

Abstract
Figuring out what is wrong in Fanconi anemia (FA) patient cells is critical to understanding the contributions of the FA pathway to DNA repair and tumor suppression. Although FA patients exhibit a wide range of disease manifestation as well as severity (asymptomatic to congenital abnormalities, bone marrow failure, and cancer), cells from FA patients share underlying defects in their ability to process DNA lesions that interfere with DNA replication. In particular, FA cells are very sensitive to agents that induce DNA interstrand crosslinks (ICLs). The cause of this pronounced ICL sensitivity is not fully understood, but has been linked to the aberrant activation of DNA damage repair proteins, checkpoints and pathways. Thus, regulation of these responses through coordination of repair processing at stalled replication forks is an essential function of the FA pathway. Here, we briefly summarize some of the aberrant DNA damage responses contributing to defects in FA cells, and detail the newly-identified relationship between FA and the mismatch repair protein, MSH2. Understanding the contribution of MSH2 and/or other proteins to the replication problem in FA cells will be key to assessing therapeutic options to improve the health of FA patients. Moreover, loss of these factors, if linked to improved replication, could be a key event in the progression of FA cells to cancer cells. Likewise, loss of these factors could synergize to enhance tumorigenesis or confer chemoresistance in tumors defective in FA-BRCA pathway proteins and provide a basis for biomarkers for disease progression and response.
Keywords: crosslink repair, DNA repair, FANCJ helicase, Fanconi Anemia, mismatch repair, MSH2
It remains a mystery why Fanconi anemia (FA) patient cells are exquisitely sensitive to agents that induce DNA interstrand crosslinks (ICLs). This sensitivity is common to cells from the 16 known FA complementation groups (FANCA through FANCQ) of which patients suffer a range of maladies including congenital abnormalities, developmental defects, anemia, and cancer (recently reviewed in1,2). Even in the absence of exogenous genotoxic stress, FA hematopoetic stem cells exhibit elevated DNA damage and abnormal checkpoint responses, explaining the increased prevalence for bone marrow failure in FA patients.3 Thus, FA cells have an underlying deficiency in repairing not only ICLs but also endogenous DNA damage.4 A basic understanding of the underlying replication problem and the resulting increased sensitivity to ICLs could therefore provide an opportunity for therapeutic intervention, not only for bone marrow failure but also for treatment of malignancy that develops in FA patients. In a larger context, this understanding could provide insight into how the FA pathway modulates the response of tumors to a broad range of crosslinking chemotherapy agents, such as platinum salts and alkylating agents.
To protect the genome from replication stress and potentially throughout the cell cycle, the FA-BRCA pathway functions with a diverse set of proteins including: structure-specific endonucleases which bind and incise DNA structures; error tolerating translesion DNA synthesis (TLS) polymerases, which bypass certain DNA lesions; and DNA recombinases. In the most basic model, the pathway is triggered when a DNA replication fork collides with a DNA lesion, such as an ICL.5 An upstream core complex functions to mono-ubiquitinate FANCD2 and FANCI (“ID” complex), thereby triggering changes in the FA complex that facilitates the coordination of the FAN endonuclease with other nucleases (XPF-ERCC1, MUS81-EME1, SLX1-SLX4, SNM1A, and SNM1B) implicated in ICL repair that incise and unhook DNA crosslinks to generate a double-stranded DNA break (DSB). The unhooked ICL is bypassed by TLS polymerases and the replication fork is restored via recombination events mediated in part by downstream FA proteins such as FANCJ/BACH1, FANCD1/BRCA2, and FANCN/PalB2. Elegant studies continue to reveal many new details of FA pathway function in ICL repair,5 including the recent finding that FANCM can traverse an ICL and therefore separate repair from replication.6 Moreover, additional functions of the FA pathway continue to emerge2,7 as well as a greater understanding of modifiers of FA phenotypes. In particular, an endogenous source of DNA crosslinks that disrupts replication in FA cells comes from aldehydes.8 Correspondingly, loss of aldehyde dehydrogenases, such as ALDH2 in an FA mouse, exacerbates many features of FA, including bone marrow failure.9-11 Here, we review other factors that contribute to defects in FA cells that could provide insight toward therapeutic intervention, balancing out defects to suppress bone marrow failure or targeting pathways in FA cancer progression to limit tumorigenesis.
Overactive Nonhomologous End-Joining Pathway
Clues to understanding the defects in FA cells have come from the identification of proteins or pathways whose inactivation improves the fitness of FA cells. One of the first pathways identified to contribute to both genomic instability and ICL hypersensitivity in FA cells is the nonhomologous end-joining (NHEJ) pathway. Aside from its role in rearranging immunoglobulin, NHEJ fixes broken DNA ends and is important for genome stability. In FA cells, several groups report that NHEJ is overactive in S phase and interferes with the preservation of replication forks. Thus, inhibition of NHEJ can improve FA cells in many respects. For example, loss of NHEJ factor, Ku in cells mutant for FA pathway protein, FANCC enhanced ICL resistance and reduced genomic instability.12 Similarly, inhibition of the NHEJ factor, DNA-PKcs, enhanced ICL resistance in cells deficient for FA pathway proteins FANCD2, FANCA or FANCC.13 One of the best examples of rescue by elimination of NHEJ comes from studies in BRCA1-deficient cells, which share common defects with FA cells. Most notably, loss of 53BP1, a DNA repair protein functioning in part with the NHEJ pathway, restores homologous recombination (HR) and rescues the lethality in Brca1 deficient mice.14-17 These promising findings suggest that elimination of NHEJ could restore fitness in FA/BRCA pathway-deficient cells by reinstating DNA repair, albeit with the cost of impaired NHEJ. However, in some FA/BRCA pathway-deficient cells, elimination of NHEJ failed to completely suppress crosslink sensitivity. In particular, Brca1 deficient mouse cells that have 53BP1 loss remain sensitive to ICL-inducing agents. Combined loss of 53BP1 and Ku was able to enhance resistance, but not to the level observed in cells with wild-type BRCA1.18 Likewise loss of NHEJ did not enhance ICL resistance in worms or human FA-J patient cells that are deficient for the BRCA1-interacting helicase FANCJ (BACH1/BRIP1).13,19 The relationship between FA and NHEJ pathways appears to be species-specific given that in contrast to the rescue in human cells,13 inactivation of NHEJ exacerbated ICL sensitivity in Fancd2-−/− mouse cells and double knockout mice had more severe developmental defects.18,20 Thus, BRCA-FA proteins likely have several functions in the repair of ICLs that extend beyond balancing HR and NHEJ pathways.
Aberrant DNA Damage Responses and Opportunistic DNA Nucleases
In addition to NHEJ, several other factors have been suggested to contribute to the proliferative impairment of FA cells. First, the p53-p21 axis has been linked to the elevated DNA damage response (DDR) in FA cells. Consistent with this interpretation, depletion of p53 suppresses haematopoietic stem and progenitor cell (HSPC) defects observed in Fancd2−/− mice.3 Again, rescue appears to fall short of a panacea for FA cells as elimination of the p53-p21 axis did not suppress the underlying replication stress and genomic instability that accumulates in FA cells.3 Second, nucleases have been linked with an overactive DDR and genomic instability in FA cells. The nuclease-helicase DNA2 over-resects ICLs in FANCD2-deficient cells and depletion of DNA2 suppresses cisplatin sensitivity.21 Moreover, the MRE11 nuclease aberrantly degrades replication forks and contributes to genomic instability in BRCA2/FANCD1-, BRCA1-, and FANCA-deficient cells.22,23 In fact, in FA cells, inhibition of the MRE11 nuclease can preserve fork integrity similar to overexpression of a mutant Rad51 that binds and coats stalled forks.22 However, given that HR is not also restored, preserving stalled forks alone may not improve ICL repair. Thus, it may be critical to both protect replication forks from nucleases and also enhance recombination to comprehensively improve the fitness of FA cells. Accordingly, overexpression of wild-type Rad51 improved recombination and resistance to DNA damaging agents in BRCA2 mutant cells.24,25 Moreover, depletion of the anti-recombinase PARI reduced chromosomal aberrations and sensitivity of FA cells to PARP inhibitors.26
MSH2 and Defects in Cells Lacking FANCJ-MLH1 Interaction
More recently, enhanced replication stress and an overactive DDR in FA cells have been linked to proteins of the mismatch repair (MMR) pathway. The link to MMR came from studies designed to understand why ICL resistance requires the direct interaction between FANCJ and the MMR protein, MLH1.27-29 We found that the MMR protein, MSH2 underlies the ICL processing defects resulting from loss of the FANCJ-MLH1 interaction.19 Consistent with this finding, depletion of MSH2 suppresses defects found in cells deficient for the FANCJ-MLH1 interaction, including ICL-induced sensitivity, chromosomal aberrations, abnormal G2/M accumulation, and as well as an over-active NHEJ pathway. Depletion of MSH2 did not appear to alter recombination. Instead, the restored ICL resistance was dependent on Rad18-dependent pathway and the Rev1 bypass polymerase suggesting MSH2 depletion enhanced TLS.19 These outcomes are not unprecedented. In fact, MSH2 deficiency reverses proliferation defects due to short telomeres by a mechanism independent of recombination.30 Moreover, deficiency in MMR is associated with fewer DSBs at stalled forks and enhanced bypass.31-35 Furthermore, loss of MMR is linked to enhanced ICL resistance.36 While the function of MMR proteins in ICL processing is not entirely clear, they bind ICL lesions and other DNA structures that form at stalled replication forks through either the heterodimer, MutSβ (composed of MSH2 and MSH3), or MutSα (composed of MSH2 and MSH6), which subsequently recruit the MutLα complex (composed of MLH1 and PMS2).34,37-40 Our data indicate that at stalled replication forks, MSH2 interferes with replication restart in cells lacking the FANCJ-MLH1 interaction.19 Therefore, the interaction between FANCJ and MLH1 could serve to coordinate the DDR and to prevent unproductive MMR processing that impedes the recovery of cells following replication stress.
A conceptual model predicts that FANCJ might serve to catalytically displace the MSH2 protein complex bound to damaged or alternatively structured DNA to facilitate ICL repair (Fig. 1). This scenario is supported by recent biochemical data showing that FANCJ partners with the single-stranded DNA binding protein Replication Protein A (RPA) to efficiently disrupt high affinity interactions of proteins bound to duplex DNA driven by FANCJ's motor ATPase.41 The co-localization of FANCJ with RPA after replication stress also suggests that FANCJ is well situated at forks to help them resume DNA synthesis potentially in an RPA-stimulated manner.42 Conceivably, FANCJ contributes to replication restart similar to other RPA-interacting factors that have DNA metabolizing functions such as the SMARCAL1 translocase or RecQ helicases BLM, WRN, and the Fanconi Anemia FANCM/FAAP24 complex.43-50 Understanding the molecular mechanism whereby FANCJ promotes ICL repair and the restart of stalled replication forks, mediated by its interaction with MLH1 in a pathway suppressed by MSH2, should provide valuable insight to how FANCJ operates with other downstream factors to enable appropriate processing of ICLs or stalled forks so that HR can proceed to preserve genomic stability.
Figure 1.
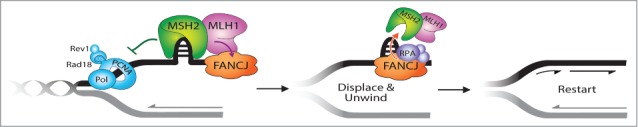
FANCJ-MLH1 interaction suppresses MSH2 to restart stalled replication forks. The timely localization of FANCJ to a fork blocking lesion is mediated by its MLH1 interaction, which places FANCJ in position to subsequently dismantle MSH2 or the DNA structures bound by MSH2 at stalled replication forks. This MSH2 displacement activity is dependent on FANCJ ATPase activity and is facilitated by FANCJ binding to RPA. Failure to displace MSH2 disrupts replication restart and underlies the “problem” in FA cells that lack FA and MMR pathway connections.
Linking MMR and FA-BRCA Pathways for Mutual Regulation
In addition to the FANCJ-MLH1 interaction, FA and MMR pathways are linked at several nodes, suggesting this crosstalk has functional importance. These interactions include BRCA1-MSH2, FANCD2-MLH1/MSH2, SLX4/FANCP-MSH2, and FANCD2-associated nuclease, FAN1-MLH1,27,51-61 but the functional importance of these interactions has not been fully addressed. A simplified FA pathway in yeast reveals an interaction between the FANCM-like helicase, Mph1, and MutSα functions to repair ICL-induced breaks through the recruitment of Rad51. Notably, this Mph1-MutSα ICL repair pathway involves Chl1, a putative FANCJ helicase.61 In human cells, it was demonstrated that MSH2 promotes FA pathway function including the chromatin localization of FA core components, the monoubiquitination of FANCD2,1,51,52 and the localization of FANCJ to sites of DNA crosslinks.62,63 Given the relationship between the FANCJ-MLH1 interaction and MSH2, a central question is whether other components of the FA pathway also function to balance the activity of MSH2 or other MMR proteins. Supporting this idea, MSH2 loss also suppresses defects in cells deficient for BRCA1 or FANCD2, and this rescue was confirmed in Msh2−/− Fancd2−/− mouse cells. Rescue included not only enhanced ICL resistance, but also suppression of an aberrant DDR including the NHEJ factor, DNA-PKcs.19 The mechanism linked to suppression of these FA defects was not fully explored, but it is worth considering that similar to the rescue in cells lacking the FANCJ-MLH1 interaction, MSH2 depletion enhances restart pathways.19 Alternatively, MSH2 loss could reduce fork collapse by enabling FANCM to traverse ICLs and ensure replication forks are uncoupled from ICL repair.6 This model could explain why MSH2 depletion did not suppress ICL sensitivity in FANCM-null chicken cells.52 Likewise, MSH2 depletion did not suppress ICL sensitivity in FANCA deficient cells.19 Since neither FANCM nor FANCA have known MMR interactions, MSH2 depletion may only rescue FA cells that have defects in FA-MMR interactions but otherwise have intact ICL processing enzymes. Collectively, these findings suggest that cross talk between the MMR and FA pathways enables not only the FA pathway to modulate the MMR pathway, but also for the MMR pathway to modulate the FA pathway and generates a full-regulatory circuit.
Modulators of FA Cell Fitness and Clinical Potential
Ideally the health of FA patients will also be improved by the inactivation or suppression of proteins or pathways that reduce the fitness of FA cells. In particular, it will be important to consider strategies that reduce sources of endogenous DNA damage. Incubation of FA cells under low oxygen conditions can enhance proliferation,64 and treatment with antioxidants delays carcinogenesis in FA mice.65 Moreover, enhancing aldehyde catabolism through agonists or balancing their loss with free radical quenchers are promising future directions.66 In addition, the expression and activity levels of factors that promote replication stress in FA cells could provide biomarkers for disease onset and progression. For example, in FA patients, MSH2 loss could signify that bone marrow failure is improbable but that leukemia is developing. Indeed, MMR-deficient mice are predisposed to hematologic malignancy67,68 and MMR loss is implicated in acute myeloid leukemia in humans.69-71 Not only does loss of MMR protein expression and function contribute to cancer susceptibility, it also contributes to chemoresistance.36,72 Given that the FA-BRCA pathway is essential for tumor suppression in a range of tumors, including breast and ovarian cancer,73 MMR loss could contribute to a diverse set of cancers and affect patient response. Significantly, if cancers evolve or develop chemoresistance through loss of MSH2 function, several therapeutic options can be considered.74 In particular, methotrexate induces oxidative DNA damage and is selectively lethal to cancer cells with defects in MSH275 and may prove efficacious on FA-associated cancers in which free radicals already reduce fitness. Developing novel and directed cancer therapies is of high priority, especially since FA patients are exquisitely sensitive to the DNA damage induced by traditional therapies. Ideally, if we are able to right what is wrong in FA patient cells, there will be hope to mitigate bone marrow failure and the development of cancer in patients.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We wish to thank Drs. Roger A. Greenberg (UPENN), William R. Kobertz (UMASS), and Jennifer Calvo (UMASS) for comments on the manuscript.
Funding
This work was supported by NCI RO1 Grant 11150917 (SC) and in part by the Intramural Research Program of the National Institutes of Health, National Institute on Aging (RMB).
References
- 1. Walden H, Deans AJ. The Fanconi anemia DNA repair pathway: structural and functional insights into a complex disorder. Annu Rev Biophys 2014; 43:257-78; PMID:24773018; http://dx.doi.org/ 10.1146/annurev-biophys-051013-022737. [DOI] [PubMed] [Google Scholar]
- 2. Nalepa G, Clapp DW. Fanconi anemia and the cell cycle: new perspectives on aneuploidy. F1000Prime Rep 2014; 6:23; PMID:24765528; http://dx.doi.org/ 10.12703/P6-23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ceccaldi R, Parmar K, Mouly E, Delord M, Kim JM, Regairaz M, Pla M, Vasquez N, Zhang QS, Pondarre C, et al. Bone marrow failure in fanconi anemia is triggered by an exacerbated p53/p21 DNA damage response that impairs hematopoietic stem and progenitor cells. Cell Stem Cell 2012; 11:36-49; PMID:22683204; http://dx.doi.org/ 10.1016/j.stem.2012.05.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pang Q, Andreassen PR. Fanconi anemia proteins and endogenous stresses. Mutat Res 2009; 668:42-53; PMID:19774700; http://dx.doi.org/ 10.1016/j.mrfmmm.2009.03.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhang J, Walter JC. Mechanism and regulation of incisions during DNA interstrand cross-link repair. DNA Repair 2014; 19:135-42; PMID:24768452; http://dx.doi.org/ 10.1016/j.dnarep.2014.03.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Huang J, Liu S, Bellani MA, Thazhathveetil AK, Ling C, de Winter JP, Wang Y, Wang W, Seidman MM. The DNA translocase FANCM/MHF promotes replication traverse of DNA interstrand crosslinks. Mol Cell 2013; 52:434-46; PMID:24207054; http://dx.doi.org/ 10.1016/j.molcel.2013.09.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kim H, D'Andrea AD. Regulation of DNA cross-link repair by the Fanconi anemia/BRCA pathway. Gene Dev 2012; 26:1393-408; PMID:22751496; http://dx.doi.org/ 10.1101/gad.195248.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Noda T, Takahashi A, Kondo N, Mori E, Okamoto N, Nakagawa Y, Ohnishi K, Zdzienicka MZ, Thompson LH, Helleday T, et al. Repair pathways independent of the Fanconi anemia nuclear core complex play a predominant role in mitigating formaldehyde-induced DNA damage. Biochem Biophys Res Commun 2011; 404:206-10; PMID:21111709; http://dx.doi.org/ 10.1016/j.bbrc.2010.11.094 [DOI] [PubMed] [Google Scholar]
- 9. Langevin F, Crossan GP, Rosado IV, Arends MJ, Patel KJ. Fancd2 counteracts the toxic effects of naturally produced aldehydes in mice. Nature 2011; 475:53-58; PMID:21734703; http://dx.doi.org/ 10.1038/nature10192 [DOI] [PubMed] [Google Scholar]
- 10. Rosado IV, Langevin F, Crossan GP, Takata M, Patel KJ. Formaldehyde catabolism is essential in cells deficient for the Fanconi anemia DNA-repair pathway. Nat Struct Mol Biol 2011; 18:1432-4; PMID:22081012; http://dx.doi.org/ 10.1038/nsmb.2173 [DOI] [PubMed] [Google Scholar]
- 11. Garaycoechea JI, Patel KJ. Why does the bone marrow fail in Fanconi anemia? Blood 2014; 123:26-34; PMID:24200684; http://dx.doi.org/ 10.1182/blood-2013-09-427740 [DOI] [PubMed] [Google Scholar]
- 12. Pace P, Mosedale G, Hodskinson MR, Rosado IV, Sivasubramaniam M, Patel KJ. Ku70 corrupts DNA repair in the absence of the Fanconi anemia pathway. Science 2010; 329:219-23; PMID:20538911; http://dx.doi.org/ 10.1126/science.1192277 [DOI] [PubMed] [Google Scholar]
- 13. Adamo A, Collis SJ, Adelman CA, Silva N, Horejsi Z, Ward JD, Martinez-Perez E, Boulton SJ, La Volpe A. Preventing nonhomologous end joining suppresses DNA repair defects of Fanconi anemia. Mol Cell 2010; 39:25-35; PMID:20598602; http://dx.doi.org/ 10.1016/j.molcel.2010.06.026 [DOI] [PubMed] [Google Scholar]
- 14. Cao L, Xu X, Bunting SF, Liu J, Wang RH, Cao LL, Wu JJ, Peng TN, Chen J, Nussenzweig A, et al. A selective requirement for 53BP1 in the biological response to genomic instability induced by Brca1 deficiency. Mol Cell 2009; 35:534-41; PMID:19716796; http://dx.doi.org/ 10.1016/j.molcel.2009.06.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bunting SF, Callén E, Wong N, Chen HT, Polato F, Gunn A, Bothmer A, Feldhahn N, Fernandez-Capetillo O, Cao L, et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell 2010; 141:243-54; PMID:20362325; http://dx.doi.org/ 10.1016/j.cell.2010.03.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bunting SF, Nussenzweig A. Dangerous liaisons: Fanconi anemia and toxic nonhomologous end joining in DNA crosslink repair. Mol Cell 2010; 39:164-66; PMID:20670885; http://dx.doi.org/ 10.1016/j.molcel.2010.07.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bouwman P, Aly A, Escandell JM, Pieterse M, Bartkova J, van der Gulden H, Hiddingh S, Thanasoula M, Kulkarni A, Yang Q, et al. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat Struct Mol Biol 2010; 17:688-95; PMID:20453858; http://dx.doi.org/ 10.1038/nsmb.1831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bunting SF, Callén E, Kozak ML, Kim JM, Wong N, López-Contreras AJ, Ludwig T, Baer R, Faryabi RB, Malhowski A, et al. BRCA1 functions independently of homologous recombination in DNA interstrand crosslink repair. Mol Cell 2012; 46:125-35; PMID:22445484; http://dx.doi.org/ 10.1016/j.molcel.2012.02.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Peng M, Xie J, Ucher A, Stavnezer J, Cantor SB. Crosstalk between BRCA-Fanconi anemia and mismatch repair pathways prevents MSH2-dependent aberrant DNA damage responses. EMBO J 2014; 33(15):1698-712; PMID:24966277; http://dx.doi.org/ 10.15252/embj.201387530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Houghtaling S, Newell A, Akkari Y, Taniguchi T, Olson S, Grompe M. Fancd2 functions in a double strand break repair pathway that is distinct from non-homologous end joining. Hum Mol Genet 2005; 14:3027-33; PMID:16135554; http://dx.doi.org/ 10.1093/hmg/ddi334 [DOI] [PubMed] [Google Scholar]
- 21. Karanja KK, Lee EH, Hendrickson EA, Campbell JL. Preventing over-resection by DNA2 helicase/nuclease suppresses repair defects in Fanconi anemia cells. Cell Cycle (Georgetown, Tex) 2014; 13:1540-50; PMID:24626199; http://dx.doi.org/ 10.4161/cc.28476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Schlacher K, Christ N, Siaud N, Egashira A, Wu H, Jasin M. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 2011; 145:529-42; PMID:21565612; http://dx.doi.org/ 10.1016/j.cell.2011.03.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Schlacher K, Wu H, Jasin M. A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell 2012; 22:106-16; PMID:22789542; http://dx.doi.org/ 10.1016/j.ccr.2012.05.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Brown ET, Holt JT. Rad51 overexpression rescues radiation resistance in BRCA2-defective cancer cells. Mol Carcinog 2009; 48:105-9; PMID:18618591; http://dx.doi.org/ 10.1002/mc.20463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lee SA, Roques C, Magwood AC, Masson J-Y, Baker MD. Recovery of deficient homologous recombination in Brca2-depleted mouse cells by wild-type Rad51 expression. DNA Repair 2009; 8:170-81; PMID:18992372; http://dx.doi.org/ 10.1016/j.dnarep.2008.10.002 [DOI] [PubMed] [Google Scholar]
- 26. Moldovan G-L, Dejsuphong D, Petalcorin MI, Hofmann K, Takeda S, Boulton SJ, D'Andrea AD, et al. Inhibition of homologous recombination by the PCNA-interacting protein PARI. Mol Cell 2012; 45:75-86; PMID:22153967; http://dx.doi.org/ 10.1016/j.molcel.2011.11.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Peng M, Litman R, Xie J, Sharma S, Brosh RM Jr, Cantor SB. The FANCJ/MutLalpha interaction is required for correction of the cross-link response in FA-J cells. EMBO J 2007; 26:3238-49; PMID:17581638; http://dx.doi.org/ 10.1038/sj.emboj.7601754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cantor SB, Xie J. Assessing the link between BACH1/FANCJ and MLH1 in DNA crosslink repair. Environ Mol Mutagen 2010; 51:500-07; PMID:20658644; http://dx.doi.org/ 10.1002/em.20568 [DOI] [PubMed] [Google Scholar]
- 29. Xie J, Guillemette S, Peng M, Gilbert C, Buermeyer A, Cantor SB. An MLH1 mutation links BACH1/FANCJ to colon cancer, signaling, and insight toward directed therapy. Cancer Prevention Res 2010; 3:1409-16; PMID:20978114; http://dx.doi.org/ 10.1158/1940-6207.CAPR-10-0118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Martinez P, Siegl-Cachedenier I, Flores JM, Blasco MA. MSH2 deficiency abolishes the anticancer and pro-aging activity of short telomeres. Aging Cell 2009; 8:2-17; PMID:18986375; http://dx.doi.org/ 10.1111/j.1474-9726.2008.00441.x [DOI] [PubMed] [Google Scholar]
- 31. Durant ST, Morris MM, Illand M, McKay HJ, McCormick C, Hirst GL, Borts RH, Brown R. Dependence on RAD52 and RAD1 for anticancer drug resistance mediated by inactivation of mismatch repair genes. Curr Biol 1999; 9 51-4; PMID:9889125; http://dx.doi.org/ 10.1016/S0960-9822(99)80047-5 [DOI] [PubMed] [Google Scholar]
- 32. Lin X, Trang J, Okuda T, Howell SB. DNA polymerase zeta accounts for the reduced cytotoxicity and enhanced mutagenicity of cisplatin in human colon carcinoma cells that have lost DNA mismatch repair. Clin Cancer Res 2006; 12:563-8; PMID:16428501; http://dx.doi.org/ 10.1158/1078-0432.CCR-05-1380 [DOI] [PubMed] [Google Scholar]
- 33. Wu F, Lin X, Okuda T, Howell SB. DNA polymerase zeta regulates cisplatin cytotoxicity, mutagenicity, and the rate of development of cisplatin resistance. Cancer Res 2004; 64:8029-35; PMID:15520212; http://dx.doi.org/ 10.1158/0008-5472.CAN-03-3942 [DOI] [PubMed] [Google Scholar]
- 34. Brown R, Hirst GL, Gallagher WM, McIlwrath AJ, Margison GP, van der Zee AG, Anthoney DA. hMLH1 expression and cellular responses of ovarian tumour cells to treatment with cytotoxic anticancer agents. Oncogene 1997; 15:45-52; PMID:9233776; http://dx.doi.org/ 10.1038/sj.onc.1201167 [DOI] [PubMed] [Google Scholar]
- 35. Moreland NJ, Illand M, Kim YT, Paul J, Brown R. Modulation of drug resistance mediated by loss of mismatch repair by the DNA polymerase inhibitor aphidicolin. Cancer Res 1999; 59:2102-6; PMID:10232595 [PubMed] [Google Scholar]
- 36. Jiricny J. The multifaceted mismatch-repair system. Nat Rev Mol Cell Biol 2006; 7:335-46; PMID:16612326; http://dx.doi.org/ 10.1038/nrm1907 [DOI] [PubMed] [Google Scholar]
- 37. Yamada M, O'Regan E, Brown R, Karran P. Selective recognition of a cisplatin-DNA adduct by human mismatch repair proteins. Nucleic Acids Res 1997; 25:491-6; PMID:9016586; http://dx.doi.org/ 10.1093/nar/25.3.491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Duckett DR, Drummond JT, Murchie AI, Reardon JT, Sancar A, Lilley DM, Modrich P. Human MutSalpha recognizes damaged DNA base pairs containing O6-methylguanine, O4-methylthymine, or the cisplatin-d(GpG) adduct. Proc Natl Acad Sci U S A 1996; 93:6443-7; PMID:8692834; http://dx.doi.org/ 10.1073/pnas.93.13.6443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhang N, Lu X, Zhang X, Peterson CA, Legerski RJ. hMutSbeta is required for the recognition and uncoupling of psoralen interstrand cross-links in vitro. Mol Cell Biol 2002; 22:2388-97; PMID:11884621; http://dx.doi.org/ 10.1128/MCB.22.7.2388-2397.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wu Q, Vasquez KM. Human MLH1 protein participates in genomic damage checkpoint signaling in response to DNA interstrand crosslinks, while MSH2 functions in DNA repair. PLoS Genet 2008; 4:e1000189; PMID:18787700; http://dx.doi.org/ 10.1371/journal.pgen.1000189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sommers JA, Banerjee T, Hinds T, Wan B, Wold MS, Lei M, Brosh RM Jr. Novel function of the Fanconi anemia group J or RECQ1 helicase to disrupt protein-DNA complexes in a replication protein A-stimulated manner. J Biol Chem 2014; 289:19928-41; PMID:24895130; http://dx.doi.org/ 10.1074/jbc.M113.542456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gupta R, Sharma S, Sommers JA, Kenny MK, Cantor SB, Brosh RM Jr. FANCJ (BACH1) helicase forms DNA damage inducible foci with replication protein A and interacts physically and functionally with the single-stranded DNA-binding protein. Blood 2007; 110:2390-8; PMID:17596542; http://dx.doi.org/ 10.1182/blood-2006-11-057273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell 2010; 40:179-204; PMID:20965415; http://dx.doi.org/ 10.1016/j.molcel.2010.09.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bachrati CZ, Hickson ID. RecQ helicases: guardian angels of the DNA replication fork. Chromosoma 2008; 117:219-33; PMID:18188578; http://dx.doi.org/ 10.1007/s00412-007-0142-4 [DOI] [PubMed] [Google Scholar]
- 45. Bansbach CE, Betous R, Lovejoy CA, Glick GG, Cortez D. The annealing helicase SMARCAL1 maintains genome integrity at stalled replication forks. Gene Dev 2009; 23:2405-14; PMID:19793861; http://dx.doi.org/ 10.1101/gad.1839909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ciccia A, Bredemeyer AL, Sowa ME, Terret ME, Jallepalli PV, Harper JW, Elledge SJ. The SIOD disorder protein SMARCAL1 is an RPA-interacting protein involved in replication fork restart. Gene Dev 2009; 23:2415-25; PMID:19793862; http://dx.doi.org/ 10.1101/gad.1832309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Huang M, Kim JM, Shiotani B, Yang K, Zou L, D'Andrea AD. The FANCM/FAAP24 complex is required for the DNA interstrand crosslink-induced checkpoint response. Mol Cell 2010; 39:259-68; PMID:20670894; http://dx.doi.org/ 10.1016/j.molcel.2010.07.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Postow L, Woo EM, Chait BT, Funabiki H. Identification of SMARCAL1 as a component of the DNA damage response. J Biol Chem 2009; 284:35951-61; PMID:19841479; http://dx.doi.org/ 10.1074/jbc.M109.048330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yuan J, Ghosal G, Chen J. The annealing helicase HARP protects stalled replication forks. Gene Dev 2009; 23:2394-9; PMID:19793864; http://dx.doi.org/ 10.1101/gad.1836409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Yusufzai T, Kong X, Yokomori K, Kadonaga JT. The annealing helicase HARP is recruited to DNA repair sites via an interaction with RPA. Gene Dev 2009; 23:2400-4; PMID:19793863; http://dx.doi.org/ 10.1101/gad.1831509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Williams SA, Wilson JB, Clark AP, Mitson-Salazar A, Tomashevski A, Ananth S, Glazer PM, Semmes OJ, Bale AE, Jones NJ, et al. Functional and physical interaction between the mismatch repair and FA-BRCA pathways. Hum Mol Genet 2011; 20:4395-410; PMID:21865299; http://dx.doi.org/ 10.1093/hmg/ddr366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Huang M, Kennedy R, Ali AM, Moreau LA, Meetei AR, D'Andrea AD, Chen CC. Human MutS and FANCM complexes function as redundant DNA damage sensors in the Fanconi Anemia pathway. DNA Repair (Amst) 2011; 10:1203-12; PMID:21975120; http://dx.doi.org/ 10.1016/j.dnarep.2011.09.006 [DOI] [PubMed] [Google Scholar]
- 53. Svendsen JM, Smogorzewska A, Sowa ME, O'Connell BC, Gygi SP, Elledge SJ, Harper JW. Mammalian BTBD12/SLX4 assembles a Holliday junction resolvase and is required for DNA repair. Cell 2009; 138:63-77; PMID:19596235; http://dx.doi.org/ 10.1016/j.cell.2009.06.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wang Y, Cortez D, Yazdi P, Neff N, Elledge SJ, Qin J. BASC, a super complex of BRCA1-associated proteins involved in the recognition and repair of aberrant DNA structures. Genes Dev 2000; 14:927-39; PMID:10783165 [PMC free article] [PubMed] [Google Scholar]
- 55. Kratz K, Schöpf B, Kaden S, Sendoel A, Eberhard R, Lademann C, Cannavó E, Sartori AA, Hengartner MO, Jiricny J. Deficiency of FANCD2-associated nuclease KIAA1018/FAN1 sensitizes cells to interstrand crosslinking agents. Cell 2010; 142:77-88; PMID:20603016; http://dx.doi.org/ 10.1016/j.cell.2010.06.022 [DOI] [PubMed] [Google Scholar]
- 56. Liu T, Ghosal G, Yuan J, Chen J, Huang J. FAN1 acts with FANCI-FANCD2 to promote DNA interstrand cross-link repair. Science 2010; 329:693-6; PMID:20671156; http://dx.doi.org/ 10.1126/science.1192656 [DOI] [PubMed] [Google Scholar]
- 57. O'Donnell L, Durocher D. DNA repair has a new FAN1 club. Mol Cell 2010; 39:167-9; PMID:20670886; http://dx.doi.org/ 10.1016/j.molcel.2010.07.010 [DOI] [PubMed] [Google Scholar]
- 58. Shereda RD, Machida Y, Machida YJ. Human KIAA1018/FAN1 localizes to stalled replication forks via its ubiquitin-binding domain. Cell Cycle 2010; 9:3977-83; PMID:20935496; http://dx.doi.org/ 10.4161/cc.9.19.13207 [DOI] [PubMed] [Google Scholar]
- 59. Smogorzewska A, Desetty R, Saito TT, Schlabach M, Lach FP, Sowa ME, Clark AB, Kunkel TA, Harper JW, Colaiácovo MP, et al. A genetic screen identifies FAN1, a Fanconi anemia-associated nuclease necessary for DNA interstrand crosslink repair. Mol Cell 2010; 39:36-47; PMID:20603073; http://dx.doi.org/ 10.1016/j.molcel.2010.06.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Yoshikiyo K, Kratz K, Hirota K, Nishihara K, Takata M, Kurumizaka H, Horimoto S, Takeda S, Jiricny J. KIAA1018/FAN1 nuclease protects cells against genomic instability induced by interstrand cross-linking agents. Proc Natl Acad Sci U S A 2010; 107:21553-7; PMID:21115814; http://dx.doi.org/ 10.1073/pnas.1011081107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ward TA, Dudášová Z, Sarkar S, Bhide MR, Vlasáková D, Chovanec M, McHugh PJ. Components of a Fanconi-like pathway control Pso2-independent DNA interstrand crosslink repair in yeast. PLoS Genet 2012; 8:e1002884; PMID:22912599; http://dx.doi.org/ 10.1371/journal.pgen.1002884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Suhasini AN, Sommers JA, Muniandy PA, Coulombe Y, Cantor SB, Masson JY, Seidman MM, Brosh RM Jr. Fanconi anemia group J helicase and MRE11 nuclease interact to facilitate the DNA damage response. Mol Cell Biol 2013; 33:2212-27; PMID:23530059; http://dx.doi.org/ 10.1128/MCB.01256-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Guillemette S, Branagan A, Peng M, Dhruva A, Schärer OD, Cantor SB. FANCJ localization by mismatch repair is vital to maintain genomic integrity after UV irradiation. Cancer Res 2014; 74:932-44; PMID:24351291; http://dx.doi.org/ 10.1158/0008-5472.CAN-13-2474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Schindler D, Hoehn H. Fanconi anemia mutation causes cellular susceptibility to ambient oxygen. Am J Hum Genet 1988; 43:429-35; PMID:3177386 [PMC free article] [PubMed] [Google Scholar]
- 65. Zhang Q-S, Eaton L, Snyder ER, Houghtaling S, Mitchell JB, Finegold M, Van Waes C, Grompe M. Tempol protects against oxidative damage and delays epithelial tumor onset in Fanconi anemia mice. Cancer Res 2008; 68:1601-8; PMID:18316625; http://dx.doi.org/ 10.1158/0008-5472.CAN-07-5186 [DOI] [PubMed] [Google Scholar]
- 66. Chen C-H, Ferreira JCB, Gross ER, Mochly-Rosen D. Targeting aldehyde dehydrogenase 2: new therapeutic opportunities. Physiol Rev 2014; 94:1-34; PMID:24382882; http://dx.doi.org/ 10.1152/physrev.00017.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Reitmair AH, et al. MSH2 deficient mice are viable and susceptible to lymphoid tumours. Nat Genet 1995; 11:64-70; PMID:7550317; http://dx.doi.org/ 10.1038/ng0995-64 [DOI] [PubMed] [Google Scholar]
- 68. Campbell MR, Nation PN, Andrew SE. A lack of DNA mismatch repair on an athymic murine background predisposes to hematologic malignancy. Cancer Res 2005; 65:2626-35; PMID:15805259; http://dx.doi.org/ 10.1158/0008-5472.CAN-04-3158 [DOI] [PubMed] [Google Scholar]
- 69. Mao G, Yuan F, Absher K, Jennings CD, Howard DS, Jordan CT, Gu L. Preferential loss of mismatch repair function in refractory and relapsed acute myeloid leukemia: potential contribution to AML progression. Cell Res 2008; 18:281-9, PMID:18227862; http://dx.doi.org/ 10.1038/cr.2008.14 [DOI] [PubMed] [Google Scholar]
- 70. Casorelli I, Offman J, Mele L, Pagano L, Sica S, D'Errico M, Giannini G, Leone G, Bignami M, Karran P. Drug treatment in the development of mismatch repair defective acute leukemia and myelodysplastic syndrome. DNA Repair 2003; 2:547-59; PMID:12713812; http://dx.doi.org/ 10.1016/S1568-7864(03)00020-X [DOI] [PubMed] [Google Scholar]
- 71. Offman J, Opelz G, Doehler B, Cummins D, Halil O, Banner NR, Burke MM, Sullivan D, Macpherson P, Karran P. Defective DNA mismatch repair in acute myeloid leukemia/myelodysplastic syndrome after organ transplantation. Blood 2004; 104:822-8; PMID:15090454; http://dx.doi.org/ 10.1182/blood-2003-11-3938 [DOI] [PubMed] [Google Scholar]
- 72. Karran P. Mechanisms of tolerance to DNA damaging therapeutic drugs. Carcinogenesis 2001; 22:1931-7; PMID:11751422; http://dx.doi.org/ 10.1093/carcin/22.12.1931 [DOI] [PubMed] [Google Scholar]
- 73. Walsh T, King MC. Ten genes for inherited breast cancer. Cancer Cell 2007; 11:103-05; PMID:17292821; http://dx.doi.org/ 10.1016/j.ccr.2007.01.010 [DOI] [PubMed] [Google Scholar]
- 74. Martin SA, Lord CJ, Ashworth A. Therapeutic targeting of the DNA mismatch repair pathway. Clin Cancer Res 2010; 16:5107-13; PMID:20823149; http://dx.doi.org/ 10.1158/1078-0432.CCR-10-0821 [DOI] [PubMed] [Google Scholar]
- 75. Martin SA, McCarthy A, Barber LJ, Burgess DJ, Parry S, Lord CJ, Ashworth A. Methotrexate induces oxidative DNA damage and is selectively lethal to tumour cells with defects in the DNA mismatch repair gene MSH2. EMBO Mol Med 2009; 1:323-37; PMID:20049736; http://dx.doi.org/ 10.1002/emmm.200900040 [DOI] [PMC free article] [PubMed] [Google Scholar]