

Mitogen activated protein kinase (MAPK) signaling pathways convert extracellular signals into specific cellular responses through a cascade of phosphorylation events. Hence, tight regulation of these signaling pathways is essential for normal cell function and deregulation leads to various diseases including tumorigenesis. Three major MAPK families have been found deregulated in cancer: ERK1 and ERK2 (ERK1/2), p38 MAPK and the Jun N-terminal kinase (JNK). In non-transformed cells, ERK1/2 are involved mainly in the mitogenesis response whereas p38 MAPK and JNK mediate cellular stress and inflammatory responses.
The early steps of colorectal cancer (CRC) are driven, among other events, by mutations in KRAS and BRAF that lead to constitutive activation of the ERK1/2 signaling cascade. In addition to controlling cell proliferation, this pathway is also involved in processes that are essential for metastasis, such as epithelial to mesenchymal transition (EMT), migration or invasion. Recent data show that activation of ERK2 but not ERK1 mediates liver metastasis in a xenograft mouse model of CRC.1 ERK1 and ERK2 are serine/threonine protein kinases that share 84% of sequence homology and are thought to have many overlapping functions, however recent evidence points out to a specific role of ERK2 as a mediator of EMT both in breast and colon tumors.2 In this line, expression of the transcription factor FRA1, an ERK2 target that can regulate EMT, has been associated with recurrence in CRC patients.1 Additionally, increased phosphorylation levels of ERK2 over ERK1 have been detected in primary human colon tumors associated with metastasis development.1
In spite of the evidence supporting a role for activated ERK2 in metastasis, the mechanisms that lead to deregulated ERK1/2 activity levels are still poorly defined. An attractive possibility is that dual-specificity protein phosphatases (DUSP) that specifically inactivate ERK2 could be implicated, but a link between decreased expression of specific phosphatases in primary tumors with increased phospho-ERK2 levels and tumor relapse has to be provided. Recently, inactivation of one of these phosphatases (DUSP6/MKP-3) was shown to correlate with ERK2-dependent EMT and contribute to colon cancer invasion. Additionally, DUSP6 downregulation was detected in invasive pancreatic carcinomas and decreased DUSP6 levels correlate with increased tumor grade in non-small-cell lung carcinoma (NSCLC).3,4 Of note, mutations in KRAS are detected in 90% of pancreatic adenocarcinoma and in 40% of NSCLC. Altogether, these data suggest that besides the RAS/RAF-induced hyperactivation of ERK1/2 signaling that occurs at the initial steps of CRC, subsequent changes modulating the balance between ERK1 and ERK2 activities may play a role in invasion and metastases. This mechanism might also apply to tumors from different origins than those driven by mutations in RAS or RAF genes.
p38 MAPK signaling can also regulate CRC at different levels.1,5 For example, downregulation of p38α, an ubiquitously expressed p38 MAPK family member, in intestinal epithelial cells accelerates colitis-associated colon tumor formation whereas p38α inhibition in established colon tumors decreases tumor burden.5 Interestingly, p38 MAPK signaling has also been implicated in CRC metastasis. In particular, the lung metastatic colonization from established liver metastasis requires decreased p38 MAPK activity, which leads to increased expression of parathyroid hormone-like hormone (PTHLH) by the CRC cells in the liver. PTHLH in turn induces endothelial cell death in the lung vasculature facilitating tumor cell extravasation and lung colonization.1 The reduced p38 MAPK activity in liver metastasis correlates with the downregulation of MKK6, a specific direct activator of p38 MAPK. Indeed, low expression of MKK6 in colon primary tumors is associated with worse prognosis underlying the importance of p38 MAPK regulation in late steps of CRC. Moreover, downregulation of MKK6 (as well as MKK4, an activator of both JNK and p38α) is also associated with ovarian cancer metastasis.6 However, it is unclear how MKK6 levels are reduced to suppress p38 MAPK activity in tumor cells and support late events of dissemination and metastasis. In fact, very little is known on the regulation of MKK6 expression, except for a negatively feed-back loop triggered by p38α that leads to MKK6 mRNA downregulation.7
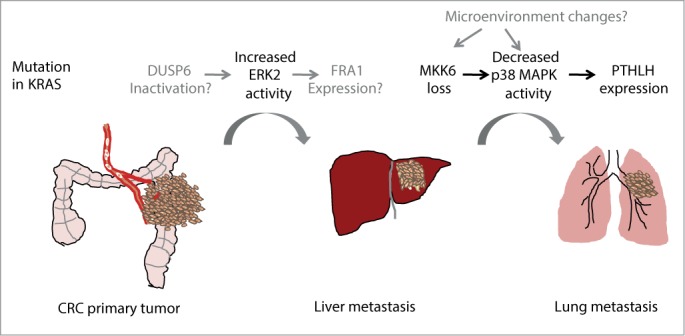
Given the importance of the p38 MAPK pathway in the response to many extracellular signals including chemokines, cytokines, hypoxia or osmotic shock, it is likely that changes in the tumor microenvironment modulate p38 MAPK activity in tumor cells. For example, changes in fibronectin signaling can trigger, via inactivation of the small GTP-ase Cdc42, the inactivation of p38 MAPK, which together with ERK1/2 activation lead to exit from metastatic cell dormancy in human head and neck carcinoma.6 Alternatively, proteins that can directly inactivate p38 MAPK and whose activity is potentially modulated during tumor progression may also be implicated in this process, for example the protein phosphatase type 2C (PP2C) family members such as WIP1.
ERK2 activation and p38 MAPK downregulation are both detected in primary tumors and contribute to CRC metastasis to the liver and the lung, respectively.1 Given the co-occurrence of both events in primary tumors, it is tantalizing to speculate that they are co-selected by means of a common regulator that interconnects the 2 pathways. Elucidation of the hierarchical regulatory mechanisms involved may identify an overarching controller central to CRC metastasis. Additionally, further studies on the dynamics of MAPK signaling during tumor formation and metastasis and the characterization of how different signaling pathways become integrated should help to understand and eventually tackle the metastatic process.
References
- 1. Urosevic J, et al. Nat Cell Biol 2014; 16:685-94; PMID:24880666; http://dx.doi.org/ 10.1038/ncb2977 [DOI] [PubMed] [Google Scholar]
- 2. Shin S, et al. . Mol Cell 2010; 38:114-27; PMID:20385094; http://dx.doi.org/ 10.1016/j.molcel.2010.02.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Okudela K, et al. . Am J Pathol 2009; 175:867-81; PMID:19608870; http://dx.doi.org/ 10.2353/ajpath.2009.080489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Furukawa T, et al. . Mod Pathol 2005; 18:1034-42; PMID:15832194; http://dx.doi.org/ 10.1038/modpathol.3800383 [DOI] [PubMed] [Google Scholar]
- 5. Gupta J, et al. . Cancer Cell 2014; 25:484-500; PMID:24684847; http://dx.doi.org/ 10.1016/j.ccr.2014.02.019 [DOI] [PubMed] [Google Scholar]
- 6. Aguirre-Ghiso JA. Nat Rev Cancer 2007; 7:834-46; PMID:17957189; http://dx.doi.org/ 10.1038/nrc2256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ambrosino C, et al. . Mol cell biol 2003; 23:370-81; PMID:12482988 [DOI] [PMC free article] [PubMed] [Google Scholar]