

Introduction
Idiopathic dilated cardiomyopathy (DCM) is a leading cause of heart failure characterized by an enlarged ventricular cavity leading to systolic dysfunction. DCM patients have a considerable annual mortality rate of 5–10%, with half of them being sudden unexpected deaths due to ventricular tachycardia (VT) or ventricular fibrillation (VF).1 Although a multifactorial disease, DCM appears to be inheritable in approximately 70% of cases.2,3 Causative gene mutations have been identified in a broad range of genes coding for proteins with a variety of function, such as cytoskeletal, sarcomeric or ion homeostasis related.4 Among the latter category, several mutations have been identified in Ca2+ handling proteins in familial and sporadic DCM cases. An increasing body of evidence indicates that abnormal intracellular Ca2+ handling underlies contractile dysfunction5,6 and contributes to ventricular arrhythmogenesis in failing myocardium.7,8 A prime example is phospholamban (PLN), which is directly involved in the uptake of Ca2+ by the sarcoplasmic reticulum (SR), on a beat-to-beat basis, thereby regulating cardiac contraction and relaxation. PLN mutations have been directly associated with the development of dilated cardiomyopathy and heart failure in patients and animal models.9 However, modifier genes are thought to influence the clinical outcome both in PLN cases, as well as DCM cases in general.10 We herein describe a DCM case which illustrates the complex genetic contribution to disease development and progression.
Clinical report
Clinical characteristics
A male patient aged 56 years presented for evaluation because of sustained ventricular tachycardia episodes (Figure 1). He had a history of dilated cardiomyopathy diagnosed at the age of 40 years. Patient also presented heart failure symptoms (NYHA class II) with left ventricular ejection fraction (LVEF) of 25%. The ECG showed atrial fibrillation with frequent ventricular extra systolic beats (Figure 2). The Echo revealed severe left ventricular dilatation and systolic dysfunction (Figure 3). An AICD was implanted and the patient presented several ventricular tachycardia episodes terminating by AICD firing (Figure 4) during the following years. Patient had also presented clinical deterioration with advance heart failure symptoms and frequent hospital admissions especially 2-3 years after his presentation. He finally died because of end stage heart failure at the age of 60.
Figure 1.
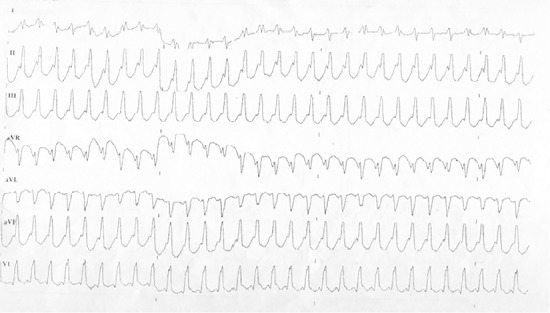
Sustained monomorphic ventricular tachycardia episode recorded in patient via ECG.
Figure 2.

Patient ECG revealing atrial fibrillation with frequent ventricular extra systolic beats.
Figure 3.
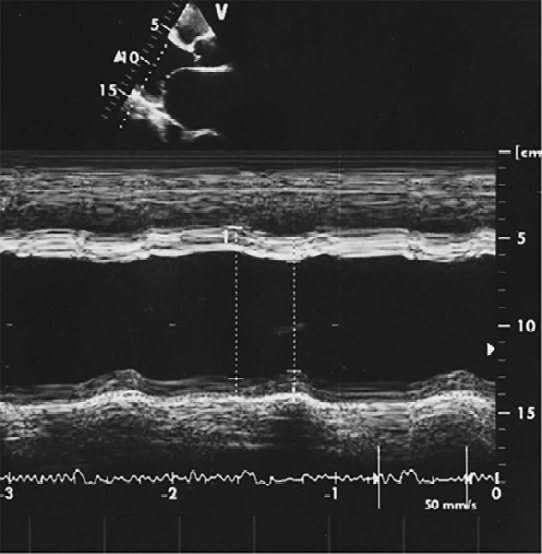
Patient echo study revealing severe left ventricular dilatation (left ventricular end diastolic diameter: 70 mm and left ventricular end systolic diameter 59 mm) and severe systolic dysfunction (left ventricular ejection fraction: 25%).
Figure 4.

Patient AICD record revealing ventricular tachycardia episode that terminated by AICD firing.
Genetics characteristics
High quality DNA was extracted from the peripheral blood of the patient, and was analysed by Next Generation Sequencing using the HiSeq Illumina platform. One hundred and seventy genes previously associated with heart disease or known to be involved in cardiac function were screened, and 163 genetic variants were detected in this patient. Among these, the vast majority were predicted to be benign variants. However, there were three heterozygous variants of interest:
(i) a known pathogenic nonsense mutation (c.116T>G) in the PLN gene, that leads to a premature stop codon (L39X) (Figure 6),9
(ii) a frameshift mutation (c.1495_1496insAGAC) in the C-terminus of CACNB2 (the beta subunit of the voltage-dependent calcium channel Ca(v)1.2) (Figure 6).
(iii) a non-synonymous single nucleotide polymorphism (SNP) (c.9217C>T; p.L3073F) in laminin 2 (LAMA2), predicted to have a deleterious according to the Sorting Intolerant From Tolerant (SIFT) bioinformatical algorithm, and a possibly damaging effect according to the Polymorphism Phenotyping v2 (Polyphen2) algorithm (Figure 6).
(iv) a non-synonymous SNP (c.6082A>G; p.T2028A) in the Alstrom Syndrome 1 (ALMS1) gene, predicted to have a deleterious effect according to SIFT.
All four genetic variants were confirmed by targeted Sanger sequencing. High-throughput sequencing is emerging as a powerful approach in revealing genotype-phenotype correlations of clinical significance.11
Figure 6.
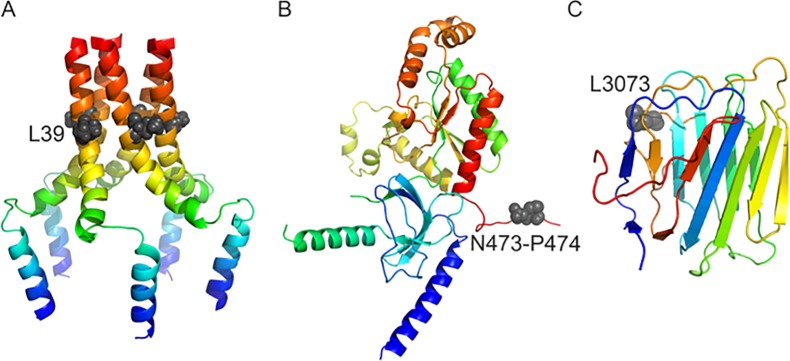
Protein structures and positions of mutations. A) The pentameric structure of PLN. B) A partial modelled structure of CACNB2 (residues 88 to 474). C) A model of the fifth module of laminin G-like domain. The 3D structures are represented as coloured cartoons, where the grey spheres indicate the key mutant residues.
Discussion
Calcium homeostasis plays a critical role in normal cardiac function, and its impairment can lead to DCM. The sarcoplasmic reticulum (SR) is the principal organelle that controls intracellular Ca2+ cycling in cardiomyocytes, and thereby regulates cardiac contraction and relaxation. For the cardiomyocyte to be in a steady state with respect to intracellular Ca2+ balance, the amount of Ca2+ released from the SR into the cytoplasm must equal that re-accumulated by the action of SERCA uptake pumps. Cytosolic Ca2+ is sequestered into the SR lumen by the Ca2+-ATPase (SERCA2a) during muscle relaxation. The stored Ca2+ is subsequently released from the SR through the ryanodine receptor channels to activate myofibrillar contraction. The activity of SERCA2a is reversibly regulated by PLN, a 52 amino acid phosphoprotein (Figure 5 and 6A). Dephosphorylated PLN interacts with SERCA2a and decreases the affinity of the Ca2+-pump for Ca2+, whereas phosphorylation of PLN through the beta-adrenergic pathway relieves this inhibition and augments relaxation.12 It has been postulated that phosphorylation of PLN at both S16 and tbl17 by PKA and CaMKII activation is the ultimate goal of sympathetic stimulation in heart, and PLN mutations can be causative for DCM.13 Homozygosity for the L39X mutation in PLN has been shown to lead to reduced PLN mRNA expression and absence of the PLN protein.9
Figure 5.

Schematic of the cardiomyocyte structure components, including PLN, laminin 2 and Ca(v)1.2 (modified from Fatkin et al Phys Rev 2012).
The phenotype of L39X mutation carriers in the PLN gene has been shown to vary considerably, ranging from severe DCM to rare reports of hypertrophic cardiomyopathy (HCM) or even normal cardiac function.9,14 In cardiomyopathies there are several reports of different mutations in a gene being associated with a different prognosis,15 as well as reports of rare cases where the same gene mutation leads to different forms of cardiomyopathy.16 These observations can be attributed at least partly to the interplay of different mutations, genetic variants, epigenetic and environmental factors, ultimately modifying molecular, cellular and cardiac functions.
The patient described herein, represents the first PLN L39X DCM case developing sustained VT, a major risk factor for sudden cardiac death.17 This is an unusual observation, considering that the absence of PLN in mice suppresses stress-induced VTs in an established model of catecholaminergic polymorphic VT.18 The development of malignant arrhythmias in the presented case study could potentially be explained by the co-existence of the CACNB2 frameshift variant at the C-terminus, the LAMA2 L3073F, and the ALMS1 tbl2028A variants. As demonstrated below, changes in the gene sequence or expression levels of CACNB2, LAMA2, and ALMS1 have been previously associated with DCM and/or ventricular arrhythmias.
The CACNB2 codes for one of the three subunits of the L-type calcium channel (LTCC), a voltage-dependent calcium channel whose activation allows Ca2+ to enter the cardiomyocyte during the action potential (AP) and constitutes the major Ca2+ entry pathway (Figure 5). Specifically, CACNB2 is the dominant isoform known to play an essential role in the voltage dependence of LTCC.19,20 Consequently, mutations in CACNB2 can lead to electrical instability of the heart, and have been described in patients with cardiac arrhythmia syndromes, namely short QT or early depolarization syndromes.21,22 CACNB2 gene expression has been reported to be downregulated in DCM patients and possibly associated to left ventricular dysfunction.23 The novel frameshift CACNB2 mutation described herein, leads to the abolishment of its C-terminal subunit where important regulatory phosphorylation sites are located.24 This could be associated with abnormal triggering of the ryanodine receptor (RyR) opening that may lead to cardiomyocyte delayed after depolarization (DAD) and ultimately, arrhythmia propagation. To visualize the mutational region of CACNB2, we used homology modelling for model building as there is a lack of 3D structure for the protein. The template structure of the voltage-dependent calcium channel beta subunit from rabbit was used to model the residues from 88 to 469.25 A loop region (470 to 476) was built to show the mutational position between N473 and P474 (c.1495_1496). The model structures were built in discovery studio (DS, Accelrys) and refined using energy minimization with CHARMm force field (Figure 6B and 7A).
Figure 7.

Sequence alignment for modeling structures. A) The calcium channel beta-2 subunit of human and rabbit share 98% sequence identity from residues 88 to 469. B) The fifth module of laminin G-like module from human and mouse share 88% sequence identity. The block box indicates the key mutational sequences that are conserved among the species.
Unlike CACNB2, mutations in LAMA2 and ALMS1, have only rarely been associated with cardiac arrhythmias. LAMA2 codes for the alpha-2 subunit, which forms the laminin 2 protein (also known as merosin) when combined with the beta-1 and gamma-1 subunits, and the laminin 4 protein when combined with the beta-2 and gamma-1 subunits. Laminins are found in the extracellular matrix, and serve as a major component of the striated muscle cytoarchitecture (Figure 5). The majority of LAMA2 mutations have been associated with the development of muscular dystrophy, however, one rare occasion also developed DCM with life-threatening ventricular arrhythmias.26 That patient carried two different dominant mutations: a missense mutation in exon 29 (c.4405 T>C, p.Cys1469Arg) and a nonsense mutation in exon 31 (c.4645 C>T, p.Arg1549Stop). In our patient, the key residue was located in Laminin G-like 5 domain (p. 2939 to 3120). Since there is no crystal structure available for this region, we modelled the domain in DS using a template structure of the fifth laminin g-like module of the mouse laminin alpha2 chain.27 The sequence of the mouse and human shared 88% identity and importantly the key residue L3073 was conserved (see sequence alignment, Figure 7B). The modelled domain was optimized by energy minimization using CHARMm force field in DS (Figure 6C).
Mutations in ALMS1 lead to Alström syndrome, a rare autosomal recessive genetic disorder characterized by metabolic, endocrine and sensory impairment (blandness and deafness) as well as liver, pulmonary and renal disease, over time.28 Approximately 60% of ALMS cases develop DCM, while recently two siblings presented with central conduction system disease and cardiac rhythm abnormalities.29 Although there are no reports on the role of ALMS1 in the heart, it is interesting to note that ALMS1 silencing in kidney epithelial cells inhibited intracellular calcium influx.30
Overall, the combination of the CM causative L39X mutation in PLN, with the predicted pathogenic genetic variants in CACNB2, LAMA2 and ALMS1 in DCM is associated with sustained VT.
What have we learned?
-
•
This is the first report of a L39X mutation carrier presenting with sustained VT, in addition to cardiomyopathy.
-
•
These observations strengthen the evidence of one mutation contributing to multiple different phenotypes, possibly under the influence of other genetic or environmental factors.
-
•
Next generation sequencing is a powerful, unbiased, spherical approach to depict genetic variants with a causative or modifying role.
-
•
Three modifier gene candidates emerge that could be implicated in the development of sustained VT in DCM patients, and specifically carriers of the PLN L39X mutation.
Acknowledgements
D.S. was supported by research grants from the Greek General Secretariat for Research and Technology (Aristeia II: CALCIRHYTHM), the Fondation Sante and the Hellenic Cardiological Society. R.B., R.W., P.B. and S.C. were supported by the NIHR Biomedical Research Unit in Cardiovascular Disease at Royal Brompton & Harefield NHS Foundation Trust and Imperial College London.
References
- 1.Grimm W, Maisch B. Sudden cardiac death in dilated cardiomyopathy – therapeutic options. Herz. 2002;27(8):750–759. doi: 10.1007/s00059-002-2425-0. [DOI] [PubMed] [Google Scholar]
- 2.Grunig E, Tasman JA, Kucherer H, Franz W, Kubler W, Katus HA. Frequency and phenotypes of familial dilated cardiomyopathy. Journal of the American College of Cardiology. 1998;31(1):186–194. doi: 10.1016/s0735-1097(97)00434-8. [DOI] [PubMed] [Google Scholar]
- 3.Yacoub MH. Decade in review–cardiomyopathies: Cardiomyopathy on the move. Nature reviews Cardiology. 2014;11(11):628–629. doi: 10.1038/nrcardio.2014.157. [DOI] [PubMed] [Google Scholar]
- 4.Shaw T, Elliott P, McKenna WJ. Dilated cardiomyopathy: a genetically heterogeneous disease. Lancet. 2002;360(9334):654–655. doi: 10.1016/S0140-6736(02)09879-3. [DOI] [PubMed] [Google Scholar]
- 5.Haghighi K, Schmidt AG, Hoit BD, Brittsan AG, Yatani A, Lester JW, Zhai J, Kimura Y, Dorn GW, MacLennan DH, Kranias EG. Superinhibition of sarcoplasmic reticulum function by phospholamban induces cardiac contractile failure. The Journal of biological chemistry. 2001;276(26):24145–24152. doi: 10.1074/jbc.M102403200. [DOI] [PubMed] [Google Scholar]
- 6.Schmidt AG, Zhai J, Carr AN, Gerst MJ, Lorenz JN, Pollesello P, Annila A, Hoit BD, Kranias EG. Structural and functional implications of the phospholamban hinge domain: impaired SR Ca2+ uptake as a primary cause of heart failure. Cardiovascular research. 2002;56(2):248–259. doi: 10.1016/s0008-6363(02)00541-2. [DOI] [PubMed] [Google Scholar]
- 7.Janse MJ. Electrophysiological changes in heart failure and their relationship to arrhythmogenesis. Cardiovascular research. 2004;61(2):208–217. doi: 10.1016/j.cardiores.2003.11.018. [DOI] [PubMed] [Google Scholar]
- 8.Pogwizd SM, Bers DM. Cellular basis of triggered arrhythmias in heart failure. Trends in cardiovascular medicine. 2004;14(2):61–66. doi: 10.1016/j.tcm.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 9.Haghighi K, Kolokathis F, Pater L, Lynch RA, Asahi M, Gramolini AO, Fan GC, Tsiapras D, Hahn HS, Adamopoulos S, Liggett SB, Dorn GW, MacLennan DH, Kremastinos DT, Kranias EG. Human phospholamban null results in lethal dilated cardiomyopathy revealing a critical difference between mouse and human. The Journal of clinical investigation. 2003;111(6):869–876. doi: 10.1172/JCI17892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Arvanitis DA, Sanoudou D, Kolokathis F, Vafiadaki E, Papalouka V, Kontrogianni-Konstantopoulos A, Theodorakis GN, Paraskevaidis IA, Adamopoulos S, Dorn GW, Kremastinos DT, Kranias EG. The Ser96Ala variant in histidine-rich calcium-binding protein is associated with life-threatening ventricular arrhythmias in idiopathic dilated cardiomyopathy. European heart journal. 2008;29(20):2514–2525. doi: 10.1093/eurheartj/ehn328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lopes LR, Syrris P, Guttmann OP, O'Mahony C, Tang HC, Dalageorgou C, Jenkins S, Hubank M, Monserrat L, McKenna WJ, Plagnol V, Elliott PM. Novel genotype-phenotype associations demonstrated by high-throughput sequencing in patients with hypertrophic cardiomyopathy. Heart. 2015;101(4):294–301. doi: 10.1136/heartjnl-2014-306387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brittsan AG, Kranias EG. Phospholamban and cardiac contractile function. Journal of molecular and cellular cardiology. 2000;32(12):2131–2139. doi: 10.1006/jmcc.2000.1270. [DOI] [PubMed] [Google Scholar]
- 13.MacLennan DH, Kranias EG. Phospholamban: a crucial regulator of cardiac contractility. Nature reviews Molecular cell biology. 2003;4(7):566–577. doi: 10.1038/nrm1151. [DOI] [PubMed] [Google Scholar]
- 14.Landstrom AP, Adekola BA, Bos JM, Ommen SR, Ackerman MJ. PLN-encoded phospholamban mutation in a large cohort of hypertrophic cardiomyopathy cases: summary of the literature and implications for genetic testing. American heart journal. 2011;161(1):165–171. doi: 10.1016/j.ahj.2010.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Roberts R, Sigwart U. New concepts in hypertrophic cardiomyopathies, part I. Circulation. 2001;104(17):2113–2116. doi: 10.1161/hc4201.097429. [DOI] [PubMed] [Google Scholar]
- 16.Osterziel KJ, Perrot A. Dilated cardiomyopathy: more genes means more phenotypes. European heart journal. 2005;26(8):751–754. doi: 10.1093/eurheartj/ehi208. [DOI] [PubMed] [Google Scholar]
- 17.Zipes DP, Wellens HJ. Sudden cardiac death. Circulation. 1998;98(21):2334–2351. doi: 10.1161/01.cir.98.21.2334. [DOI] [PubMed] [Google Scholar]
- 18.Bai Y, Jones PP, Guo J, Zhong X, Clark RB, Zhou Q, Wang R, Vallmitjana A, Benitez R, Hove-Madsen L, Semeniuk L, Guo A, Song LS, Duff HJ, Chen SR. Phospholamban knockout breaks arrhythmogenic Ca(2)(+) waves and suppresses catecholaminergic polymorphic ventricular tachycardia in mice. Circulation research. 2013;113(5):517–526. doi: 10.1161/CIRCRESAHA.113.301678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Foell JD, Balijepalli RC, Delisle BP, Yunker AM, Robia SL, Walker JW, McEnery MW, January CT, Kamp TJ. Molecular heterogeneity of calcium channel beta-subunits in canine and human heart: evidence for differential subcellular localization. Physiological genomics. 2004;17(2):183–200. doi: 10.1152/physiolgenomics.00207.2003. [DOI] [PubMed] [Google Scholar]
- 20.Lao QZ, Kobrinsky E, Harry JB, Ravindran A, Soldatov NM. New Determinant for the CaVbeta2 subunit modulation of the CaV1.2 calcium channel. The Journal of biological chemistry. 2008;283(23):15577–15588. doi: 10.1074/jbc.M802035200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Antzelevitch C, Pollevick GD, Cordeiro JM, Casis O, Sanguinetti MC, Aizawa Y, Guerchicoff A, Pfeiffer R, Oliva A, Wollnik B, Gelber P, Bonaros EP, Jr, Burashnikov E, Wu Y, Sargent JD, Schickel S, Oberheiden R, Bhatia A, Hsu LF, Haïssaguerre M, Schimpf R. Loss-of-function mutations in the cardiac calcium channel underlie a new clinical entity characterized by ST-segment elevation, short QT intervals, and sudden cardiac death. Circulation. 2007;115(4):442–449. doi: 10.1161/CIRCULATIONAHA.106.668392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Burashnikov E, Pfeiffer R, Barajas-Martinez H, Delpon E, Hu D, Desai M, Borggrefe M, Häissaguerre M, Kanter R, Pollevick GD, Guerchicoff A, Laiño R, Marieb M, Nademanee K, Nam GB, Robles R, Schimpf R, Stapleton DD, Viskin S, Winters S, Wolpert C. Mutations in the cardiac L-type calcium channel associated with inherited J-wave syndromes and sudden cardiac death. Heart rhythm: the official journal of the Heart Rhythm Society. 2010;7(12):1872–1882. doi: 10.1016/j.hrthm.2010.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Molina-Navarro MM, Rosello-Lleti E, Ortega A, Tarazon E, Otero M, Martinez-Dolz L, Lago F, González-Juanatey JR, España F, García-Pavía P, Montero JA, Portolés M, Rivera M. Differential gene expression of cardiac ion channels in human dilated cardiomyopathy. PloS one. 2013;8(12):e79792. doi: 10.1371/journal.pone.0079792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brandmayr J, Poomvanicha M, Domes K, Ding J, Blaich A, Wegener JW, Moosmang S, Hofmann F. Deletion of the C-terminal phosphorylation sites in the cardiac beta-subunit does not affect the basic beta-adrenergic response of the heart and the Ca(v)1.2 channel. The Journal of biological chemistry. 2012;287(27):22584–22592. doi: 10.1074/jbc.M112.366484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Opatowsky Y, Chen CC, Campbell KP, Hirsch JA. Structural analysis of the voltage-dependent calcium channel beta subunit functional core and its complex with the alpha 1 interaction domain. Neuron. 2004;42(3):387–399. doi: 10.1016/s0896-6273(04)00250-8. [DOI] [PubMed] [Google Scholar]
- 26.Carboni N, Marrosu G, Porcu M, Mateddu A, Solla E, Cocco E, Maioli MA, Oppo V, Piras R, Marrosu MG. Dilated cardiomyopathy with conduction defects in a patient with partial merosin deficiency due to mutations in the laminin-alpha2-chain gene: a chance association or a novel phenotype? Muscle & nerve. 2011;44(5):826–828. doi: 10.1002/mus.22228. [DOI] [PubMed] [Google Scholar]
- 27.Hohenester E, Tisi D, Talts JF, Timpl R. The crystal structure of a laminin G-like module reveals the molecular basis of alpha-dystroglycan binding to laminins, perlecan, and agrin. Molecular cell. 1999;4(5):783–792. doi: 10.1016/s1097-2765(00)80388-3. [DOI] [PubMed] [Google Scholar]
- 28.Marshall JD, Hinman EG, Collin GB, Beck S, Cerqueira R, Maffei P, Milan G, Zhang W, Wilson DI, Hearn T, Tavares P, Vettor R, Veronese C, Martin M, So WV, Nishina PM, Naggert JK. Spectrum of ALMS1 variants and evaluation of genotype-phenotype correlations in Alstrom syndrome. Human mutation. 2007;28(11):1114–1123. doi: 10.1002/humu.20577. [DOI] [PubMed] [Google Scholar]
- 29.Czosek RJ, Goldenberg P, Miller EM, Spicer R, Towbin JA, Ware SM. Cardiac electrical system involvement in Alström syndrome: uncommon causes of dilated cardiomyopathies. Cardiogenetics. 2012;2(e2):6–10. [Google Scholar]
- 30.Li G, Vega R, Nelms K, Gekakis N, Goodnow C, McNamara P, Wu H, Hong NA, Glynne R. A role for Alstrom syndrome protein, alms1, in kidney ciliogenesis and cellular quiescence. PLoS genetics. 2007;3(1):e8. doi: 10.1371/journal.pgen.0030008. [DOI] [PMC free article] [PubMed] [Google Scholar]