleep pressure (Riedner et al., 2007). For sleep slow waves detection, the recordings were down-sampled to 100 Hz (low-pass anti-aliasing finite impulse response filter of order 100, pass band 0–17 Hz, transition band 17–83 Hz, 60 dB stop band attenuation), and the band from 0.3–4 Hz was obtained with zero-phase infinite impulse response Butterworth filters. The low-pass and high-pass filters were of sixth order each, resulting from a third order forward and reverse filtering. Negative half-waves were detected as consecutive zero crossings separated by 0.25 to 1 s, as in existing detectors (Riedner et al., 2007; Valderrama et al., 2012). The detection of the slow waves and all the ensuing computations were done in Matlab.
The negative peaks in scalp slow waves were considered as to be associated with the ‘down’ or hyperpolarizing state of neuronal populations (Steriade, 2006). They were of higher amplitude, better defined than the positive peaks, and, hence, more stable and easier to detect (Massimini et al., 2004). Consequently, in the scalp channels we restricted the detection of slow waves to negative half-waves, with their corresponding preceding and following positive half-waves. To keep only physiological slow waves, we excluded any slow waves overlapping with segments one second before and after artefacts and interictal epileptic spikes visible on the scalp channels.
As we needed a clearly defined polarity, we excluded all slow waves showing discordant polarity in the scalp channels, i.e. slow waves in which the negative half-wave of one of the scalp channels overlapped with the trailing positive half-wave in the other scalp channel for more than 5% of the positive half-wave duration. To include only the most prominent half-waves in each channel we only kept the 25% with highest absolute amplitude. By using this relative threshold instead of a fixed amplitude threshold we could use a single value for scalp and intracerebral channels, and it was not necessary to adjust the threshold for each patient (Clemens et al., 2007), as it was shown that the amplitude of slow waves is dependent on gender and age (Carrier et al., 2011). The last step was to select only the high amplitude ‘widespread slow waves’, i.e. slow waves that were simultaneously detected in at least one of the two scalp channels and 50% of the intracerebral frontal channels with normal EEG activity.
To compare the rate of occurrence of interictal activity during high amplitude widespread slow waves, control segments of equal length to those with slow waves were randomly selected in every patient during intervals of the same sleep stages. They consisted of slow waves of low amplitude (among the 50% with lowest amplitude), non-widespread slow waves or no slow waves.
Assessment of epileptic spikes in intracerebral EEG
The depth-recorded montages were bipolar from one depth electrode contact to the neighbouring contact. Isolated epileptic spikes were manually identified in all frontal lobe intracerebral electrodes during the sleep slow waves and the corresponding control sections by a scorer who was blinded to the scalp EEG and to the presence of widespread slow waves. A time scale of 30 mm/s was chosen. Spikes had to be separated from neighbouring spikes by an interval of >1.5 s to fulfil the criterion for isolated spikes; trains of spikes were not marked for this study. A maximum of five different spike sets was marked for each patient. A spike set was defined by its difference in localization or morphologic appearance. For each spike, the duration of the highest amplitude spike as identified in the referential montage was marked in the bipolar montage. For the analysis, the onset of the marked events was used. For marking, a time scale of 150 mm/s was chosen.
Assessment of HFOs in the intracerebral EEG
For each subject, HFOs were visually marked in the first minute of the concatenated high amplitude sleep slow waves and the corresponding control sections using a bipolar montage, by two separate scorers. The concordance between marked HFOs was assessed using Cohen’s kappa coefficient (Cohen, 1960), computed for each bipolar channel. If Cohen’s kappa coefficient was <0.6, both scorers re-analysed the corresponding channel and established a consensus. The remaining time was then marked by one of the scorers based on the consensus reached.
For the identification of HFOs, channels were displayed with a maximum time resolution (∼0.6 s) across the computer monitor using an 80 Hz high pass finite impulse response filter. Only clear events containing at least four consecutive oscillations were marked as HFOs, and two events were considered distinct when separated by at least two non-HFO oscillations (Jacobs et al., 2010).
Computation of power in different bands
To corroborate the presumption that the negative scalp half-waves reflect the intracerebral ‘down’ states, we computed the power in the gamma (30–80 Hz) and ripple (80–250 Hz) bands around the negative peak of the slow waves. First, the same types of filter as previously described for the delta band were used to obtain the band-pass filtered signals. Their squared amplitude was then low-pass filtered with a zero-phase low pass infinite impulse response filter (second order, cut-off frequency 0.5, 4 and 5 Hz, respectively) to obtain the time course of the power in each frequency band. This quasi-instantaneous power was averaged time-locked to the negative peaks of the slow waves, in 3-s long sections centred at the negative peak. The resulting power evolution was averaged for each patient in all intracerebral channels exhibiting normal EEG and epileptic EEG activity, and then further averaged among patients.
Hypothesis testing
We tested whether during non-REM sleep the spikes and HFOs occur more often at the time of high amplitude widespread slow waves than during control segments obtained from the same sleep period. We pooled the data from all patients. As the number of channels and the length of the first non-REM cycle varied among patients, we limited the number of events per patient to a maximum equal to three times the minimum number of events per patient. This was done to avoid biased results based on a few patients with a large number of events. In addition, we also report results without limiting the number of events in order to illustrate the robustness of the data. We performed a single sample t-test, with an equal number of spikes in slow wave segments and control segments (of equal length) as the null hypothesis.
We also tested whether the density of spikes and HFOs was different between the negative half-wave and the preceding and following positive half-waves. A single sample t-test was performed, the null hypothesis being equal densities (i.e. number of events proportional to the duration of the negative and positive half-waves).
Results
We first show that the high amplitude frontal slow waves marked on the scalp have simultaneous correlates in the frontal intracerebral channels, and that these high amplitude widespread slow waves modulate the interictal epileptic activity. Then, we analyse the differences between the ‘up’ and ‘down’ states of the slow waves, regarding both physiological and epileptic activity. Finally, we show differences between intracerebral channels with normal and epileptic EEG activity.
Analysed channels and investigated time period
A total of 259 bipolar channels (12–59 per patient) gathered from 34 electrodes in the frontal lobe (2–7 per patient) were included in the final analysis (for electrode positions see Supplementary Table 1). Of these 259 channels, 129 (49.8%) had interictal epileptic discharges (4–56 per patient), and 130 (50.2%) were classified as channels with normal EEG activity (3–24 per patient).
The median duration of N2 and N3 non-REM sleep of the first sleep cycle was 56 min [range 36.5–118 min; mean 62.4 min, standard deviation (SD) 27.8 min]. During this time, the total number of high amplitude widespread slow waves was 3964 (median 480.5, range 180–1236, mean 495.5, SD 336.5 per patient). The frequency of the detected high amplitude slow waves was ∼1 Hz as shown in Supplementary Fig. 1. These high amplitude widespread slow waves were compared to 3964 matched control segments, which consisted of slow waves of low amplitude (among the 50% with lowest amplitude), non-widespread slow waves or no slow waves during the same sleep stages. The total duration of time analysed in this study was 98 min (49 min of slow waves and 49 min of control segments).
Intracerebral correlates of scalp slow waves
To investigate whether intracerebral slow waves peaked at the time of the detected scalp slow waves, we computed the power in the delta band in scalp and intracerebral channels. This demonstrated the presence of intracerebral slow waves peaking at the same time as scalp slow waves, confirming that slow waves investigated in this study occurred synchronously over wide regions. Similar results were found for intracerebral channels with normal EEG activity and intracerebral channels with interictal epileptic spikes (Fig. 2).
Figure 2.
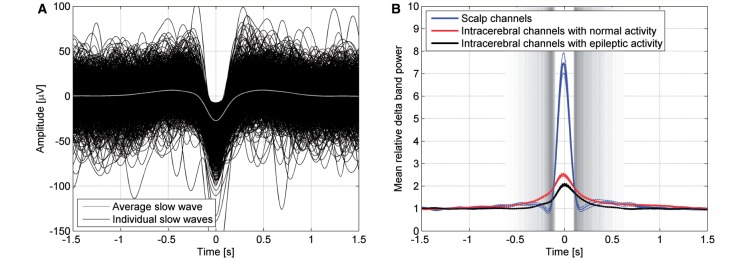
Intracerebral correlates of scalp slow waves. (A) Superimposed individual scalp slow waves and their average. The averaging was done after aligning the peaks of the negative half-waves (shown as time zero; negative down). (B) Power in the delta band in scalp and intracerebral channels demonstrated the presence of intracerebral slow waves peaking at the same time as scalp slow waves (blue line). Similar results were found for channels with normal EEG activity (red line) and channels with interictal epileptic spikes (black line). The standard error of the channels' mean is depicted in broken lines for 16 scalp channels, 130 intracerebral channels with normal EEG activity, and 129 intracerebral channels with epileptic EEG activity. The grey bands indicate the time of the transitions from positive to negative (left) and negative to positive (right) half-waves, the grey level indicating the density of the 3964 individual transitions from positive to negative and negative to positive half-waves, darker when more transitions occur.
High amplitude widespread slow waves modulate epileptic activity
A total of 442 isolated epileptic spikes (18–100 per patient) and 8487 HFOs (427–3325 per patient) were marked during the selected high amplitude widespread slow wave and control segments. After limiting the number of spikes to a maximum equal to three times the minimum (54 per patient), the total number of spikes was 273. The majority of spikes (209, 77%) were observed during the high amplitude widespread slow waves, the remaining spikes occurring during the control segments (P ≈ 0). On an individual level, all patients showed higher numbers of spikes during high amplitude widespread slow waves (range, 58–94%).
The distribution of HFOs was similar. After limiting the HFOs to a maximum of 1080 per patient, equal to three times the minimum per patient, the total number of HFOs was 5811. Sixty-five per cent (3768) occurred during high amplitude widespread slow waves, the remaining HFOs occurring during control periods (P ≈ 0). On an individual level, seven of eight subjects had a higher number of HFOs during the widespread slow waves (range, 54–84%), whereas only one patient showed fewer HFOs during the slow waves (43%) compared to the control segments.
We investigated the influence of the amplitude of the widespread slow waves on the probability of coinciding with epileptic discharges. We found that, among the high amplitude widespread slow waves, slow waves of higher amplitude facilitated spikes and HFOs more than slow waves of lower amplitude (Fig. 3). In addition, an important proportion of the spikes and HFOs occurring in the ‘control segments’ were associated with the low amplitude slow waves that make part of these segments (72% of spikes and 66% of HFOs in control segments). The remaining events occurred independently of the slow waves, or were associated with local slow waves not visible in the scalp.
Figure 3.
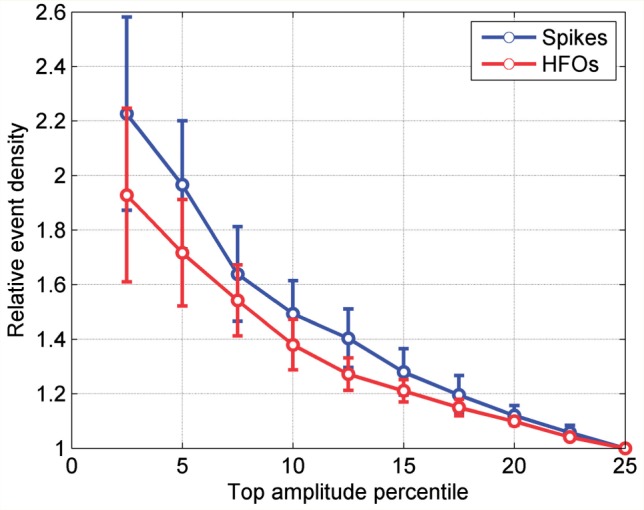
Density of spikes and HFOs as a function of the percentage of the highest amplitude slow waves analysed. The density is relative to the density found for the frontal widespread slow waves with the 25% highest amplitude. Error bars depict the standard error of the mean among the eight patients.
If we did not limit the number of spikes and HFOs per patient, similar percentages were found: 79% of spikes and 66% of ripples occurred during the slow waves, indicating the robustness of our results.
Physiological rhythms and coupling to the ‘up’ state in normal channels
A sleep slow wave comprises a surface negative half-wave (presumptive hyperpolarizing ‘down’ state), and the preceding and following surface positive waves (presumptive depolarizing ‘up’ state) (Steriade et al., 1993b). To confirm previous findings (Grenier et al., 2001; Mukovski et al., 2007), we examined the distribution of the gamma and ripple band power in relation to the negative slow half-wave in channels with normal EEG activity. We found that both the intracranial gamma and ripple band power show an increase before and after the negative half-wave, with suppression during the negative half-wave (Fig. 4A and B) suggesting that gamma and ripple band power are coupled to the ‘up’ state of the slow wave.
Figure 4.
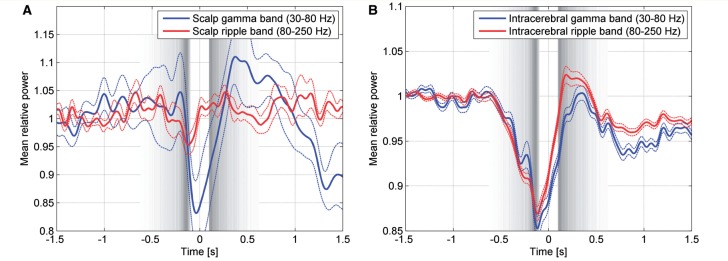
Gamma (blue line) and ripple band (red line) power for scalp channels (A) and for intracerebral channels with normal EEG activity (B). Broken lines indicate the standard error of the mean among the 16 scalp channels and the 130 intracerebral channels with normal EEG activity. Time zero corresponds to the peak of the negative half-waves, and their beginning and end are indicated by the grey bands, defined as in Fig. 2. As expected the ripple band in scalp channels is not very informative, because its amplitude is very low and hence confounded by the noise associated to the equipment (electrodes and amplifier). Of note, power in the gamma and ripple bands is higher after the negative half-wave, at the time of the cortical ‘up’ state.
Epileptic activity and coupling to the transition from ‘up’ to ‘down’ state
Next, we compared the density of spikes and HFOs between the negative half-wave (‘down’ state) and positive half-wave (‘up’ state) of the slow wave. As illustrated in Fig. 5, and contrary to our initial hypothesis, the spike density was significantly lower during the positive half-waves compared to the negative half-waves (P = 0.04). Forty-three per cent (n = 88) of the spikes occurring during slow waves had their onset during the scalp negative half-waves, which comprised 35.7% of the total slow wave time. More specifically, as can be seen in Fig. 5, most spikes occurred at the transition from the preceding positive to the negative half-wave. On an individual level, in six of the eight subjects the spike density was higher during the negative half-waves.
Figure 5.
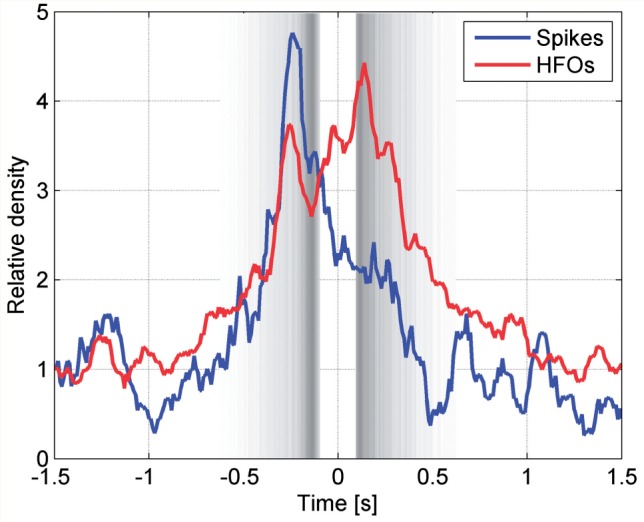
Variation in time of the spike and HFO density. Time zero corresponds to the peak of the negative half-waves, and their beginning and end are indicated by the grey bands, as defined in Fig. 2. An increase in the spike rate is seen at the transition between the negative half-wave and the preceding positive half-wave (blue line). An increase in HFO density is seen at the transition between the negative half-wave and the preceding positive half-wave as well as during the negative half-wave and first part of the following positive half-wave (red line).
The HFO density was also significantly lower during the positive scalp half-waves than during the negative half-waves (P ∼ 0). Forty-three per cent (n = 1602) of the HFOs occurring during slow waves had their onset during the negative scalp half-wave which comprised 35.7% of the total slow wave time. More specifically, as can be seen in Fig. 5, HFOs had a bimodal distribution peaking at the transition from the preceding positive half-wave to the negative half-wave as well as the transition from the negative half-wave to the following positive half-wave. In seven of the eight subjects the HFO density was higher during the negative half-waves. Representative examples for the coupling of epileptic spikes and HFOs across the slow wave cycle are provided in Fig. 6.
Figure 6.

Representative examples for the coupling of epileptic spikes and HFOs across the slow wave cycle. Example of a slow wave and an epileptic spike (left), a slow wave and an HFO in a channel with epileptic activity (middle) and an HFO in a channel with normal EEG activity (right). The top row shows the slow wave in a scalp channel, the second row shows the same time period for an intracerebral channel with normal EEG activity, and the third row an intracerebral channel with epileptic EEG activity. The bottom row shows the HFO signal with a different time and amplitude scale, corresponding to the shaded periods in the intracerebral channels. All the channels are in the left frontal region [anterior cingulate gyrus (LCA1–2), orbitofrontal area (LOF1–2, LOF2–3), second frontal gyrus (LCA7–8), third frontal gyrus (LOF9–10)], each example corresponds to a different patient. The scalp slow wave on the right panel is of shorter duration than the scalp slow waves on the left or middle panel. *In this example a normal sleep slow wave and no epileptic spike is seen in a channel called ‘epileptic’ because it has spikes at other times. Note that the spike and the HFO in the intracerebral channel with epileptic activity (middle) occurs prior to the peak of the scalp negative half-wave, whereas the HFO in the channel with normal EEG activity (right) occurs after the peak of the scalp negative half-wave.
If the numbers of spikes and HFOs per patient were not limited, similar percentages (44% of spikes and 42% of HFOs) occurred during the negative half-waves, indicating the robustness of these results.
Distribution pattern of HFOs is different in channels with epileptic and normal activity
As the distribution of HFOs was bimodal compared to the single peak in epileptic spike distribution (Fig. 5), we investigated whether this bimodal distribution (with a second peak at the beginning of the ‘up’ state) might be due to potentially physiological HFOs as suggested by the results we found for physiological EEG rhythms (Fig. 4). Therefore, we computed the HFO density in channels with epileptic spikes and in channels with normal activity. The variation in the timing of the HFO density is given in Fig. 7. An increase in HFO density was observed at the transition between the negative half-wave and the preceding positive one in channels with epileptic activity but not in channels with normal activity. In contrast, channels with normal activity showed a peak which occurred at the transition, between the negative half-wave and the following positive half-wave.
Figure 7.
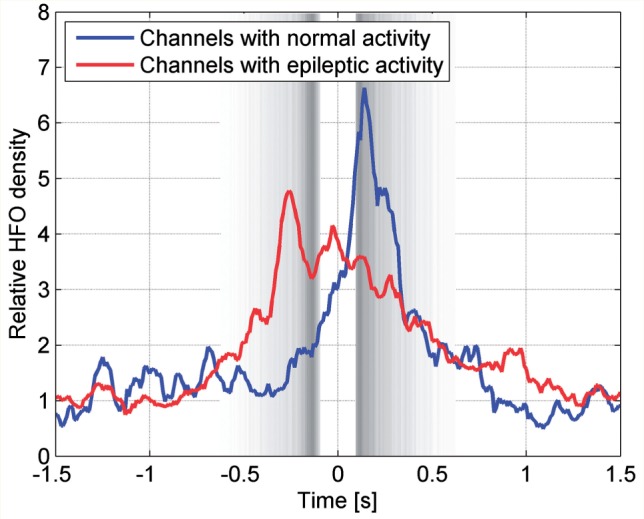
Variation in time of the HFO density. Time zero corresponds to the peak of the negative half-waves, and their beginning and end are indicated by the grey bands, as defined in Fig. 2. An increase in HFO density is observed at the transition between the negative half-wave and the preceding positive one in channels with epileptic EEG activity (red dotted line), but not in channels with normal EEG activity (blue solid line). In contrast, there is an increase in HFO density at the transition between the negative half-wave and the following positive one in channels with normal EEG activity.
We then determined if the pattern of the ripple band power (representing ongoing ripple-band activity, including but not restricted to individual brief and high amplitude HFO events) was similar to that of HFOs. The mean ripple power in channels with epileptic activity shows a peak at the same time as the peak in HFO density (Fig. 8A). However, HFOs are discrete events associated with an important increase in power for frequencies >80 Hz occurring only occasionally. The median ripple band power is more robust than the mean to these occasional outliers associated with individual HFOs, and showed the same general pattern in channels with normal and epileptic activity (Fig. 8B). This indicates that in most channels with epileptic activity, beyond the presence of occasional HFOs there is not a substantial difference in the ripple band behaviour compared to channels with normal activity, with preserved physiological associations to ‘up’ and ‘down’ states.
Figure 8.
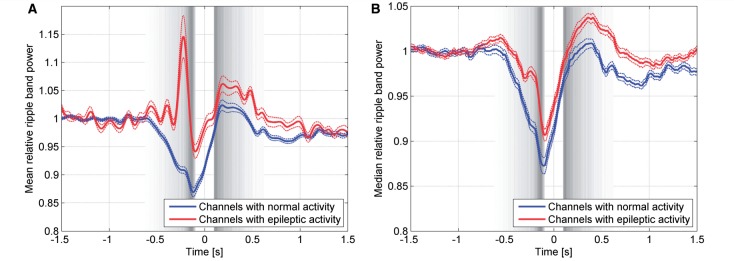
Average ripple band power distributed over channels with normal EEG activity (blue line), and channels with interictal spikes (red line). (A) The pattern of the ripple band power in intracerebral channels with normal EEG activity is different from that in channels with epileptic EEG activity, due to the presence of HFOs. (B) The median, less influenced by outliers associated with individual HFOs, shows the same general pattern in channels with normal and epileptic EEG activity. Broken lines indicate the standard error of the mean in A and standard error of the median in B among the 130 intracerebral channels with normal EEG activity and the 129 intracerebral channels with epileptic EEG activity.
Discussion
In this study we were interested to investigate the mechanisms by which activation of epileptic discharges occur during non-REM sleep, and hypothesized that cortical sleep slow waves, which have been experimentally identified as ‘up’ and ‘down’ states (Steriade, 2006; Haider and McCormick, 2009; Crunelli and Hughes, 2010), play a facilitating role for the expression of the epileptic activity. The main findings of this study are that: (i) high amplitude widespread slow waves are characterized by a significantly higher epileptic spike and HFO density than control segments gathered from the same sleep stages; and (ii) both epileptic events appear to predominate during the deactivated ‘down’ state of the slow waves, being most prominent during the transition from the preceding ‘up’ to the ‘down’ state, a finding somewhat unexpected and suggesting a mechanism distinct from the physiological EEG rhythms known to be activated by the ‘up’ state of the slow waves.
Widespread high amplitude slow waves facilitate epileptic activity
Several studies showed that non-REM sleep facilitates epileptic activity in focal epilepsy (Montplaisir et al., 1987; Sammaritano et al., 1991; Malow et al., 1998; Staba et al., 2004; Bagshaw et al., 2009). In this study, we demonstrated that these observations do not reflect a uniform activation by non-REM sleep, but that high amplitude widespread slow waves present during this state are specific modulators of epileptic activity. We found that interictal epileptic spikes and HFOs are more frequent during the largest amplitude widespread slow waves than in control periods with no or with low amplitude slow waves. This finding corresponds to and extends the current literature showing that the expression of interictal epileptiform potentials during sleep is related to the cyclic alternating pattern and more specifically pattern A1 which consists of a synchronous EEG pattern such as present in recurrent slow wave activation periods (for reviews see Parrino et al., 2000; Halász et al., 2013).
The increase in epileptic spike and HFO density during high amplitude slow waves is in line with the hypothesis that high temporal and spatial synchronization is an important factor facilitating epileptic activity. In a rodent study modelling mesiotemporal lobe epilepsy, Nazer and Clayton (2009) found that the number and amplitude of epileptiform discharges differed according to the state of slow wave sleep, and that they were higher during hippocampal slow than during hippocampal theta oscillations. We did not address if the high amplitude slow waves also facilitate ictal events, but several studies using scalp EEG showed that synchronous EEG delta activity or K-complexes are associated with the occurrence of sleep-related ictal episodes (Sforza et al., 1993; Terzano et al., 1997; Provini et al., 1999; Terzaghi et al., 2007, 2008; Perucca et al., 2013).
Physiological EEG rhythms are coupled to the ‘up’ state
Cortical slow waves play an important role in grouping physiological sleep rhythms such as spindles (7–14 Hz) (Steriade et al., 1993a). These findings were reproduced and extended to the broad gamma (20–100 Hz) and higher frequency bands (>80 Hz) for the rodent (Valencia et al., 2013) and feline cortex (Grenier et al., 2001; Steriade, 2006; Mukovski et al., 2007). In humans, the evidence that slow waves modulate physiological rhythms remains rather scarce. In the present study we confirmed that in intracerebral EEG channels with normal EEG activity, gamma and high frequency >80 Hz (ripple) band power are more pronounced during the cortical ‘up’ state. This corresponds well to a study in epileptic patients showing that gamma activity in the ‘IN-phase’ pattern is correlated to the cortical ‘up’ state (Valderrama et al., 2012).
Epileptic activity is coupled to the transition from ‘up’ to ‘down’ state
To assess whether epileptic activity is also more pronounced during the depolarizing ‘up’ state favouring hyperexcitability as facilitating trigger, we investigated the epileptic spike and HFO density in relation to ‘up’ and ‘down’ states. Unexpectedly, this analysis revealed that both spike and HFO densities in epileptic tissue are not coupled to the ‘up’ state, but to the transition from the preceding ‘up’ to the ‘down’ state. How can one interpret these surprising findings? As synchronization in the CNS is achieved primarily through inhibitory mechanisms (Farrant and Kaila, 2007; Mann and Paulsen, 2007; Engel, 2012), dysfacilitation during the hyperpolarizing ‘down’ state could predispose to neuronal hypersynchronization resulting in interictal epileptiform discharges. This hypothesis is supported by different experimental studies pointing to the paradoxical role played by GABAA receptor-mediated inhibitory mechanisms in synchronizing neuronal networks and generating epileptic activity (for a review, see Avoli and de Curtis, 2011).
However, one must also consider that both excitatory (principal) neurons and inhibitory (fast spiking) cells fire during the ‘up’ state, indicating that inhibition may actively contribute to the ‘up’ state (Steriade et al., 1993b; Sanchez-Vives and McCormick, 2000). Progressive removal of inhibition induced by GABAA receptor antagonists shortens the ‘up’ state duration and prolongs the ‘down’ state while causing a gradual increase in the network firing rate during ‘up’ states (Sanchez-Vives et al., 2010). Interestingly, these authors have obtained experimental and modeling evidence suggesting that epileptiform bursts tend to appear in the transitions between ‘up’ and ‘down’ states, which is in line with our findings. Whether a potential difference in the patterns of the ‘up’–’down’ state in the healthy versus epileptic brain (Bragin et al., 2012) contributes to our findings is speculative.
HFOs in channels with normal activity peak differently from those in channels with epileptic activity during the slow wave cycle
The distribution pattern of HFOs in relation to the slow wave was bimodal with a first peak in HFO density at the transition between the preceding ‘up’ and the following ‘down’ state and a second smaller peak at the transition to the following ‘up’ state. Interestingly, this distribution depended on the type of activity: whereas the HFO density peaked at the beginning of the ‘down’ state in case of channels with epileptic activity, it occurred at the transition to the ‘up’ state in channels with normal activity. This result suggests that the distribution pattern of HFOs in relation to the slow wave cycle may help to distinguish between physiological and pathological HFOs.
We are not aware of any experimental study investigating interictal HFOs in chronic epilepsy in relation to the slow wave cycle. In case of acute seizures in the feline model, seizure-related HFOs occurred during the ‘up’ state, as was also shown for physiological sleep- or anaesthesia-related HFOs (Grenier et al., 2001, 2003). The reason for this discrepancy with our data is not clear and awaits further examination.
This difference in the distribution pattern of HFOs is of importance and could be used to improve the seizure onset zone localization especially in brain areas in which physiological HFOs are present such as the paracentral areas, the hippocampus and the occipital cortex (Buzsáki et al., 1992; Grenier et al., 2001; Axmacher et al., 2008; Nagasawa et al., 2012; Melani et al., 2013). Only few papers focused on the differentiation between physiological and pathological HFOs, and a definite differentiating marker between both entities has not yet been established (Nagasawa et al., 2012; Melani et al., 2013; Wang et al., 2013; Kerber et al., 2014).
Potential limitations
One limitation of our study is that we have only indirect evidence for the cortical states of the slow waves as we did not use simultaneous intracellular recordings, which would be the proof for depolarization and hyperpolarization. However, we could confirm that physiological EEG rhythms, especially power in the gamma band, which was shown in the feline model to be an indirect proxy marker for the ‘up’ and ‘down’ state (Mukovski et al., 2007), appeared predominantly during the positive half-wave representing the presumed ‘up’ state. Hence we are confident that the detected half-waves correctly reflect the ‘up’ and ‘down’ states of the slow wave. The real onset and offset of the respective states, however, cannot be precisely determined with this indirect approach. Moreover, as we used a common average reference, we cannot totally exclude that some highly synchronous slow waves may have been missed. Also, we carefully excluded every segment with spikes on the scalp, therefore we believe that the presence of post-spike slow waves did not influence our findings (Urrestarazu et al., 2006). Finally, we decided a priori to focus on widespread slow waves detected on frontal scalp channels during the first sleep cycle and investigated their coupling with physiological and pathological brain activity within the same lobe; by doing so, we could presumably avoid the contribution of more focal slow waves, which occur particularly during later sleep cycles (Nir et al., 2011), and a potential spreading effect of the slow wave (Massimini et al., 2004). Nevertheless it cannot be completely ruled out that, despite this procedure, this spread could make the exact timing of epileptic spikes and HFOs dependent on the distance from the frontal scalp electrodes to the electrode that recorded the epileptic activity.
Conclusion
This study investigated the influence of cortical slow waves (and the role of the ‘up’ and ‘down’ states) on human focal epileptic activity. Our results point to high amplitude widespread slow waves facilitating the occurrence of focal interictal epileptic activity compared to control segments from the same sleep stages in patients with pharmaco-resistant focal epilepsy. These findings provide a mechanistic explanation of the role of non-REM sleep in facilitating epileptic activity. Moreover, and in contrast to physiological EEG rhythms in normal channels peaking during the ‘up’ state, epileptic spikes and HFOs occur mostly at the transition from the preceding ‘up’ to the ‘down’ state. This evidence suggests that in the epileptic brain periods coinciding with high synchronization rather than periods coinciding with hyperexcitability are associated with increased occurrence of interictal spikes and HFOs. Finally, our study demonstrates that HFOs in channels with normal activity peak differently during the slow wave cycle compare to HFOs in channels with epileptic activity, which might allow for a differentiation between physiological and pathological HFOs.
Supplementary Material
Acknowledgements
The authors are grateful for the assistance of the staff and technicians at the EEG Department at the Montreal Neurological Institute and Hospital, particularly Ms. Lorraine Allard and Ms. Nicole Drouin. The authors are also grateful to Dr Jeffery Hall and Dr André Olivier from the Department of Neurosurgery at the Montreal Neurological Institute and Hospital for electrode implantation during epilepsy surgery evaluation.
Glossary
Abbreviations
- HFO
high frequency oscillation
- REM
rapid eye movement
Funding
This work was supported by the Canadian Institutes of Health Research (grant MOP-102710), the Austrian Sleep Research Association and the Austrian Science Fund (Schrödinger fellowship abroad J3485 to Dr. Birgit Frauscher).
Supplementary material
Supplementary material is available at Brain online.
References
- Avoli M, de Curtis M. GABAergic synchronization in the limbic system and its role in the generation of epileptiform activity. Prog Neurobiol. 2011;95:104–32. doi: 10.1016/j.pneurobio.2011.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Axmacher N, Elger CE, Fell J. Ripples in the medial temporal lobe are relevant for human memory consolidation. Brain. 2008;131:1806–17. doi: 10.1093/brain/awn103. [DOI] [PubMed] [Google Scholar]
- Bagshaw AP, Jacobs J, LeVan P, Dubeau F, Gotman J. Effect of sleep stage on interictal high-frequency oscillations recorded from depth macroelectrodes in patients with focal epilepsy. Epilepsia. 2009;50:617–28. doi: 10.1111/j.1528-1167.2008.01784.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battaglia FP, Sutherland GR, McNaughton BL. Hippocampal sharp wave bursts coincide with neocortical ‘up-state’ transitions. Learn Mem. 2004;11:697–704. doi: 10.1101/lm.73504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltramo R, D’Urso G, Dal Maschio M, Farisello P, Bovetti S, Clovis Y, et al. Layer-specific excitatory circuits differentially control recurrent network dynamics in the neocortex. Nat Neurosci. 2013;16:227–34. doi: 10.1038/nn.3306. [DOI] [PubMed] [Google Scholar]
- Berry RB, Brooks R, Gamaldo CE, Harding SM, Marcus CL. Vaughn BV for the American Academy of Sleep Medicine. The AASM Manual for the Scoring of Sleep and Associated Events: Rules, Terminology and Technical Specifications, version 2.0. Darien, Illinois: American Academy of Sleep Medicine, 2012. www.aasmnet.org. [Google Scholar]
- Bragin A, Benassi SK, Engel J. Patterns of the up-down state in normal and epileptic mice. Neuroscience. 2012;225:76–87. doi: 10.1016/j.neuroscience.2012.08.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzsáki G, Horváth Z, Urioste R, Hetke J, Wise K. High-frequency network oscillation in the hippocampus. Science. 1992;256:1025–7. doi: 10.1126/science.1589772. [DOI] [PubMed] [Google Scholar]
- Carrier J, Viens I, Poirier G, Robillard R, Lafortune M, Vandewalle G, et al. Sleep slow wave changes during the middle years of life. Eur J Neurosci. 2011;33:758–66. doi: 10.1111/j.1460-9568.2010.07543.x. [DOI] [PubMed] [Google Scholar]
- Chauvette S, Volgushev M, Timofeev I. Origin of active states in local neocortical networks during slow wave oscillation. Cereb Cortex. 2010;20:2660–74. doi: 10.1093/cercor/bhq009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemens Z, Mölle M, Eröss L, Barsi P, Halász P, Born J. Temporal coupling of parahippocampal ripples, sleep spindles and slow oscillations in humans. Brain. 2007;130:2868–78. doi: 10.1093/brain/awm146. [DOI] [PubMed] [Google Scholar]
- Clemens Z, Mölle M, Eros L, Jakus R, Rasonyi G, Halasz P, et al. Fine-tuned coupling between human parahippocampal ripples and sleep spindles. Eur J Neurosci. 2011;33:511–20. doi: 10.1111/j.1460-9568.2010.07505.x. [DOI] [PubMed] [Google Scholar]
- Cohen J. A coefficient of agreement for nominal scales. Educ Psychol Meas. 1960;20:37–46. [Google Scholar]
- Crunelli V, Hughes SW. The slow (<1 Hz) rhythm of non-REM sleep: a dialogue between three cardinal oscillations. Nat Neurosci. 2010;13:9–17. doi: 10.1038/nn.2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel J. Chapter 4: Basic mechanisms of seizures and epilepsy. In: Engel J, editor. Seizures and epilepsy. 2nd edn. Oxford University Press; 2012. pp. 99–156. [Google Scholar]
- Farrant M, Kaila K. The cellular, molecular and ionic basis of GABA(A) receptor signalling. Prog Brain Res. 2007;160:59–87. doi: 10.1016/S0079-6123(06)60005-8. [DOI] [PubMed] [Google Scholar]
- Frauscher B, von Ellenrieder N, Dubeau F, Gotman J. Scalp spindles are associated with widespread intracranial activity with unexpectedly low synchrony. Neuroimage. 2015;105:1–12. doi: 10.1016/j.neuroimage.2014.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grenier F, Timofeev I, Steriade M. Focal synchronization of ripples (80-200 Hz) in neocortex and their neuronal correlates. J Neurophysiol. 2001;86:1884–98. doi: 10.1152/jn.2001.86.4.1884. [DOI] [PubMed] [Google Scholar]
- Grenier F, Timofeev I, Steriade M. Neocortical very fast oscillations (ripples, 80-200 Hz) during seizures: intracellular correlates. J Neurophysiol. 2003;89:841–52. doi: 10.1152/jn.00420.2002. [DOI] [PubMed] [Google Scholar]
- Haider B, McCormick DA. Rapid neocortical dynamics: cellular and network mechanisms. Neuron. 2009;62:171–89. doi: 10.1016/j.neuron.2009.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halász P, Kelemen A, Szűcs A. The role of NREM sleep micro-arousals in absence epilepsy and in nocturnal frontal lobe epilepsy. Epilepsy Res. 2013;107:9–19. doi: 10.1016/j.eplepsyres.2013.06.021. [DOI] [PubMed] [Google Scholar]
- Isomura Y, Sirota A, Ozen S, Montgomery S, Mizuseki K, Henze DA, et al. Integration and segregation of activity in entorhinal-hippocampal subregions by neocortical slow oscillations. Neuron. 2006;52:871–82. doi: 10.1016/j.neuron.2006.10.023. [DOI] [PubMed] [Google Scholar]
- Ives JR. New chronic EEG electrode for critical/intensive care unit monitoring. J Clin Neurophysiol. 2005;22:119–123. doi: 10.1097/01.wnp.0000152659.30753.47. [DOI] [PubMed] [Google Scholar]
- Jacobs J, Zijlmans M, Zelmann R, Chatillon CE, Hall J, Olivier A, et al. High-frequency electroencephalographic oscillations correlate with outcome of epilepsy surgery. Ann Neurol. 2010;67:209–20. doi: 10.1002/ana.21847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerber K, Duempelmann M, Schelter B, Le Van P, Korinthenberg R, Schulze-Bonhage A, et al. Differentiation of specific ripple patterns help to identify epileptogenic areas for surgical procedures. Clin Neurophysiol. 2014;125:1339–45. doi: 10.1016/j.clinph.2013.11.030. [DOI] [PubMed] [Google Scholar]
- Malow BA, Lin X, Kushwaha R, Aldrich MS. Interictal spiking increases with sleep depth in temporal lobe epilepsy. Epilepsia. 1998;39:1309–16. doi: 10.1111/j.1528-1157.1998.tb01329.x. [DOI] [PubMed] [Google Scholar]
- Mann EO, Paulsen O. Role of GABAergic inhibition in hippocampal network oscillations. Trends Neurosci. 2007;30:343–9. doi: 10.1016/j.tins.2007.05.003. [DOI] [PubMed] [Google Scholar]
- Massimini M, Huber R, Ferrarelli F, Hill S, Tononi G. The sleep slow oscillation as a traveling wave. J Neurosci. 2004;24:6862–70. doi: 10.1523/JNEUROSCI.1318-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melani F, Zelmann R, Mari F, Gotman J. Continuous high frequency activity: a peculiar SEEG pattern related to specific brain regions. Clin Neurophysiol. 2013;124:1507–16. doi: 10.1016/j.clinph.2012.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metherate R, Cox CL, Ashe JH. Cellular bases of neocortical activation: modulation of neuronal oscillations by the nucleus basalis and endogenous acetylcholine. J Neurosci. 1992;12:4701–11. doi: 10.1523/JNEUROSCI.12-12-04701.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mölle M, Marshall L, Gais S, Born J. Grouping of spindle activity during slow oscillations in human non-rapid eye movement sleep. J Neurosci. 2002;22:10941–7. doi: 10.1523/JNEUROSCI.22-24-10941.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mölle M, Yeshenko O, Marshall L, Sara SJ, Born J. Hippocampal sharp wave-ripples linked to slow oscillations in rat slow-wave sleep. J Neurophysiol. 2006;96:62–70. doi: 10.1152/jn.00014.2006. [DOI] [PubMed] [Google Scholar]
- Montplaisir J, Laverdière M, Saint-Hilaire JM, Rouleau I. Nocturnal sleep recording in partial epilepsy: a study with depth electrodes. J Clin Neurophysiol. 1987;4:383–8. doi: 10.1097/00004691-198710000-00003. [DOI] [PubMed] [Google Scholar]
- Mukovski M, Chauvette S, Timofeev I, Volgushev M. Detection of active and silent states in neocortical neurons from the field potential signal during slow-wave sleep. Cereb Cortex. 2007;17:400–14. doi: 10.1093/cercor/bhj157. [DOI] [PubMed] [Google Scholar]
- Nagasawa T, Juhasz C, Rothermel R, Hoechstetter K, Sood S, Asano E. Spontaneous and visually-driven high-frequency oscillations in the occipital cortex: intracranial recordings in epileptic patients. Hum Brain Mapp. 2012;33:569–83. doi: 10.1002/hbm.21233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nazer F, Dickson CT. Slow oscillation state facilitates epileptiform events in the hippocampus. J. Neurophysiol. 2009;102:1880–9. doi: 10.1152/jn.90795.2008. [DOI] [PubMed] [Google Scholar]
- Nir Y, Staba RJ, Andrillon T, Vyazovskiy VV, Cirelli C, Fried I, et al. Regional slow waves and spindles in human sleep. Neuron. 2011;70:153–69. doi: 10.1016/j.neuron.2011.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrino L, Smerieri A, Spaggiari MC, Terzano MG. Cyclic alternating pattern (CAP) and epilepsy during sleep: how a physiological rhythm modulates a pathological event. Clin Neurophysiol. 2000;111(Suppl 2):S39–46. doi: 10.1016/s1388-2457(00)00400-4. [DOI] [PubMed] [Google Scholar]
- Perucca P, Dubeau F, Gotman J. Widespread EEG changes precede focal seizures. PLoS One. 2013;8:e80972. doi: 10.1371/journal.pone.0080972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provini F, Plazzi G, Tinuper P, Vandi S, Lugaresi E, Montagna P. Nocturnal frontal lobe epilepsy. A clinical and polygraphic overview of 100 consecutive cases. Brain. 1999;122:1017–31. doi: 10.1093/brain/122.6.1017. [DOI] [PubMed] [Google Scholar]
- Riedner BA, Vyzazovsky VV, Huber R, Massimini M, Esser S, Murphy M, et al. Sleep homeostasis and cortical synchronization: III. A high density EEG study of sleep slow waves in humans. Sleep. 2007;30:1643–57. doi: 10.1093/sleep/30.12.1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sammaritano M, Gigli GL, Gotman J. Interictal spiking during wakefulness and sleep and the localization of foci in temporal lobe epilepsy. Neurology. 1991;41:290–7. doi: 10.1212/wnl.41.2_part_1.290. [DOI] [PubMed] [Google Scholar]
- Sanchez-Vives MV, Mattia M, Compte A, Perez-Zabalza M, Winograd M, Descalzo VF, et al. Inhibitory modulation of cortical up states. J Neurophysiol. 2010;104:1314–24. doi: 10.1152/jn.00178.2010. [DOI] [PubMed] [Google Scholar]
- Sanchez-Vives MV, McCormick DA. Cellular and network mechanisms of rhythmic recurrent activity in neocortex. Nat Neurosci. 2000;3:1027–34. doi: 10.1038/79848. [DOI] [PubMed] [Google Scholar]
- Sforza E, Montagna P, Rinaldi R, Tinuper P, Cerullo A, Cirignotta F, et al. Paroxysmal periodic motor attacks during sleep: clinical and polygraphic features. Electroencephalogr Clin Neurophysiol. 1993;86:161–6. doi: 10.1016/0013-4694(93)90003-e. [DOI] [PubMed] [Google Scholar]
- Sirota A, Csicsvari J, Buhl D, Buzsáki G. Communication between neocortex and hippocampus during sleep in rodents. Proc Natl Acad Sci USA. 2003;100:2065–9. doi: 10.1073/pnas.0437938100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staba RJ, Wilson CL, Bragin A, Jhung D, Fried I, Engel J., Jr High-frequency oscillations recorded in human medial temporal lobe during sleep. Ann Neurol. 2004;56:108–15. doi: 10.1002/ana.20164. [DOI] [PubMed] [Google Scholar]
- Steriade M. Grouping of brain rhythms in corticothalamic systems. Neurosci. 2006;137:1087–106. doi: 10.1016/j.neuroscience.2005.10.029. [DOI] [PubMed] [Google Scholar]
- Steriade M, Contreras D, Amzica F. Synchronized sleep oscillations and their paroxysmal developments. Trends Neurosci. 1994;17:199–208. doi: 10.1016/0166-2236(94)90105-8. [DOI] [PubMed] [Google Scholar]
- Steriade M, Contreras D, Curró Dossi R, Nuñez A. The slow oscillation in reticular thalamic and thalamocortical neurons: scenario of sleep rhythm generation in interacting thalamic and neocortical networks. J Neurosci. 1993a;13:3284–99. doi: 10.1523/JNEUROSCI.13-08-03284.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steriade M, Nuñez A, Amzica F. A novel slow (<1 Hz) oscillation of neocortical neurons in vivo: depolarizing and hyperpolarizing components. J Neurosci. 1993b;13:3252–65. doi: 10.1523/JNEUROSCI.13-08-03252.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terzaghi M, Sartori I, Mai R, Tassi L, Francione S, Cardinale F, et al. Sleep-related minor motor events in nocturnal frontal lobe epilepsy. Epilepsia. 2007;48:335–41. doi: 10.1111/j.1528-1167.2006.00929.x. [DOI] [PubMed] [Google Scholar]
- Terzaghi M, Sartori I, Mai R, Tassi L, Francione S, Cardinale F, et al. Coupling of minor motor events and epileptiform discharges with arousal fluctuations in NFLE. Epilepsia. 2008;49:670–6. doi: 10.1111/j.1528-1167.2007.01419.x. [DOI] [PubMed] [Google Scholar]
- Terzano MG, Monge-Strauss MF, Mikol F, Spaggiari MC, Parrino L. Cyclic alternating pattern as a provocative factor in nocturnal paroxysmal dystonia. Epilepsia. 1997;38:1015–25. doi: 10.1111/j.1528-1157.1997.tb01485.x. [DOI] [PubMed] [Google Scholar]
- Timofeev I, Steriade M. Low-frequency rhythms in the thalamus of intact-cortex and decorticated cats. J Neurophysiol. 1996;76:4152–68. doi: 10.1152/jn.1996.76.6.4152. [DOI] [PubMed] [Google Scholar]
- Urrestarazu E, Jirsch JD, Le Van P, Hall J, Avoli M, Dubeau F, et al. High-frequency intracerebral EEG activity (100-500 Hz) following interictal spikes. Epilepsia. 2006;47:1465–76. doi: 10.1111/j.1528-1167.2006.00618.x. [DOI] [PubMed] [Google Scholar]
- Valderrama M, Crépon B, Botella-Soler V, Martinerie J, Hasboun D, Alvarado-Rojas C, et al. Human gamma oscillations during slow wave sleep. PloS One. 2012;7:e33477. doi: 10.1371/journal.pone.0033477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valencia M, Artieda J, Bolam JP, Mena-Segovia J. Dynamic interaction of spindles and gamma activity during cortical slow oscillations and its modulation by subcortical afferents. PLoS One. 2013;8:e67540. doi: 10.1371/journal.pone.0067540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Wang IZ, Bulacio JC, Mosher JC, Gonzalez-Martinez J, Alexopoulos AV, et al. Ripple classification helps to localize the seizure-onset zone in neocortical epilepsy. Epilepsia. 2013;54:370–6. doi: 10.1111/j.1528-1167.2012.03721.x. [DOI] [PubMed] [Google Scholar]
- Zijlmans M, Jiruska P, Zelmann R, Leijten FS, Jefferys JG, Gotman J. High-frequency oscillations as a new biomarker in epilepsy. Ann Neurol. 2012;71:169–78. doi: 10.1002/ana.22548. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.