

Abstract
Mutations in genes encoding regulators of mTOR, the mammalian target of rapamycin, commonly provide survival signals in cancer cells. Rapamycin and analogs of rapamycin have been used with limited success in clinical trials to target mTOR-dependent survival signals in a variety of human cancers. Suppression of mTOR predominantly causes G1 cell cycle arrest, which likely contributes to the ineffectiveness of rapamycin-based therapeutic strategies. While rapamycin causes the accumulation of cells in G1, its effect in other cell cycle phases remains largely unexplored. We report here that when synchronized MDA-MB-231 breast cancer cells are allowed to progress into S-phase from G1, rapamycin activates the apoptotic machinery with a concomitant increase in cell death. In Calu-1 lung cancer cells, rapamycin induced a feedback increase in Akt phosphorylation at Ser473 in S-phase that mitigated rapamycin-induced apoptosis. However, sensitivity to rapamycin in S-phase could be reestablished if Akt phosphorylation was suppressed. We recently reported that glutamine (Gln) deprivation causes K-Ras mutant cancer cells to aberrantly arrest primarily in S-phase. Consistent with observed sensitivity of S-phase cells to rapamycin, interfering with Gln utilization sensitized both MDA-MB-231 and Calu-1 K-Ras mutant cancer cells to the apoptotic effect of rapamycin. Importantly, rapamycin induced substantially higher levels of cell death upon Gln depletion than that observed in cancer cells that were allowed to progress through S-phase after being synchronized in G1. We postulate that exploiting metabolic vulnerabilities in cancer cells such as S-phase arrest observed with K-Ras-driven cancer cells deprived of Gln, could be of great therapeutic potential.
Keywords: cell cycle, glutamine, mTOR, rapamycin, synthetic lethality
Abbreviations
- 4E-BP1
eIF4E binding protein-1
- eIF4E
eukaryotic initiation factor 4E
- Gln
glutamine
- GOT
glutamate-oxaloacetate-transaminase
- mTOR
mammalian target of rapamycin
- mTORC1/2
mTOR complex 1/2
- PARP
poly-ADP-ribose polymerase
- PI3K
phosphatidylinositol-3-kinase
- S6K
S6 kinase
- TGF-β
transforming growth factor-β.
Introduction
mTOR – the mammalian target of rapamycin, plays a key role in the control of cell proliferation. mTOR is responsive to the presence of both growth factors that instruct a cell to divide; and nutrients that instruct as to whether there is sufficient raw material for the cell to double its mass and divide. Hence, it has been proposed that mTOR integrates growth factor and nutrient cues to control cell cycle progression and proliferation.1,2 Given this central role in cell proliferation, it is of no surprise that mTOR is active in what may be most human cancers.3,4 In addition to its role as an integrator of growth factor and nutrient signals, mTOR suppresses apoptotic programs that represent what is likely the first line of defense against cancer; and thusly, mTOR signals have commonly been referred to as cancer cell survival signals.1,5 The role that mTOR plays in promoting cancer cell survival has generated strong interest in targeting mTOR in order to reverse the survival effect of mTOR and induce apoptosis. There have been a large number of clinical trials involving rapamycin or more commonly rapamycin analogs (rapalogs). Although there has been some modest improvement with renal cancers, the impact of rapalogs has been largely disappointing.6
We reported previously that rapamycin at high doses induces apoptosis in several human cancer cell lines in the absence, but not in the presence of serum.7 The factor in serum that protected the cells from the apoptotic effect of rapamycin was TGF-β. Rapamycin treatment elevated TGF-β signals leading to G1 arrest, which in combination with suppression of Rb phosphorylation prevented apoptosis.7,8 Cancer cells with defective TGF-β signals could be killed by rapamycin in the presence of serum/TGF-β.7,9 This led us to speculate that if cells progressed past a late G1 mTOR-dependent checkpoint,10 then once in S-phase, the suppression of mTOR signals would trigger default apoptotic programs.11
An under-appreciated aspect of rapamycin treatment is the different doses needed to suppress the phosphorylation of different substrates of mTOR. mTOR exists in 2 complexes – mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2). mTORC2 is largely resistant to rapamycin, however prolonged rapamycin has been shown to suppress mTORC2 in some cancer cell lines.12 mTORC2 can also be suppressed with short-term treatment by suppressing the level of the co-activator phosphatidic acid,13 which binds mTOR in a manner that is competitive with rapamycin.14,15 The effect of rapamycin on mTORC1 substrates varies in that there are substantial differences in the doses needed for different substrates. Phosphorylation of ribosomal subunit S6 kinase (S6K) is suppressed in the nano-molar range; whereas, suppression of phosphorylation of eukaryotic initiation factor 4E (eIF4E)-binding protein 1 (4E-BP1) requires micro-molar doses.16,17 This is an important point in that micro-molar doses are required to induce apoptosis.7,16 When 4E-BP1 is phosphorylated, it dissociates from eIF4E and eIF4E can then initiate cap-dependent translation.18 The key factor for rapamycin-induced apoptosis in MDA-MB-231 breast cancer cells is eIF4E. Knockdown of 4E-BP1, which liberates eIF4E prevented the apoptotic effect of rapamycin,16 and knockdown of eIF4E was sufficient to induce apoptosis in MDA-MB-231 cells.17
In this report, we have investigated the apparent cell cycle specificity for the apoptotic effects of rapamycin. We have determined that the apoptotic programs activated by rapamycin occur specifically after cells have passed through G1 into S-phase. The apoptotic effect of rapamycin was substantially enhanced if the cancer cells were arrested in S-phase. This could be especially important for K-Ras driven cancer cells because they bypassed a glutamine (Gln)-dependent G1 cell cycle checkpoint and arrested in S-phase when Gln utilization was suppressed. Thus, K-Ras-driven cancers may be susceptible to strategies that involve interfering with Gln utilization in combination with suppression of mTOR.
Results
G1 cell cycle progression into S-phase for MDA-MB-231 and Calu-1 cells
We previously reported that in the absence of TGF-β, many human cancer lines are killed by rapamycin.7,9,16,17 In the presence of TGF-β, rather than killing the cancer cells, rapamycin treatment had a cytostatic effect and these cells arrested in late G1.7,8,10 This led us to propose that rapamycin preferentially killed cancer cells that passed through G1 and entered S-phase.11 We reported recently that Ras-driven cancer cells are more sensitive to treatments that target cells that have entered S-phase.19 We chose to investigate 2 K-Ras-driven cancer cells – MDA-MB-231 breast and Calu-1 lung cancer cells, which we have used previously to address the vulnerability of Ras-driven cancer cells that had entered S-phase.19,20 We wanted to establish a G1 or G0 cell cycle arrest that was upstream from the late G1 site where rapamycin arrests cells.10 Using serum withdrawal to arrest cancer cells in G0 is problematic because most cancer cells have acquired a mutation – such as Ras, which permits passage through the growth factor-dependent restriction point.21 We therefore arrested the cancer cells with lovastatin, which causes G1 cell cycle arrest in both normal and cancer cells.22,23 Lovastatin arrests cells relatively early in G1 and the arrest can be overcome with mevalonic acid. MDA-MB-231 and Calu-1 cells were treated lovastatin for 36 or 48 hr respectively. The G1 arrested cells were then released from cell cycle arrest by placing in fresh medium containing mevalonic acid. Mevalonic acid is the product of β-hydroxy-β-methylglutaryl-CoA (HMG-CoA) reductase – the target of lovastatin and the rate-limiting step in cholesterol biosynthesis.24 As shown in Figure 1A, treatment of asynchronous MDA-MB-231 and Calu-1 cells with lovastatin resulted in a shift to a predominantly G1 population of cells. Upon changing to fresh medium containing mevalonic acid to release from the lovastatin-induced G1 arrest, there was a shift to a predominantly S-phase population of cells between 22 and 24 hr (Fig. 1A). We also examined the level of cyclin A, which is elevated as cells enter S-phase25 during the transition from lovastatin arrest in G1 to S-phase. As shown in Figure 1B, there was an increase in cyclin A levels that correlated with an increased population of S-phase cells. Thus, lovastatin induces G1 arrest in both MDA-MB-231 and Calu-1 cells, and upon restoring fresh medium containing mevalonic acid, the cells proceed to S-phase. Given that the time course was longer than the time required for progressing from mitosis to S, there is clearly a recovery period needed to reprogram progression into S-phase.
Figure 1.
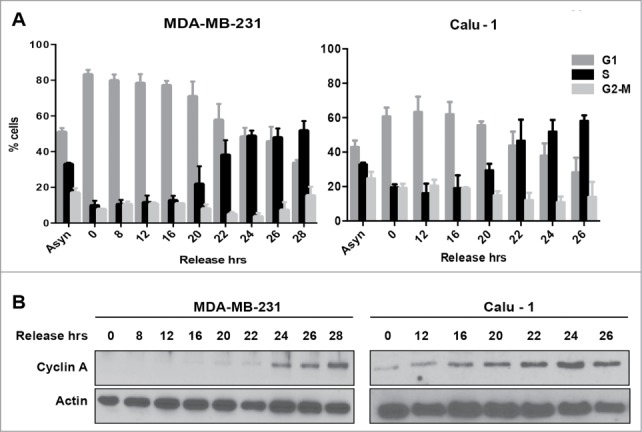
Cell cycle progression from G1 into S-phase for MDA-MB-231 and Calu-1 cells. (A) MDA-MB-231 breast cancer cells and Calu-1 lung cancer cells were plated at 30% confluence in medium containing 10% serum. After 24 hours, cells were synchronized using lovastatin as described in Materials and Methods. Upon release from G1 block, cells were collected at indicated time points and analyzed for cell cycle distribution by measuring DNA content using flow cytometry. Error bars represent the standard deviation for experiments repeated at least 3 times. (B) Western blot analysis performed to determine the levels of cyclin A, and actin. These data shown are representative of experiments repeated at least 3 times.
mTOR inhibition by rapamycin enhances apoptosis in S phase of the cell cycle in MDA-MB-231 cells but not in Calu-1 cells
Having established a time course for progression from lovastatin-induced G1 arrest to S-phase, we next investigated the ability of rapamycin to induce apoptosis in G1 relative to S-phase. We previously reported that high (20 μM) doses of rapamycin that suppress phosphorylation of 4E-BP1 were required for rapamycin-induced apoptosis.16,17 MDA-MB-231 (Fig. 2A) and Calu-1 (Fig. 2B) cells were arrested in G1 with lovastatin as in Figure 1 and then released with fresh medium containing mevalonic acid. Upon release from lovastatin block, rapamycin was added at the times indicated. Twenty-four hr after addition of rapamycin, cells were collected and the levels of cleaved PARP were evaluated. At 22 hr after release when the cells were entering S-phase, there was an increase in PARP cleavage with rapamycin in MDA-MB-231 cells (Fig. 2A), but not in Calu-1 cells (Fig. 2B). Cell viability correlated with the levels of PARP cleavage induced by rapamycin for both the MDA-MB-231 and Calu-1 cells (Fig. 2C). The Calu-1 cells survived even though phosphorylation of 4E-BP1 was inhibited by rapamycin (Fig. 2B). Thus, rapamycin, at doses that suppress 4E-BP1 phosphorylation, induced PARP cleavage in MDA-MB-231 cells when they entered S-phase, whereas Calu-1 cells were resistant to the apoptotic effects of rapamycin.
Figure 2.
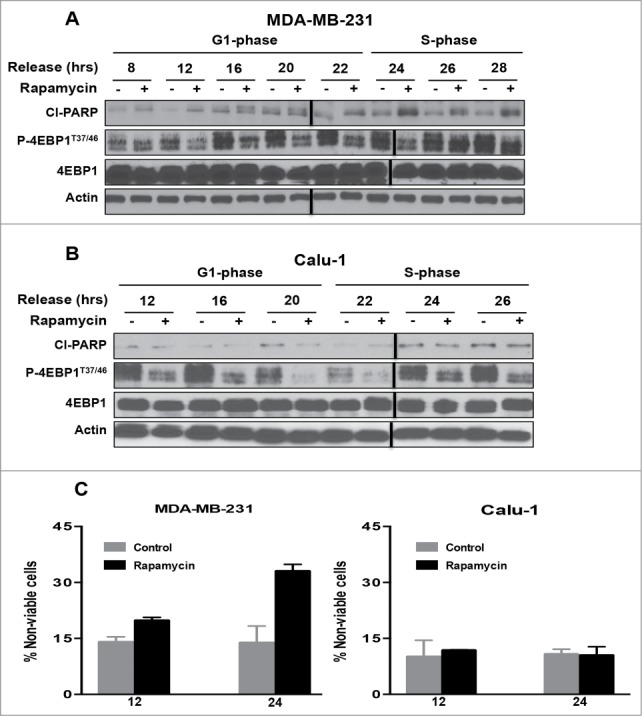
mTOR inhibition by rapamycin enhances apoptosis in S-phase of the cell cycle in MDA-MB-231 cells but not in Calu-1 cells. MDA-MB-231 (A) and Calu-1 (B) cells were synchronized in G1 phase of the cell cycle using lovastatin as in Figure 1. Upon release from G1 block, rapamycin (20μM) was added at indicated time points. After 24 hours, cells were collected and Western blot analysis was performed for cleaved PARP (Cl−PARP), P-4E-BP1T37/46, 4E-BP1, and actin. These data shown are representative of experiments repeated at least 3 times. (C) MDA-MB-231 and Calu-1 cells were synchronized using lovastatin as in A and B. Upon release from G1 block, the cells were treated with rapamycin at 12 and 24 hr. Cells were collected 24 hr later and cell viability assays were performed using trypan blue exclusion as described in Materials and Methods. Error bars represent the standard deviation for experiment at least repeated 3 times.
Combined inhibition of mTORC1 and Akt phosphorylation induces PARP cleavage in Calu-1 cells
The data in Figure 2 reveal that while rapamycin induced an apoptotic program in MDA-MB-231 cells in S-phase, Calu-1 cells were resistant to this treatment. We reported previously that elevated Akt phosphorylation at Ser473 can suppress rapamycin-induced apoptosis in pancreatic cancer cells9. Previous reports have indicated that inhibition of mTORC1 with rapamycin elevates Akt phosphorylation in some cancer cells including Calu-1.26,27 Since activation of Akt is well known to confer survival in cancer cells,28,29 we investigated the effect of suppressing Akt phosphorylation at Ser473, along with mTORC1 inhibition in Calu-1 cells. To determine whether elevated Akt phosphorylation was responsible for the lack of apoptosis seen in S-phase Calu-1 cells treated with rapamycin, we investigated the effect of rapamycin in combination with the phosphatidylinositol-3-kinase (PI3K) inhibitor LY294002, which suppresses AKt phosphorylation.9 Calu-1 cells were arrested in G1 with lovastatin and then released with fresh medium containing mevalonic acid as in Figure 1. As shown in Figures. 3A and B, rapamycin treatment led to elevated levels of Akt phosphorylation at Ser473 at both 12 and 24 hr post release from lovastatin block. There was a more dramatic increase seen 24 hr post release with rapamycin treatment, which is consistent with 2 recent reports indicating that phosphorylation Akt at Ser473 is more prominent in S-phase.30,31 LY294002 suppressed the rapamycin-induced increase in Akt phosphorylation in both G1 and S-phase (Fig. 3A and B). Along with the suppression of Akt phosphorylation, there was a corresponding increase in PARP cleavage (Fig. 3B) and cell death (Fig. 3C) in the S-phase Calu-1 cells. The elevated PARP cleavage and cell death seen with LY294002 alone (Figs. 3B and C) is likely because PI3K and mTOR are related kinases and LY294002 partially inhibits mTOR as well as PI3K.32 These data demonstrate that the feedback activation of Akt via suppression of mTORC126,27 inhibits the apoptotic machinery activated by rapamycin in Calu-1 cells. However, suppression of Akt phosphorylation restores the sensitivity of S-phase cells to rapamycin.
Figure 3.
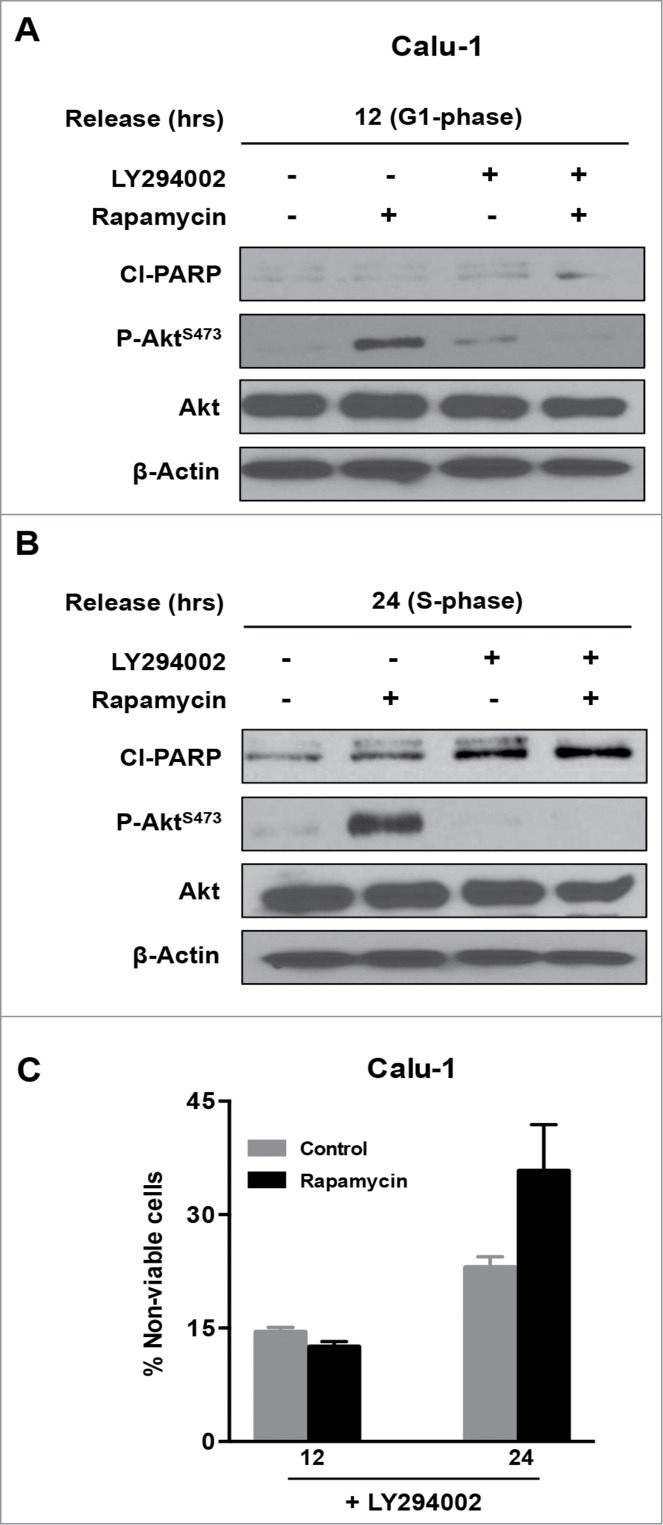
Combined inhibition of mTORC1 and Akt phosphorylation induces PARP cleavage in Calu-1 cells. Calu-1 cells were synchronized using lovastatin as in Figure 1. Upon release from G1 block, rapamycin and LY294002 (20 μM each) were added at 12 hours (G1 phase) (A) and at 24 hours (S phase) (B). Twenty-four hours later, cells were collected and Western blot analysis was performed for cleaved PARP (Cl−PARP), P-AktS-473, Akt and actin. The data shown are representative of experiments repeated at least 2 times. (C) Calu-1 cells were synchronized using Lovastatin as above. Upon release from G1 block, the cells were treated with rapamycin and LY294002 at indicated times for 24 hours. Cells were then collected and cell viability assays were performed as in Figure 2C. Error bars represent the standard deviation for experiment at least repeated 2 times.
Arresting K-Ras-driven cancer cells in S-phase by suppressing Gln utilization enhances rapamycin-induced apoptosis
Treating MDA-MB-231 cells with rapamycin and Calu-1 cells with rapamycin plus LY294002 when these cells were progressing through S-phase resulted in elevated levels of cleaved PARP and an increase in the percentage of non-viable cells (Figs. 2 and 3). However, the level of PARP cleavage and cell death was not as robust as was observed if cells were deprived of serum or if they were arrested in S-phase with aphidicolin – an inhibitor of DNA synthesis.7 We recently reported that K-Ras-driven cancer cells bypass a late G1 Gln-dependent checkpoint10 and instead arrested in S-phase.19 We therefore examined whether rapamycin, which apparently induces apoptosis specifically in S-phase cells, induces apoptosis in K-Ras-driven cancer cells where Gln utilization is suppressed. Gln is an important nutrient and both a nitrogen and carbon source, and although it can be synthesized in mammalian cells, it is considered a “conditionally essential” amino acid because it is required by dividing cells.33 We used 2 means to suppress Gln utilization: 1) using medium lacking Gln; and 2) blocking anaplerotic utilization of Gln with aminooxyacetate (AOA), which interferes with the transamination reaction whereby glutamate is deaminated to α-ketoglutarate while oxaloacetate is aminated to aspartic acid by the enzyme glutamate-oxaloacetate-transaminase (GOT).34 Whereas MCF7 breast cancer cells arrest in G1 in response to Gln deprivation, the MDA-MB-231 and Calu-1 K-Ras-driven cancer cells arrested largely in S-phase (Fig. 4A). AOA treatment arrested the MDA-MB-231 and Calu-1 cells in either S-phase or G2/M (Fig. 4A). We next examined the effect of rapamycin on MCF7 and MDA-MB-231 cells that had been subjected to Gln deprivation or AOA treatment. As shown in Figure 4B, treatment of MCF7 cells, which were largely in G1 with rapamycin under all conditions, did not result in significant cell death although there was some PARP cleavage upon rapamycin treatment by itself – a phenomenon we have observed previously.35 This presumably reflects a subpopulation of S-phase cells in that both Gln deprivation and AOA, which arrest cells in G1, prevented the rapamycin-induced PARP cleavage. However, in the MDA-MB-231 cells, rapamycin induced PARP cleavage in cells that had either been deprived of Gln, or where Gln utilization via GOT was blocked by AOA (Fig. 4B). For the Calu-1 cells, we employed LY294002 along with rapamycin. What was observed was somewhat unexpected in that both Gln deprivation and AOA elevated Akt phosphorylation at Ser473 and surprisingly rapamycin no longer stimulated Akt phosphorylation and, in fact, suppressed the phosphorylation stimulated by Gln deprivation and AOA (Fig. 4C). Importantly, rapamycin stimulated PARP cleavage and loss of cell viability in the absence of LY294002 (Fig. 4C). The PARP cleavage and the loss of cell viability were substantially stronger in the cells that were arrested in S-phase (Fig. 4) than those that were passing through S-phase (Figs. 2 and 3). The data in Figure 4 demonstrate that K-Ras-driven cancer cells that are arrested in S-phase and perhaps G2/M by interfering with Gln utilization are highly sensitive to the apoptotic effects of high dose rapamycin.
Figure 4.
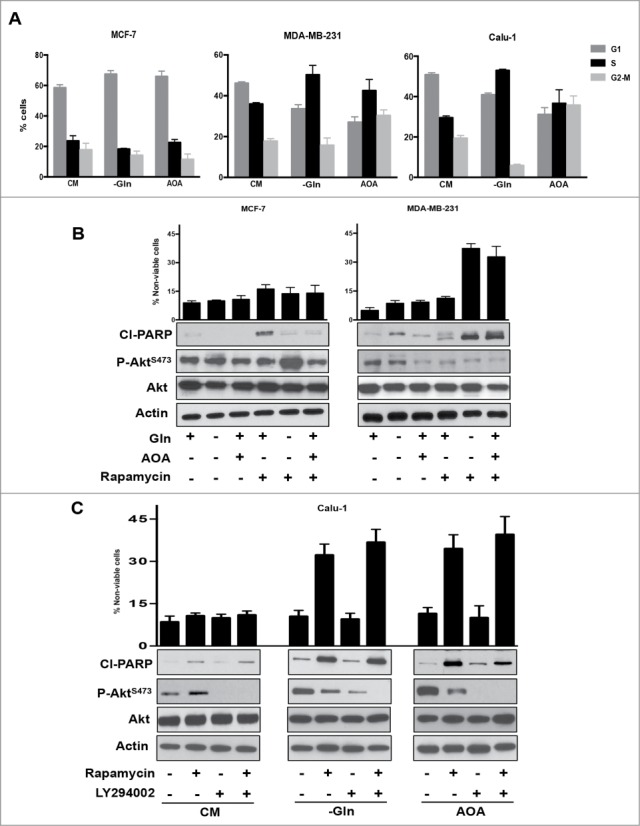
Glutamine starvation causes S phase arrest in K-Ras mutant cell lines and sensitizes them to rapamycin. (A) MCF7, MDA-MB-231, and Calu-1 cells were plated at 30% confluence. After 24 hours, cells were shifted to medium lacking Gln or complete medium containing AOA (0.5 mM) for 48 hours. Cells were collected and analyzed for cell cycle distribution by measuring DNA content using FACS analysis. Error bars represent the standard deviation for experiments repeated at least repeated 3 times. (B) MCF-7 and MDA-MB-231 cells were arrested in S phase as described in A. After 48 hours, cells were additionally treated with Rapamycin for 24 hours. Cells were collected and Western blot analysis was performed for cleaved PARP, P-Akt S473 phosphorylation and actin. Cell viability was determined as in Figure 2C. Error bars represent the standard deviation for experiments repeated 3 times. (C) Calu-1 cells were arrested in S phase as in A. After 48 hours, cells were treated with Rapamycin and LY294002 for 24 hours where indicated. Cells were then collected and Western blot analysis and cell viability assays were performed as in B. Error bars represent the standard deviation for experiments repeated 3 times.
Discussion
In this study, we have provided evidence that the apoptotic effects of rapamycin occur after transition from G1 into S-phase of the cell cycle. Prior to this transition, rapamycin causes a TGF-β-dependent cell cycle arrest at a site late in G1.8,10 While the apoptotic machinery was clearly activated by rapamycin when cells synchronized in G1 were allowed to progress synchronously to S-phase, cell viability was reduced much more substantially if the cells were arrested in S-phase. Of significance, S-phase arrest could be accomplished in cancer cells harboring K-Ras mutations by interfering with Gln utilization. Thus, a synthetic lethality for rapamycin can be created by exploiting the ability of K-Ras-driven cancer cells to override a late G1 Gln checkpoint and arrest in S-phase.19 Thus, the cell cycle specificity for the apoptotic effects of rapamycin offers novel therapeutic options for the large number of K-Ras-driven cancers.
There was a complication with the rapamycin strategy resulting from a feedback activation of mTORC2 and the phosphorylation of Akt at Ser473. In response to suppressing the phosphorylation of S6K by mTORC1, there is an IGF1 receptor- dependent increase in phosphorylation of Akt at Ser473.26,27 This was observed in the Calu-1 cells used in this study where the feedback stimulation of Akt phosphorylation suppressed activation of the S-phase apoptotic program. Rapamycin did not cause the feedback stimulation of Akt phosphorylation in the MDA-MB-231 cells. We have seen a similar resistance to rapamycin-induced apoptosis created by stimulation of Akt phosphorylation in pancreatic cancer cells with defective TGF-β signaling.9 In the previous study with pancreatic cancer cells, like the Calu-1 cells, suppressing Akt phosphorylation restored the apoptotic effect of rapamycin. Thus, activated Akt can overcome the apoptotic effect of rapamycin on S-phase cells – consistent with its known role as a survival kinase.28 It will therefore be important to know whether the feedback is active in a given cancer or cancer cell line for a rapamycin-based therapeutic strategy. However, a combination of suppressing both mTORC1 and mTORC2 could work. We used a catalytic mTOR inhibitor to achieve suppression both mTORC1 and mTORC2 and induce apoptosis in pancreatic cancer cells.9 Torin1 could be used to suppress both Akt and 4E-BP1 as has been reported previously,9,16,36 however the catalytic inhibitors do not provide the same level of specificity provided by rapamycin, which even at the high micro-molar doses is still highly specific for mTOR.13,16 It was also of interest that in Calu-1 cells deprived of Gln, rapamycin inhibited, rather than stimulated Akt phosphorylation and was able to induce the apoptotic effect in these cells that were arrested in S-phase. Thus, the feedback stimulation of Akt phosphorylation may not be an issue if a strategy that employs Gln deprivation is used. The different synthetic lethalities involving Gln deprivation and Akt suppression that can be created for rapamycin are summarized in Fig. 5.
Figure 5.
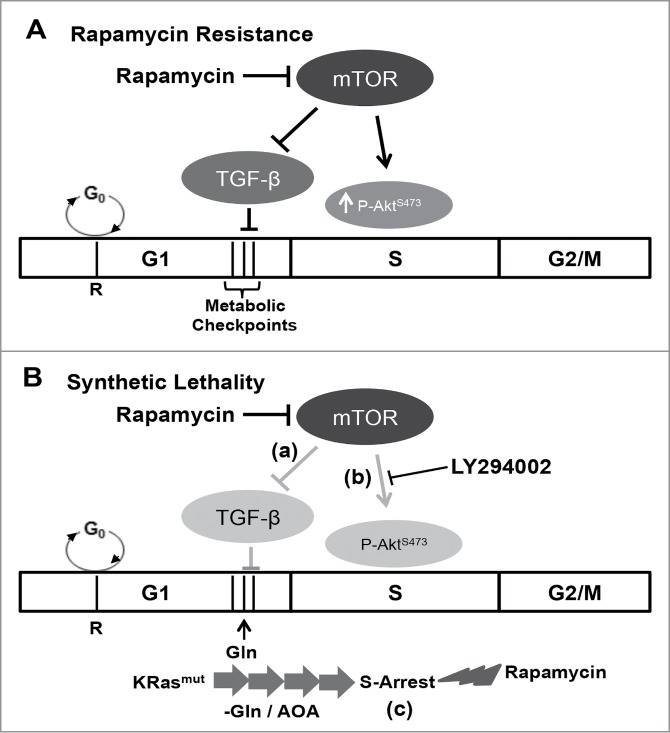
Model for cell cycle-dependent sensitivity to rapamycin. (A) Rapamycin resistance. In most cells, the apoptotic effect of rapamycin is negated by a TGF-β-dependent late G1 cell cycle arrest.7,10 Additionally, a feedback dependent increase in Akt phosphorylation at Ser473 mitigates S-phase cytotoxicity of rapamycin.9,16 (B) Synthetic lethality. A synthetic lethality for rapamycin could be created via one of the 3 mechanisms: (a) in cells with defective TGF-β signaling, rapamycin treatment fails to arrest the cells in G1, and instead the cells progress into S-phase where rapamycin causes apoptosis;7 (b) feedback activation of AktS473 phosphorylation in S-phase is suppressed with LY294002, and in the absence of Akt-dependent survival signals, rapamycin induces apoptotic cell death;9,16 and lastly (c) in K-Ras mutant cancer cells, blockade of Gln utilization causes the cells to aberrantly arrest in S-phase.19 Importantly, in this case, S-phase arrest is not accompanied with an increase in AktS473 phosphorylation upon rapamycin treatment, and as a consequence, rapamycin induces apoptotic cell death in the absence of Akt inhibition.
It is not clear at this point how inhibition of mTORC1 leads to apoptosis in cancer cells when they are arrested in S-phase, but it is clear that micro-molar doses of rapamycin that suppress phosphorylation of 4E-BP1 are required.16 Consistent with this hypothesis, suppression of 4E-BP1 expression prevented the apoptotic effect of rapamycin.16 Since suppression of 4E-BP1 phosphorylation leads to the sequestration of eIF4E and repressed cap-dependent translation,18 the observations reported here implicate eIF4E as being critical for progression through S-phase. In this regard, it is of note that eIF4E can induce tumorigenesis when dysregulated and overexpressed.37 eIF4E promotes the translation of proteins critical for cell cycle progression and survival.38 Thus, when mTORC1 is suppressed in S-phase the cell is getting a signal that nutrients are sparse and since the cell cycle is not reversible, default apoptotic signals are activated. It appears that if cells are traversing S-phase, the majority of cells are able to survive rapamycin treatment and the activation of the apoptotic machinery. However, if cells are arrested in S-phase, then there is substantial cell death caused by rapamycin. The arrest of K-Ras-driven cancer cells in S-phase caused by interfering with Gln utilization creates synthetic lethal phenotype for rapamycin. Thus, a therapeutic approach that blocks the anaplerotic utilization of Gln along with rapamycin represents a plausible approach for treating the large number of human cancers driven by K-Ras mutation.
Methods
Cells and cell culture conditions
The human cancer cell lines MDA-MB-231, MCF-7 and Calu-1 cells were obtained from the American Tissue Type Culture Collection (ATCC). All the cells were maintained in Dulbecco's modified Eagle medium (Sigma, Saint Louis, MO, D6429) supplemented with 10% fetal bovine serum (Sigma F4135).
Antibodies and reagents
Antibodies against cleaved poly-ADP-ribose polymerase (PARP) (catalog #9541), Akt, phospho-Akt (S473) (9271), 4EBP1 (9452), phospho-4EBP1 (T37/46) (9459), cyclin A (4656), and actin (8457) were obtained from Cell Signaling; anti-mouse and anti-rabbit HRP conjugated secondary antibodies were obtained from Promega. For Gln deprivation; Dulbecco's modified Eagle medium without Gln (D5546), dialyzed fetal bovine serum (F0392), and L-Gln (G7513) were obtained from Sigma. Rapamycin (R-5000) was obtained from LC Laboratories; Torin1 (4247) and lovastatin (1530) were obtained from Tocris Biosciences; mevalonate (M4667) was from Sigma.
Cell cycle synchronization and flow cytometric analysis
Activated 10 mM lovastatin stock solution was prepared by dissolving 10 mg lovastatin in 200 μl of 95% ethanol, 156 μl of 1N NaOH was then added, pH adjusted to 7.2 using 1M HCl, and finally diluted to 2.5 ml with sterile-filtered water. After lovastatin treatment, cells were released from lovastatin block by providing medium containing mevalonic acid.39 Cell cycle distribution was determined by flow cytometry as described previously.10 Briefly, cells were fixed in 70% ethanol, stained using propidium iodide, and passed through 70-µm meshes to remove cell aggregates. Fluorescence intensity corresponding to DNA content in different phase of cell cycle was measured by flow cytometry (FACSCalibur; Becton Dickinson), and analyzed using WinCycle software (Phoenix Flow Systems).
Western blot analysis
Proteins were extracted from cultured cells in M-PER (Thermo Scientific 78501). Equal amounts of proteins were subjected to SDS-PAGE on polyacrylamide separating gels. Electrophoresed proteins were then transferred to nitrocellulose membrane. After transfer, membranes were blocked in an isotonic solution containing 5% non-fat dry milk in phosphate-buffered saline. Membranes were then incubated with primary antibodies as described in the text. Depending on the origin of the primary antibody, either anti-mouse or anti-rabbit HRP conjugated IgG was used for detection using ECL system (Thermo Scientific 34080).
Cell viability
Cell viability was determined by trypan blue exclusion. Cells were harvested, washed, and treated with trypan blue (Sigma-T8154) at a concentration of 0.4% v/v. After 5 min, trypan blue uptake (dead cells) was scored using a hemocytometer.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This study was supported by National Institute of Health grants R01-CA046677 and R01-CA179542 to D.A. Foster, and a pilot project award from the Research Centers in Minority Institutions award RP-03037 from the National Center for Research Resources of the NIH.
References
- 1.Hung CM, Garcia-Haro L, Sparks CA, Guertin DA. mTOR-dependent cell survival mechanisms. Cold Spring Harb Perspect Biol 2012; 4:a008771; PMID:23124837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fingar DC, Blenis J. Target of rapamycin (TOR): an integrator of nutrient and growth factor signals and coordinator of cell growth and cell cycle progression. Oncogene 2004; 23:3151-71; PMID:15094765; http://dx.doi.org/ 10.1038/sj.onc.1207542 [DOI] [PubMed] [Google Scholar]
- 3.Ward PS, Thompson CB. Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer Cell 2012; 21:297-308; PMID:22439925; http://dx.doi.org/ 10.1016/j.ccr.2012.02.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blagosklonny MV. Molecular damage in cancer: an argument for mTOR-driven aging. Aging 2011; 3:1130-41; PMID:22246147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Foster DA. Phosphatidic acid signaling to mTOR: signals for the survival of human cancer cells. Biochim Biophys Acta 2009; 1791:949-55; PMID:19264150; http://dx.doi.org/ 10.1016/j.bbalip.2009.02.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sun SY. mTOR kinase inhibitors as potential cancer therapeutic drugs. Cancer Lett 2013; 340:1-8; PMID:23792225; http://dx.doi.org/ 10.1016/j.canlet.2013.06.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gadir N, Jackson DN, Lee E, Foster DA. Defective TGF-b signaling sensitizes human cancer cells to rapamycin. Oncogene 2008; 27:1055-62; PMID:17700525; http://dx.doi.org/ 10.1038/sj.onc.1210721 [DOI] [PubMed] [Google Scholar]
- 8.Chatterjee A, Mukhopadhyay S, Tung K, Patel D, Foster DA. Rapamycin-induced G1 cell cycle arrest employs both TGF-b and Rb pathways. Cancer Lett 2015; 360:134-40; PMID:25659819; http://dx.doi.org/ 10.1016/j.canlet.2015.01.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Le Gendre O, Sookdeo A, Duliepre SA, Utter M, Frias M, Foster DA. Suppression of AKT Phosphorylation restores rapamycin-based synthetic lethality in SMAD4-defective pancreatic cancer cells. Mol Cancer Res 2013; 11:474-81; PMID:23443316; http://dx.doi.org/ 10.1158/1557-3125.ADVBC-A113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Saqcena M, Menon D, Patel D, Mukhopadhyay S, Chow V, Foster DA. Amino acids and mTOR mediate distinct metabolic checkpoints in mammalian G1 cell cycle. PLoS One 2013; 8:e74157; PMID:23977397; http://dx.doi.org/ 10.1371/journal.pone.0074157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Foster DA, Gadir N. Can defective TGF-b signaling be an Achilles heel in human cancer?. Chinese J Cancer 2008; 27:882-4; PMID:18710627 [PubMed] [Google Scholar]
- 12.Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, Markhard AL, Sabatini DM. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell 2006; 22:159-68; PMID:16603397 http://dx.doi.org/ 10.1016/j.molcel.2006.03.029 [DOI] [PubMed] [Google Scholar]
- 13.Toschi A, Lee E, Xu L, Garcia A, Gadir N, Foster DA. Regulation of mTORC1 and mTORC2 complex assembly by phosphatidic acid: competition with rapamycin. Mol Cell Biol 2009; 29:1411-20; PMID:19114562; http://dx.doi.org/ 10.1128/MCB.00782-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen Y, Zheng Y, Foster DA. Phospholipase D confers rapamycin resistance in human breast cancer cells. Oncogene 2003; 22:3937-42; PMID:12813467; http://dx.doi.org/ 10.1038/sj.onc.1206565 [DOI] [PubMed] [Google Scholar]
- 15.Fang Y, Vilella-Bach M, Bachmann R, Flanigan A, Chen J. Phosphatidic acid-mediated mitogenic activation of mTOR signaling. Science 2001; 294:1942-5; PMID:11729323; http://dx.doi.org/ 10.1126/science.1066015 [DOI] [PubMed] [Google Scholar]
- 16.Yellen P, Saqcena M, Salloum D, Feng J, Preda A, Xu L, Rodrik-Outmezguine V, Foster DA. High-dose rapamycin induces apoptosis in human cancer cells by dissociating mTOR complex 1 and suppressing phosphorylation of 4E-BP1. Cell Cycle 2011; 10:3948-56; PMID:22071574; http://dx.doi.org/ 10.4161/cc.10.22.18124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yellen P, Chatterjee A, Preda A, Foster DA. Inhibition of S6 kinase suppresses the apoptotic effect of eIF4E ablation by inducing TGF-b-dependent G1 cell cycle arrest. Cancer Lett 2013; 333:239-43; PMID:23376634; http://dx.doi.org/ 10.1016/j.canlet.2013.01.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Richter JD, Sonenberg N. Regulation of cap-dependent translation by eIF4E inhibitory proteins. Nature 2005; 433:477-80; PMID:15690031; http://dx.doi.org/ 10.1038/nature03205 [DOI] [PubMed] [Google Scholar]
- 19.Saqcena M, Mukhopadhyay S, Hosny C, Alhamed A, Chatterjee A, Foster DA. Blocking anaplerotic entry of glutamine into the TCA cycle sensitizes K-Ras mutant cancer cells to cytotoxic drugs. Oncogene 2015; 34:2672-80; PMID:25023699; http://dx.doi.org/ 10.1038/onc.2014.207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Salloum D, Mukhopadhyay S, Tung K, Polonetskaya A, Foster DA. Mutant Ras elevates dependence on serum lipids and creates a synthetic lethality for rapamycin. Mol Cancer Ther 2014; 13:733-41; PMID:24435447; http://dx.doi.org/ 10.1158/1535-7163.MCT-13-0762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Foster DA, Yellen P, Xu L, Saqcena M. Regulation of G1 cell cycle progression: distinguishing the restriction point from a nutrient-sensing cell growth checkpoint(s). Genes Cancer 2010; 1:1124-31; PMID:21779436; http://dx.doi.org/ 10.1177/1947601910392989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Javanmoghadam-Kamrani S, Keyomarsi K. Synchronization of the cell cycle using lovastatin. Cell Cycle 2008; 7:2434-40; PMID:18677105; http://dx.doi.org/ 10.4161/cc.6364 [DOI] [PubMed] [Google Scholar]
- 23.Keyomarsi K, Sandoval L, Band V, Pardee AB. Synchronization of tumor and normal cells from G1 to multiple cell cycles by lovastatin. Cancer Res 1991; 51:3602-9; PMID:1711413 [PubMed] [Google Scholar]
- 24.Alberts AW. Discovery, biochemistry and biology of lovastatin. Am J Cardiology 1988; 62:10J-5J; PMID:3055919; http://dx.doi.org/ 10.1016/0002-9149(88)90002-1 [DOI] [PubMed] [Google Scholar]
- 25.Pagano M, Pepperkok R, Verde F, Ansorge W, Draetta G. Cyclin A is required at two points in the human cell cycle. EMBO J 1992; 11:961-71; PMID:1312467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.O'Reilly KE, Rojo F, She QB, Solit D, Mills GB, Smith D, Lane H, Hofmann F, Hicklin DJ, Ludwig DL. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res 2006; 66:1500-8; PMID:16452206; http://dx.doi.org/ 10.1158/0008-5472.CAN-05-2925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sun SY, Rosenberg LM, Wang X, Zhou Z, Yue P, Fu H, Khuri FR. Activation of Akt and eIF4E survival pathways by rapamycin-mediated mammalian target of rapamycin inhibition. Cancer Res 2005; 65:7052-8; PMID:16103051; http://dx.doi.org/ 10.1158/0008-5472.CAN-05-0917 [DOI] [PubMed] [Google Scholar]
- 28.Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell 2007; 129:1261-74; PMID:17604717; http://dx.doi.org/ 10.1016/j.cell.2007.06.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wendel HG, De Stanchina E, Fridman JS, Malina A, Ray S, Kogan S, Cordon-Cardo C, Pelletier J, Lowe SW. Survival signalling by Akt and eIF4E in oncogenesis and cancer therapy. Nature 2004; 428:332-7; PMID:15029198; http://dx.doi.org/ 10.1038/nature02369 [DOI] [PubMed] [Google Scholar]
- 30.Liu P, Begley M, Michowski W, Inuzuka H, Ginzberg M, Gao D, Tsou P, Gan W, Papa A, Kim BM. Cell-cycle-regulated activation of Akt kinase by phosphorylation at its carboxyl terminus. Nature 2014; 508:541-5; PMID:24670654; http://dx.doi.org/ 10.1038/nature13079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stumpf CR, Moreno MV, Olshen AB, Taylor BS, Ruggero D. The translational landscape of the mammalian cell cycle. Mol Cell 2013; 52:574-82; PMID:24120665; http://dx.doi.org/ 10.1016/j.molcel.2013.09.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Andrs M, Korabecny J, Jun D, Hodny Z, Bartek J, Kuca K. Phosphatidylinositol 3-linase (PI3K) and phosphatidylinositol 3-kinase-related kinase (PIKK) inhibitors: importance of the morpholine ring. J Med Chem 2014; 8:41-71; PMID:25387153; http://dx.doi.org/ 10.1021/jm501026z [DOI] [PubMed] [Google Scholar]
- 33.DeBerardinis RJ, Cheng T. Q's next: the diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene 2010; 29:313-24; PMID:19881548; http://dx.doi.org/ 10.1038/onc.2009.358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hensley CT, Wasti AT, DeBerardinis RJ. Glutamine and cancer: cell biology, physiology, and clinical opportunities. J Clin Invest 2013; 123:3678-84; PMID:23999442; http://dx.doi.org/ 10.1172/JCI69600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen Y, Rodrik V, Foster DA. Alternative phospholipase D/mTOR survival signal in human breast cancer cells. Oncogene 2005; 24:672-9; PMID:15580312; http://dx.doi.org/ 10.1038/sj.onc.1208099 [DOI] [PubMed] [Google Scholar]
- 36.Leontieva OV, Blagosklonny MV. Tumor promoter-induced cellular senescence: cell cycle arrest followed by geroconversion. Oncotarget 2014; 5:12715-27; PMID:25587030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bitterman PB, Polunovsky VA. eIF4E-mediated translational control of cancer incidence. Biochim Biophys acta 2014; Epub ahead of print; PMID:25263391; http://dx.doi.org/ 10.1016/j.bbagrm.2014.09.007 [DOI] [PubMed] [Google Scholar]
- 38.Sonenberg N. eIF4E, the mRNA cap-binding protein: from basic discovery to translational research. Biochem Cell Biol 2008; 86:178-83; PMID:18443631; http://dx.doi.org/ 10.1139/O08-034 [DOI] [PubMed] [Google Scholar]
- 39.Jackman J, O'Connor PM. Methods for synchronizing cells at specific stages of the cell cycle. Curr Protoc Cell Biol 2001; 8:8.3; PMID:18228388;; 10.1002/0471143030.cb0803s00 [DOI] [PubMed] [Google Scholar]