

Abstract
Drugs capable of specifically recognizing and killing cancer cells while sparing healthy cells are of great interest in anti-cancer therapy. An example of such a drug is edelfosine, the prototype molecule of a family of synthetic lipids collectively known as antitumor lipids (ATLs). A better understanding of the selectivity and the mechanism of action of these compounds would lead to better anticancer treatments. Using Caenorhabditis elegans, we modeled key features of the ATL selectivity against cancer cells. Edelfosine induced a selective and direct killing action on C. elegans embryos, which was dependent on cholesterol, without affecting adult worms and larvae. Distinct ATLs ranked differently in their embryonic lethal effect with edelfosine > perifosine > erucylphosphocholine >> miltefosine. Following a biased screening of 57 C. elegans mutants we found that inactivation of components of the insulin/IGF-1 signaling pathway led to resistance against the ATL edelfosine in both C. elegans and human tumor cells. This paper shows that C. elegans can be used as a rapid platform to facilitate ATL research and to further understand the mechanism of action of edelfosine and other synthetic ATLs.
Keywords: alkylphospholipid analogs, antitumor lipids, Caenorhabditis elegans, cholesterol, edelfosine, embryo, insulin/IGF-1 signaling, mechanism of action, miltefosine, tumor cell
Abbreviations
- APL
alkylphospholipid analog; ATL, antitumor lipid; DIC, differential interference contrast; IIS, insulin/IGF-1-like signaling; MCD, methyl-β-cyclodextrin; NGM, nematode growth medium
Introduction
Most drugs currently used against cancer are anti-proliferative, targeting various steps in the cell division process. A major downside to anti-proliferative chemotherapies is that they target all dividing cells, both malignant and healthy, thus strong detrimental side effects are common. Finding treatments that directly and selectively target cancer cells is a priority in cancer research.
An example of a drug capable of directly and selectively inducing cancer cell death is edelfosine (1-O-octadecyl-2-O-methyl-rac-glycero-3-phosphocholine, ET-18-OCH3).1 Edelfosine is an alkyl-lysophospholipid analog and the prototype molecule of a family of synthetic compounds collectively known as antitumor lipids (ATLs) or alkylphospholipid analogs (APLs).1-3 ATLs act on cell membranes rather than on DNA.2,4,5 They have amphipathic structures that resemble those of phospholipids and they are incorporated preferentially in tumor cells,6-8 thus apparently displaying selectivity to tumor cell membranes.1 This selectivity has been shown to depend, at least in part, on the accumulation of the drug in the cholesterol-rich lipid rafts of cancer cell membranes.6,9,10 Once ATLs incorporate into tumor cells, they induce signaling events that lead to cancer cell death by apoptosis.3,4 ATLs have been shown to act through different signaling pathways in different tumor cell types. Haematopoietic cancer cells incorporate edelfosine in cholesterol-rich lipid rafts, which in turn leads to the recruitment and clustering of the Fas/CD95 death receptor in lipid rafts, and triggers the activation of the extrinsic apoptotic pathway4,6,7,9-14 Edelfosine incorporation in lipid rafts elicits local accumulation of apoptotic molecules in lipid rafts, altering the apoptosis versus survival signaling balance.6,7,11,12,15,16 In a number of solid tumors ATLs can promote endoplasmic reticulum stress response, which triggers apoptosis.17-19 ATLs have also been shown to inhibit phosphatidylcholine biosynthesis, which can eventually lead to cell cycle arrest and apoptosis.18-22
So far the clinical applications of ATLs in cancer therapy have been limited to the use of edelfosine in the purging of the bone marrow in acute leukemia and the use of miltefosine as topical treatment of skin metastases of breast cancer and cutaneous lymphoma.23-26 Other ATLs with potential clinical applications are erucylphosphocholine, which shows promise for brain tumor treatment, and perifosine, which is currently in clinical trials.27,28 Recently, combined therapies using ATLs as well as traditional chemotherapies or radiotherapies have shown preliminary promising results.29,30 The factors that confer ATL selectivity to cancer cells and the molecular mechanisms that lead to cancer cell death are not yet well understood. Unveiling such mechanisms as well as understanding why some tumors are more sensitive than others to ATL treatment could lead to the development of more and improved clinical applications of ATLs as anticancer chemotherapy drugs.
Using tumor tissue culture cell lines or murine models to study the ATL mechanism of action is technically challenging. Simpler model organisms can be very useful to uncover general aspects of the molecular mechanism elicited by ATL treatment. For example, Saccharomyces cerevisae was recently used in chemogenomic screens that identified edelfosine resistant and sensitive mutants involved in drug uptake and cytotoxicity.31,32 However, unicellular models like S. cerevisiae have limitations because a fundamental aspect of ATL research is understanding what causes specificity of these drugs to cancer cells but not to healthy cells. A simple laboratory model that could recreate the ATL selective killing of cancer cells while sparing healthy cells would be ideal to determine why ATLs display this selectivity and how cancer cells die when treated with these synthetic lipids. In this paper we modeled the nematode C. elegans to use it in ATL research. C. elegans, which consists of 959 cells grouped into differentiated tissues,33 is extensively used as a model in cancer research, and has played a crucial role in the understanding of the mechanism of action of cancer altered pathways such as apoptosis, Ras, Notch, AKT, etc.34-39
Here we report that edelfosine treatment of the nematode C. elegans induces a selective lethal effect to embryonic cells without causing adverse effects to the worm. As described for some haematopoietic tumor cells,9,10 we show that edelfosine activity in C. elegans depends, at least partially, on cholesterol. We also show that the relative efficiency of different ATLs in C. elegans is comparable to the efficiency of the same ATLs when used in human tumor cells. Furthermore, we demonstrate that resistance and sensitivity to edelfosine in C. elegans can be genetically regulated and that the signaling pathways involved in the mechanism of action of edelfosine can be conserved in C. elegans and human tumor tissue culture cells. Our results indicate that C. elegans can be used as a discovery platform to understand the selectivity of ATLs against cancer cells as well as to study the underlying mechanism of toxicity of ATLs.
Results
Edelfosine selectively kills C. elegans embryos without affecting the physiological integrity of the worm
Edelfosine selectively kills tumor cells while sparing healthy cells.1,2,4,6-8,10,40 We tested the effect of edelfosine in C. elegans in order to check its suitability as a model for the study of edelfosine toxicity. We first assayed the effect of edelfosine upon larval growth and development. Synchronized arrested C. elegans L1 larvae reared on C. elegans laboratory medium (NGM) plates containing edelfosine doses of up to 62 μM grew at the same rate as control worms reared without edelfosine (Fig. 1A). In contrast, a very high dose of edelfosine (125 μM) completely blocked both growth as well as visible foraging activity of larvae. Animals treated at this dose never progressed to later larval stages (data not shown). C. elegans L1 larvae developed to adults in the same time period independently of whether larvae had been maintained on NGM plates with 20 μM edelfosine or on control plates (Fig. 1B). Importantly, all the edelfosine-treated L1 larvae survived development in the presence of 20 μM edelfosine (n = 155).
Figure 1.
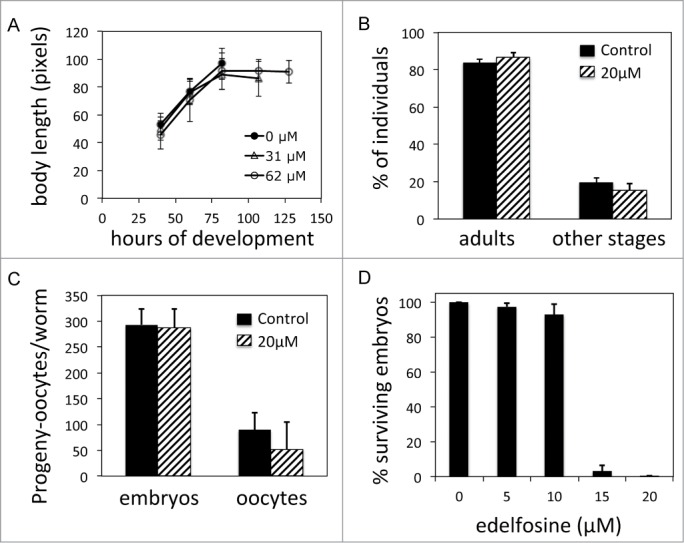
Edelfosine displays selective embryonic lethality without affecting growth or fertility. (A) Shown are growth dynamics of N2 larvae over time when larvae are maintained on NGM plates with or without edelfosine. Each data point represents the mean values of 14 to 69 individual animals ± SD. (B) Percentage of L1 larvae that became adults after 2.7 d on NGM plates with or without edelfosine. n = 427 (control) and n = 155 (edelfosine). Data shown represent mean values ± SD of 3 experiments. (C) Embryonic progeny and oocytes laid by N2 worms during 4 d for worms treated with edelfosine and for untreated worms. Data shown represent mean values for 5 independent worms ± SD. (D) Percentage of surviving embryos (n ≥ 400 per treatment) laid by L3-L4 worms treated with different concentrations of edelfosine using standard NGM plates. Each bar in the histogram represents the mean values ± SD of at least 3 independent experiments.
Since larvae treated with edelfosine did not lead to growth differences with respect to controls, we next investigated whether worms treated with edelfosine showed other physiological alterations. The elevated offspring production of C. elegans is a high energy demanding process and thus a good indicator of the physiological status of the worm. For example, C. elegans individuals with genetic and/or environmental modifications that alter their metabolism have severely reduced progeny.41 We monitored the number of embryos produced until sperm was exhausted and unfertilized oocytes started being laid on the medium, but did not observe progeny differences between edelfosine-treated and control worms (Fig. 1C). However, although progeny number was unaffected, edelfosine treatment led to severe embryonic lethality. Treating L4 larvae with 20 μM edelfosine led to virtually all their progeny embryos dying before embryogenesis could be completed. The embryonic lethality was also clearly noticeable using a lower dose of edelfosine (15 μM) as more than 90% of fertilized embryos died before embryogenesis could be completed (Fig. 1D).
Altogether, these data indicate that edelfosine has a selective lethal effect to the developing embryo without affecting the overall physiological status of the worm and without causing larval developmental lethality, larval to adult growth differences, or progeny production reduction. Thus, similar to the selective killing effect of edelfosine in tumor cells, we observe a selective killing effect of edelfosine to C. elegans embryos.
Edelfosine exerts a direct killing of C. elegans embryos
We wanted to investigate whether the selective edelfosine C. elegans embryonic lethality was due to a maternally induced effect by the drug or to a specific lethal effect to the embryo. We tested this using 2 approaches. Firstly, we determined at what developmental stage edelfosine leads to embryonic lethality. We synchronized worms at different stages of development and treated them with 20 μM edelfosine, a dosis that virtually kills all the embryos (Fig. 1D). Edelfosine provoked embryonic lethality even when the drug was administered to fully developed adults that already had embryos in their uterus (Fig. 2A). Secondly, we pre-treated L3-L4 worms with edelfosine for 24 hours and then transferred them to plates without edelfosine. Worms were able to produce viable embryos 24 hours after discontinuing the edelfosine treatment, indicating that edelfosine embryonic lethality is not persistent upon edelfosine withdrawal (Fig. S1). These 2 results suggest that edelfosine-induced embryonic lethality is not caused by a maternal effect but rather by a rapid and selective effect of the drug to the developing embryo.
Figure 2.
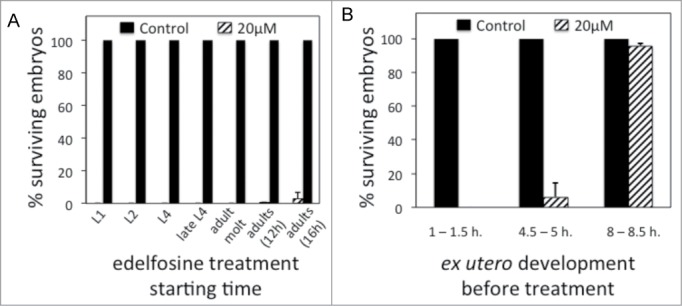
Edelfosine targets early embryonic development. (A) Percentage of surviving embryos (n ≥ 100 per treatment) laid by control worms or by worms treated with edelfosine. Different groups of age-synchronized worms are treated at different points of development or adulthood. Each bar in the histogram represents the mean values ± SD of at least 3 independent experiments. (B) Percentage of surviving embryos (n = 60–130 per treatment) when embryos are treated with edelfosine at different times of “ex utero” embryogenesis. Each bar in the histogram represents the mean values ± SD of at least 3 independent experiments.
Edelfosine targets the first stages of embryogenesis
To characterize the critical time during embryogenesis at which edelfosine induces embryonic lethality, we isolated embryos at different times of “ex-utero” development and subjected them to edelfosine treatment. Embryos at the earlier stages of “ex-utero” development were not able to survive edelfosine treatment, whereas embryos that had undergone 4.5–5 hours of “ex-utero” development started showing resistance to edelfosine, and embryos that had undergone 8–8.5 hours of “ex-utero” development were almost fully resistant to edelfosine (Fig. 2B). The aproximately 700 cells produced during embryogenesis are generated 5–6 hours upon fertilization.33 Therefore, edelfosine induces its selective embryonic lethality at the first stages of embryogenesis, before morphogenesis occurs and when rapid embryonic cell division takes place.
Edelfosine induces structural and morphological embryonic defects that lead to embryonic death
To understand the nature of embryonic lethality upon edelfosine treatment, we made differential interference contrast (DIC) microscopy recordings of edelfosine-treated embryos and followed their development over time until these died. Death was defined as permanent arrest of embryonic development. The recordings showed that upon edelfosine treatment embryos would undergo death at markedly different stages of embryogenesis (Fig. 3). The recordings also revealed different types of defects from embryo to embryo preceeding their death. These include defects in morphogenesis, structural defects, and ventral enclosure defects (GEX phenotype). However, DIC microscopy recordings as well as multi-dimensional imaging and retrospective animated comparisons between normal and drug-affected embryos revealed that embryogenesis of edelfosine-treated embryos displayed early cleavage divisions, gastrulation, and other cell movements that were normal both, in relative timing and positioning when compared to untreated controls (Movie S1; Fig. S2). We also investigated whether edelfosine treatment induced supernumerary apoptotic events during embryogenesis. To do this, we analyzed the 13 apoptosis events that take place in the lineage of AB (the anterior blastomere of the 2 cell stage embryo) in 2 edelfosine-treated embryos that developed after the first round of cell death took place. We did not observe abnormal apoptotic events (data not shown). Apoptotic cells underwent all the characteristic steps of typical C. elegans apoptosis (asymmetric division, condensation, engulfment and clearance).
Figure 3.

Edelfosine treatment produces embryonic lethality due to cell structural and embryonic morphological defects. Early embryos were mounted under the microscope using a 20 μM edelfosine solution and their development was recorded for up to 17 h. Time (minutes) is indicated at the top of the figure. Seven independent experiments containing 10 embryos are shown in the different rows and the time of death is shown. The observed defects are indicated: loss of normal cell structure (2nd, 6th and 7th-right row embryos) and morphogenesis defects at ventral enclosure (1st, 7th-left rows embryos).
In summary, the microscopy recordings showed that edelfosine-treated embryos developed normally up to the point that suffered a sudden lethal crisis, which could appear at different times during embryogenesis. Once this lethal crisis occured both, cell divisions and normal developmental cell movements ceased and various types of embryonic defects were perceived.
Long-term exposure of edelfosine to C. elegans can provoke detrimental effects
Edelfosine exerts a rapid and selective effect on early developing embryos without causing physiological alterations or other apparent phenotypes to larvae and adults. We wanted to investigate whether long-term edelfosine exposure would cause detrimental side effects to worms. We maintained worms from the point they became L4 larvae on NGM plates supplemented with various doses of edelfosine, and observed that while edelfosine did not produce detrimental effects during the first days of age, doses above 5 μM edelfosine caused an overall life span decrease, which was directly proportional to the edelfosine dose used (Fig. S3). We also subjected arrested starved L1 larvae to different concentrations of edelfosine during 5 d and quantified the percentage of larvae surviving the treatment. Edelfosine doses higher than 5 μM and up to 20 μM of edelfosine displayed a moderate yet significant decrease in the viability of starved L1 larvae. However, edelfosine doses of 40 μM and above led to a low degree of larval survival (Fig. S4). These data indicate that although short-term edelfosine treatment has a rapid and selective effect over early developing embryos without causing apparent negative phenotypes, long-term edelfosine exposure can lead to detrimental side effects in worms.
Edelfosine-induced embryonic lethality depends on cholesterol
There is evidence indicating that edelfosine selectivity to haematopoietic cancer cells can be mediated by cholesterol enrichment in cancer cell membrane lipid rafts.9,10 Additionally, biophysics studies have demonstrated a strong affinity of edelfosine for sterols.9,42 On these grounds, we wanted to investigate the importance of cholesterol in mediating the edelfosine selective lethality to C. elegans embryos. C. elegans cannot synthesize cholesterol de novo,43,44 thus standard NGM needs to be supplemented with cholesterol.45 Matyash et al. reported that yolk proteins are transported into the oocyte along with large amounts of cholesterol, making the C. elegans embryo very enriched in cholesterol.46 One possibility is that edelfosine might interact with the large amounts of cholesterol stored in the embryo, thus affecting the normal functioning of embryonic cells, which in turn could lead to embryonic lethality. We tested this possibility by adding high concentrations of cholesterol to the C. elegans NGM plates as well as by eliminating the cholesterol from the NGM plates.
As mentioned above, edelfosine has a high affinity for cholesterol.9,42 Thus, NGM prepared with abundant cholesterol should buffer a large number of edelfosine molecules before these could exert their effect to the embryos. Therefore a high concentration of cholesterol in the NGM should lead to a reduction of embryonic lethality upon edelfosine treatment. Conversely, NGM plates lacking cholesterol should lead to higher edelfosine-induced embryonic lethality as the edelfosine would not be buffered by the NGM. Therefore, a given dose of edelfosine should lead to a higher embryonic lethality when worms were treated on NGM plates lacking cholesterol.
We added different edelfosine concentrations to NGM plates that lacked cholesterol and showed that indeed the edelfosine dose that caused 50% of embryonic lethality (LD50) diminished from 12.4 μM when using standard cholesterol-supplemented (5 μg/ml) NGM plates to 3.8 μM when using NGM plates without cholesterol. Consistently, increasing the concentration of cholesterol in the NGM plates (from 5 μg/ml to 20 μg/ml) led to an almost 3-fold increase in the edelfosine dose needed to reach the LD50 (Fig. 4A). The decreased longevity of worms treated with edelfosine was not fully rescued when NGM plates were supplemented with a high concentration of cholesterol (Fig. S5). These results indicate a strong affinity between edelfosine and cholesterol and suggest an interaction with the cholesterol stored in the embryo.
Figure 4.
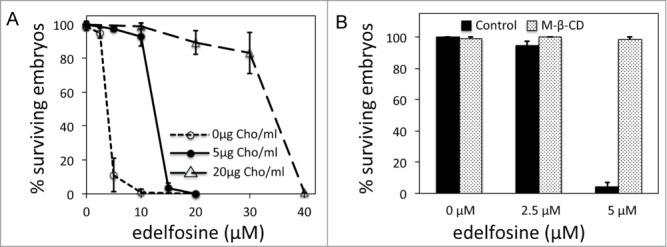
Edelfosine-induced embryonic lethality depends on cholesterol. (A) Percentage of surviving embryos laid by worms treated with edelfosine when maintained on NGM plates without or with different concentrations of cholesterol. Each data point represents the mean values ± SD of at least 3 independent experiments (see Table S1 for details). (B) Percentage of surviving embryos (n = 144–205 per treatment) when worms are treated with or without edelfosine and in the presence or absence of methyl-β-cyclodextrin (MCD). NGM treatment plates are prepared without cholesterol. Each bar in the histogram represents the mean values ± SD of at least 3 independent experiments.
It has been reported for human tumor cell lines that cholesterol sequestration by methyl-β-cyclodextrin (MCD) inhibits edelfosine-induced apoptosis.7,10,11 If embryonic cholesterol is important in mediating the edelfosine-induced embryonic lethality, then MCD cholesterol sequestration in the worm should lead to inhibition of edelfosine-induced embryonic lethality. We cultured L3-L4 larvae in plates containing either MCD, edelfosine, or edelfosine as well as MCD. As expected, embryos from edelfosine-treated worms did not survive. However, embryos from worms treated with edelfosine in combination with MCD were able to survive (Fig. 4B). Molting of C. elegans through the different larval stages requires cholesterol as the biosynthetic precursor of steroid hormones involved in this process.47 Progeny of MCD-treated worms arrested growth at different larval stages (Fig. S6), indicating that MCD is indeed sequestering cholesterol in C. elegans. Altogether the data indicate that, similar to the edelfosine dependance of cholesterol in mediating the killing of tumor cells, embryonic cholesterol is determinant in mediating edelfosine-induced embryonic lethality.
Comparative affinity of edelfosine for lipid-rafts lipids
As indicated above, cholesterol is an essential component of lipid rafts and it has been shown to play an important role for edelfosine to exert selective killing of tumor cells7,10,11 as well as C. elegans embryonic lethality (Fig. 4). We wanted to investigate whether other lipidic components of lipid rafts such as sphingomyelin and ganglioside GM-1 might also be important in mediating edelfosine selective cellular killing. If other components of lipid rafts were important for the edelfosine selectivity, we would expect edelfosine to have a strong affinity for them. We compared the affinity of edelfosine for sphingomyelin and ganglioside GM-1 with respect to the affinity of edelfosine for cholesterol. We also compared the affinity of edelfosine for phosphatidylcholine, the most abundant phospholipid of biological membranes. To do this, we prepared NGM plates lacking cholesterol and added either sphingomyelin, ganglioside GM-1, or phosphatidylcholine without or with different concentrations of edelfosine. The higher affinity edelfosine displayed for one of these components, the more buffered the edelfosine would be by its presence in the culture plate, and thus the less embryonic lethality we should observe for a given dosis of edelfosine. According to our assay, we observed a poor affinity of edelfosine for phosphatidylcholine and sphingomyelin, whereas the affinity for ganglioside GM-1 was much higher (Fig. 5). After adjusting by number of molecules per gram of compound, we determined that edelfosine had 1.4-, 6.8-, and 9-fold less affinity to ganglioside GM-1, phosphatidylcholine and sphingomyelin, respectively, than the affinity to cholesterol.
Figure 5.
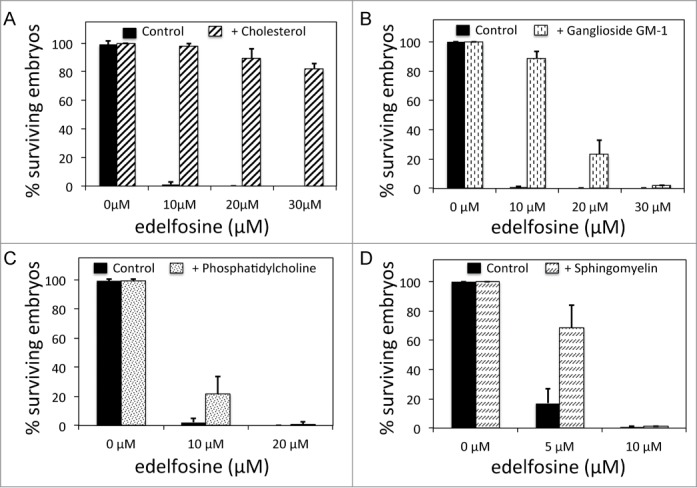
Comparative affinity of edelfosine for lipid-raft lipids. (A) Percentage of surviving embryos laid by worms untreated or treated with different concentrations of edelfosine when NGM plates are supplemented with 20 μg of cholesterol/ml of medium. (B) Percentage of surviving embryos laid by worms untreated or treated with different concentrations of edelfosine when NGM plates are supplemented with 30 μg of ganglioside GM-1/ml of medium. (C) Percentage of surviving embryos laid by worms untreated or treated with different concentrations of edelfosine when NGM plates are supplemented with 60 μg of phosphatidylcholine/ml of medium. (D) Percentage of surviving embryos laid by worms untreated or treated with different concentrations of edelfosine when NGM plates are supplemented with 60 μg of sphingomyelin/ml of medium. Each bar in all the histograms represents the mean values ± SD of at least 3 independent experiments (see Table S2 for details).
Comparative efficiency of different ATLs
Edelfosine is the prototype member of the ATL family of synthetic lipids, however 3 other analogs, perifosine, miltefosine and erucylphosphocholine, are also actively studied as putative clinically relevant drugs.27,29 We wanted to compare the relative efficiency of these 4 ATLs in inducing C. elegans embryonic lethality to assess whether their efficiency is similar to that observed in tumor cells. We compared the efficiency of the above ATLs using standard C. elegans NGM plates and showed that edelfosine and perifosine were more efficient than erucylphosphocholine and much more than miltefosine in inducing embryonic lethality (Fig. 6A). This mimicked the differential sensitivity to those ATLs observed in various tumor cell lines.17,40
Figure 6.
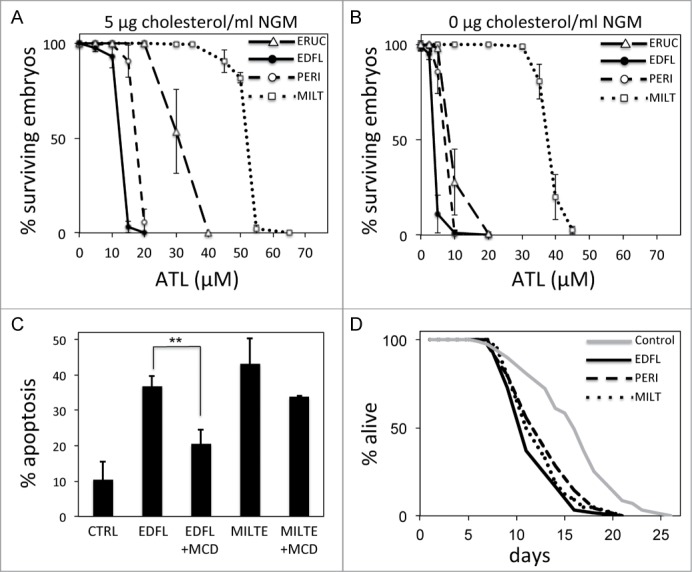
Comparative efficiency of different ATLs. (A) Percentage of surviving embryos laid by worms treated with different concentrations of ATLs when NGM plates are supplemented with 5 μg of cholesterol/ml of medium. Each data point represents the mean values ± SD of at least 3 independent experiments (see Table S3 for details). (B) Percentage of surviving embryos laid by worms treated with different concentrations of ATLs when NGM plates are prepared without cholesterol. Each data point represents the mean values ± SD of at least 3 independent experiments (see Table S4 for details). (C) Edelfosine versus miltefosine-induced apoptosis of human tumor Z-138 cells in the presence or absence of the cholesterol sequester methyl-β-cyclodextrin. Z-138 cells were untreated or pretreated with 2.5 mg/ml MCD and then incubated in the absence or in the presence of 10 μM edelfosine or 40 μM miltefosine for 24 hours. The percentage of apoptotic cells was determined by flow cytometry. Data shown are means ± SD of tree independent experiments (**P < 0.01, Student's t test). (D) Lifespan curves for worms treated with edelfosine, perifosine or miltefosine compared to control (untreated) worms. Life spans were done at 20°C and using standard NGM plates supplemented with the respective ATL at the following concentrations: perifosine = 17.5 μM; edelfosine = 12.4 μM; and miltefosine = 52 μM (see Table S5 for details). y-axis indicates % of worms that are alive. x-axis indicates day of adulthood.
Miltefosine has less affinity to cholesterol than other ATLs
We used C. elegans to investigate the efficiency of the 4 ATL analogs when cholesterol is absent from the media. As explained earlier, edelfosine has a strong affinity for cholesterol9,42 (Fig. 4), thus we wanted to study whether the other ATLs had a similar degree of affinity for cholesterol. We found that when using NGM plates without cholesterol we needed a much lower dose of edelfosine, perifosine or erucylphosphocholine to induce the same degree of embryonic lethality (Fig. 6B) than the dose we needed when the media was supplemented with cholesterol (Fig. 6A). Specifically, we found that the lethal dose to reach 50% of embryonic lethality (LD50) when using NMG plates with or without cholesterol for the case of edelfosine, perifosine and erucylphosphocholine was 3.2-, 2.5- and 3.3-fold lower for each ATL, respectively. However, in the case of miltefosine we only needed 40% more drug to obtain the same LD50 when using NGM plates with cholesterol vs. NGM plates without cholesterol (Figs. 6A and B; Table 1). We also compared the LD50 of miltefosine and edelfosine when using NGM plates with a very high concentration of cholesterol (20 μg/ml) and plates without cholesterol, and observed an 8.9-fold difference in the case of edelfosine but only 2-fold difference in the case of miltefosine (Fig. S7). These results indicate that miltefosine has less affinity to cholesterol than the other ATLs tested, and suggests that its activity is not as dependent on cholesterol as the other ATLs.
Table 1.
Miltefosine has less affinity to cholesterol than other ATLs
| ATL used | LD50 in media with cholesterola | LD50 in media without cholesterolb | Fold differencec | Expected LD50d |
|---|---|---|---|---|
| Edelfosine | 12.4 μM | 3.8 μM | 3.2 fold | 12.4 μM |
| Perifosine | 17.5 μM | 7.0 μM | 2.5 fold | 22.4 μM |
| Erucylphosphocholine | 30.0 μM | 9.0 μM | 3.3 fold | 28.8 μM |
| Miltefosine | 52.0 μM | 37.0 μM | 1.4 fold | 118.4 μM |
plates supplemented with 5 μg cholesterol/ml of NGM.
NGM plates without cholesterol.
fold difference between LD50 in media with cholesterol and LD50 in media without cholesterol.
expected LD50 relative to LD50 fold difference observed for edelfosine.
We tested whether miltefosine activity was also less cholesterol-dependent in human tumor cells. As mentioned above, MCD inhibits edelfosine-induced apoptosis.7,10,11 We treated human tumor Z-138 cells with edelfosine or miltefosine and observed that, as expected, in the presence of MCD there was inhibition of edelfosine-induced apoptosis. However, the apoptotic activity of miltefosine in the presence of MCD was not significantly inhibited (Fig. 6C). A lower MCD concentration and shorter incubation times led to comparable results (Fig. S8) demonstrating that as observed in C. elegans, miltefosine activity in human cancer cells does not depend on cholesterol as much as edelfosine activity does.
Comparative toxicity of edelfosine to miltefosine and perifosine
Our results from C. elegans embryonic lethality indicate that edelfosine is the most efficient ATL. This result is consistent with what has been observed in the great majority of human tumor cell lines tested.10,17,40 Since less edelfosine than other ATLs is needed to induce the same effect, we wanted to test whether the relative toxicity of edelfosine compared to miltefosine and to perifosine is also lower. To do this, we synchronized worm populations and cultured them on NGM plates during their whole lives after adding either edelfosine, miltefosine or perifosine at their respective embryonic LD50-inducing doses. We did not observe major differences in the life span of worms when these were treated with either ATL (Fig. 6D), indicating that the degree of toxicity for a given functional dose was similar for the 3 ATLs.
Screening of C. elegans mutants with sensitivity and resistance to edelfosine
As a proof of principle to validate that edelfosine-induced embryonic lethality in C. elegans can be genetically regulated and that the signaling pathways involved in the mechanism of action of edelfosine can be conserved in C. elegans and human tumor cells, we performed a small-scale biased screening to find C. elegans mutants with resistance and sensitivity to the embryonic lethality induced by edelfosine.
We selected 57 C. elegans strains of mutants with potential involvement in mediating ATL-induced embryonic lethality. The mutants belonged to the following categories: cholesterol transport; longevity; proteins associated to C. elegans lipid rafts; sphingolipid biosynthesis; homologs of Saccharomyces cerevisae gene deletions that induce sensitivity and resistance to edelfosine; apoptosis; and stress response (Table 2).
Table 2.
Screening of C. elegans mutants with sensitivity and resistance to edelfosine
| Strain (allele) | Human homolog | Gene function | n | % hatched (control)a | n | % hatched (10 μM)b | n | % hatched (20 μM)c | ratio 10 μM vs. control | ratio 20 μM vs. control |
|---|---|---|---|---|---|---|---|---|---|---|
| N2 wild type | - | - | 225 | 100.0 | 251 | 96.8 | 245 | 1.2 | 0.97 | 0.01 |
| N2 wild type | - | - | 244 | 98.8 | 281 | 93.2 | 358 | 0.8 | 0.94 | 0.01 |
| N2 wild type | - | - | 421 | 99.3 | 469 | 93.4 | 479 | 0.2 | 0.94 | 0.00 |
| N2 wild type | - | - | 587 | 100.0 | 651 | 91.2 | 611 | 2.6 | 0.91 | 0.03 |
| N2 wild type | - | - | 333 | 99.7 | 294 | 97.6 | 264 | 1.1 | 0.98 | 0.01 |
| N2 wild type | - | - | 296 | 99.3 | 292 | 94.5 | 290 | 0.0 | 0.95 | 0.00 |
| Cholesterol transport | ||||||||||
| rme-2(b1008) | LDL-like receptor | Nematode egg yolk uptake | 679 | 19.6 | 491 | 7.5 | 667 | 0.0 | 0.38 | 0.00 |
| cav-1(ok2089) | CAV1 | Caveolin-mediated endocytosis | 348 | 97.1 | 367 | 94.0 | 367 | 0.0 | 0.97 | 0.00 |
| cav-2(hc191) | CAV2 | Caveolin-mediated endocytosis | 396 | 99.5 | 270 | 97.8 | 413 | 0.0 | 0.98 | 0.00 |
| Longevity | ||||||||||
| daf-2(e1370) | IGF1R | Insulin-like pathway receptor | 172 | 100.0 | 279 | 97.1 | 107 | 30.8 | 0.97 | 0.31 |
| age-1(hx546) | PIK3CA | 1-phosphatidylinositol-3-kinase | 347 | 97.1 | 349 | 96.9 | 402 | 1.7 | 1.00 | 0.02 |
| daf-16(mu86) | FOXO | Insulin-like signaling pathway transcription factor | 360 | 98.6 | 405 | 95.8 | 396 | 0.2 | 0.97 | 0.00 |
| daf-18(e1375) | PTEN | Lipid phosphatase | 257 | 100.0 | 244 | 93.4 | 243 | 3.7 | 0.93 | 0.04 |
| akt-1(mg306) | AKT1 | Serine-threonine protein kinase | 127 | 96.9 | 145 | 97.9 | 338 | 4.4 | 1.01 | 0.05 |
| akt-2(tm812); sgk-1(ft15) | AKT2; SGK1 | Serine-threonine protein kinases | 267 | 98.5 | 173 | 98.3 | 262 | 1.2 | 1.00 | 0.01 |
| pdk-1(sa680) | PDK1 | Serine-threonine/tyrosine-protein kinase | 165 | 84.5 | 65 | 81.5 | 280 | 58.9 | 0.97 | 0.70 |
| clk-1(qm30) | COQ7 | Ubiquinone biosynthesis | 165 | 98.2 | 150 | 97.3 | 129 | 0.0 | 0.99 | 0.00 |
| isp-1(qm150) | UQCR5 | Ubiquinol-cytochrome C reductase | 158 | 90.5 | 127 | 74.0 | 134 | 0.0 | 0.82 | 0.00 |
| nuo-6(qm200) | MTND1 | NADH:ubiquinone oxidoreductase | 188 | 97.3 | 171 | 95.3 | 259 | 0.0 | 0.98 | 0.00 |
| eat-2(ad465) | nAChR | Pharyngeal pumping rate regulation | 293 | 98.9 | 270 | 59.3 | 371 | 0.3 | 0.60 | 0.00 |
| Proteins associated to lipid rafts | ||||||||||
| pho-1(ca101ca102) | ACPP | Phosphatase | 318 | 79.3 | 377 | 79.3 | 338 | 0.9 | 1.00 | 0.01 |
| tag-10(ok246) | MUC2 | Oligomeric gel-forming | 192 | 100.0 | 322 | 97.1 | 220 | 0.0 | 0.99 | 0.00 |
| gfi-1(ok2669) | FBN2 | Development | 295 | 98.3 | 337 | 95.6 | 322 | 0.0 | 0.97 | 0.00 |
| dod-19(ok2679) | GPR112 | G protein-coupled receptor | 159 | 96.9 | 213 | 85.0 | 232 | 0.0 | 0.88 | 0.00 |
| tre-3(ok394) | TREH | Trehalase activity | 144 | 100.0 | 158 | 84.8 | 305 | 0.0 | 0.85 | 0.00 |
| lec-1(ok1597) | Galectin | Sugar binding | 279 | 99.3 | 311 | 97.4 | 311 | 0.6 | 0.98 | 0.01 |
| daf-21(p673) | HSP90 | Heat shock protein | 211 | 98.6 | 245 | 99.2 | 203 | 29.6 | 1.01 | 0.30 |
| Sphingolipid biosynthesis | ||||||||||
| sptl-2(ok2753) | SPTLC2 | Serine Palmitoyl Transferase | 138 | 96.4 | 108 | 86.1 | 240 | 1.3 | 0.89 | 0.01 |
| sptl-3(ok1927) | SPTLC3 | Serine Palmitoyl Transferase | 319 | 98.8 | 286 | 94.1 | 292 | 0.3 | 0.95 | 0.00 |
| C23H3.4(ok1693) | SPTLC1 | Serine Palmitoyl Transferase | 280 | 98.2 | 337 | 92.6 | 395 | 0.5 | 0.94 | 0.01 |
| S. cerevisae deletion screening | ||||||||||
| tat-6(ok1984) | ATP9B | Aminophospholipid translocase | 263 | 85.6 | 218 | 80.3 | 320 | 0.0 | 0.94 | 0.00 |
| apc-10 and tag314(gk143) | ANAPC-10 | Fission yeast anaphase promoting complex | 663 | 14.9 | 640 | 13.8 | 766 | 4.7 | 0.92 | 0.31 |
| ZK370.3(ok1081) | HIP1R | Huntingtin interacting protein 1 related | 486 | 63.0 | 535 | 50.8 | 612 | 0.0 | 0.81 | 0.00 |
| ehs-1(ok146) | EPS15L1 | Calcium ion binding | 564 | 100.0 | 517 | 94.2 | 573 | 0.9 | 0.94 | 0.01 |
| hum-1(ok634) | MYO1E | ATP binding | 558 | 99.3 | 441 | 84.4 | 484 | 1.7 | 0.85 | 0.02 |
| Y65B4A.3(ok2425) | CHMP6 | Charged multivesicular body protein 6 | 347 | 70.0 | 427 | 62.3 | 430 | 2.1 | 0.89 | 0.03 |
| alx-1(gk338) | PDCD6IP | Notch binding | 429 | 98.1 | 445 | 98.2 | 369 | 2.7 | 1.00 | 0.03 |
| vps-35(ok1880) | VPS35 | Receptor-mediated endocytosis | 52 | 92.3 | 43 | 93.2 | 42 | 66.7 | 1.01 | 0.72 |
| vps-52(ok853) | VPS52 | Rab GTPase binding | 184 | 91.9 | 117 | 41.9 | 149 | 0.0 | 0.46 | 0.00 |
| max-2(ok1904) | PAK3 | Rac GTPase binding | 25 | 92.0 | 51 | 76.5 | 57 | 1.75 | 0.83 | 0.02 |
| tam-1(ok2635) | MLL2 | Larval development | 312 | 97.8 | 340 | 94.4 | 249 | 0.8 | 0.97 | 0.01 |
| F42G9.1(ok540) | PPM1G | Phosphatase | 85 | 76.5 | 102 | 77.5 | 76 | 0.0 | 1.01 | 0.00 |
| elo-6(gk233) | ELOVL3 | Regulation of growth rate | 576 | 98.4 | 620 | 94.7 | 515 | 2.1 | 0.96 | 0.02 |
| pld-1(ok2222) | PLD1 | Phospholipase D | 411 | 99.0 | 417 | 81.0 | 402 | 0.5 | 0.82 | 0.01 |
| T04C9.1a(ok1510) | ARHGAP42 | Rho GTPase activating protein 42 | 427 | 99.3 | 434 | 97.0 | 420 | 0.0 | 0.98 | 0.00 |
| vha-12(ok821) | ATP6-V1B2 | Proton-transporting ATPase | 188 | 97.3 | 121 | 95.9 | 287 | 0.4 | 0.98 | 0.00 |
| F54A3.4(ok666) | CBS | Cystathionine-beta-synthase | 314 | 98.7 | 326 | 95.4 | 270 | 0.0 | 0.97 | 0.00 |
| Apoptosis | ||||||||||
| egl-1(n487) | BH3 | Apoptosis activator | 255 | 99.2 | 209 | 99.0 | 365 | 59.7 | 1.00 | 0.60 |
| ced-9(n1950) | BCL2 | Apoptosis inhibitor | 284 | 78.9 | 294 | 80.6 | 255 | 0.0 | 1.02 | 0.00 |
| ced-4(n1162) | APAF1 | BH3 domain binding | 261 | 91.2 | 173 | 71.7 | 265 | 0.0 | 0.79 | 0.00 |
| ced-3(n717) | caspase 3/9 | Cysteine-aspartate protease | 329 | 96.4 | 288 | 78.1 | 375 | 0.0 | 0.81 | 0.00 |
| cps-6(ok1718) | EndoG | Mito-endonuclease G | 337 | 98.2 | 312 | 91.0 | 360 | 0.3 | 0.93 | 0.00 |
| Stress response | ||||||||||
| hif-1(ia4) | HIF1 | Hypoxia-induced factor | 411 | 99.5 | 467 | 93.4 | 486 | 0.0 | 0.94 | 0.00 |
| hsf-1(sy441) | HSF1 | Heat-shock transcription factor | 258 | 99.6 | 330 | 97.0 | 390 | 34.5 | 0.97 | 0.35 |
| jnk-1(gk7) | JNK1 | c-Jun N-terminal kinase | 370 | 100.0 | 374 | 97.3 | 405 | 0.0 | 0.97 | 0.00 |
| hsp-4(gk514) | HSP70 | Heat-shock response | 269 | 91.8 | 269 | 81.4 | 258 | 1.9 | 0.89 | 0.02 |
| hsp-16.2(gk249) | HSP16 | Heat-shock response | 278 | 100.0 | 265 | 98.1 | 285 | 0.0 | 0.98 | 0.00 |
| hsp-16.48(ok577) | HSP16 | Heat-shock response | 205 | 98.1 | 215 | 91.6 | 207 | 0.0 | 0.93 | 0.00 |
| hsp-43(ok647) | HSP-20 | Heat-shock response | 213 | 100.0 | 250 | 94.0 | 241 | 0.4 | 0.94 | 0.00 |
| hsp-12.6(ok1077) | HSP | Heat-shock response | 386 | 99.7 | 296 | 65.9 | 406 | 0.0 | 0.66 | 0.00 |
| hsp-3(ok1083) | HSP70 | Heat-shock response | 214 | 99.1 | 230 | 95.7 | 245 | 0.4 | 0.97 | 0.00 |
| hsp-12.3(ok3095) | HSP | Heat-shock response | 110 | 59.1 | 162 | 52.5 | 345 | 0.0 | 0.89 | 0.00 |
| hsp-12.2(ok3638) | HSP | Heat-shock response | 258 | 93.0 | 222 | 92.3 | 172 | 2.9 | 0.99 | 0.03 |
Highlighted in bold are the mutant strains that displayed resistance or sensitivity to edelfosine.
percentage of embryos that are able to hatch into larvae when maintained in control plates without edelfosine.
percentage of embryos that are able to hatch into larvae when maintained in plates containing 10 μM of edelfosine.
percentage of embryos that are able to hatch into larvae when maintained in plates containing 20 μM of edelfosine.
To screen for mutants with resistance and sensitivity to edelfosine we used standard NGM plates supplemented with either 20 μM or 10 μM of edelfosine. 20 μM was used because it is the minimal edelfosine dose that yields to virtually complete embryonic lethality, and 10 μM because it is the minimal dose that induces a significantly noticeable embryonic lethality (see Fig. 1D and Table 2).
Under control conditions, many mutant strains displayed a lower embryonic survival rate than that of wild type worms, therefore we normalized the screening results obtained for each strain relative to their percentage of embryonic survival when edelfosine was not used. As controls we performed 6 independent embryonic survival experiments treating wild type N2 worms with 0 μM, 10 μM and 20 μM of edelfosine. We used the N2 control results to set up the cut-off threshold for the selection of the resistant as well as the sensitive mutant strains. Specifically, we used the normalized embryonic survival average induced upon 10 μM and 20 μM edelfosine treatment of the 6 control experiments plus and minus 3 standard deviations as the confidence interval to select mutant strains with resistance or sensitivity to edelfosine, respectively. From the 57 mutant strains tested, we found 8 that showed resistance and 12 that showed sensitivity to edelfosine (Table 2). The resistant strains include 3 components of the insulin/IGF-1-like signaling pathway (daf-2, pdk-1, akt-1);35 the stress response genes hsf-1 and daf-2148 (daf-21 is also a lipid raft associated protein);49 the drug uptake related gene apc-10;31 the vacuolar sorting gene vps-35;31 and the apoptosis activator egl-1.36 The sensitive strains include the embryonic cholesterol transporter rme-2;46 the longevity genes isp-1 and eat-2; the protein associated to lipid rafts tre-3;49 the phospholipase D pld-1 gene; the apoptosis factors ced-3 and ced-4;36 the heat-shock response gene hsp-12.6;48 and the vesicular traffic homologs from the edelfosine-induced S. cerevisae toxicity gene deletion screening ZK370.3, hum-1, vps-52, and max-2.31,32
Inhibition of the insulin/IGF-1 signaling pathway leads to edelfosine resistance in C. elegans as well as in human tumor cells
Three of the 8 mutant strains that displayed resistance to edelfosine were components of the IIS pathway. These include the daf-2 insulin-like growth factor 1 receptor and the serine-threonine protein kinases pdk-1 and akt-1.35 For the remainder of this work we focused our attention on the IIS pathway in conferring edelfosine resistance.
We first verified that loss of function of these genes indeed led to edelfosine-induced embryonic lethality resistance. We tested 3 independent pdk-1 mutant alleles, pdk-1(sa680), pdk-1(sa709), and pdk-1(mg142), which had previously been characterized as a strong loss of function, a weaker loss of function, and gain of function allele, respectively.50 pdk-1(sa680) worms (strongest loss of function allele) displayed a greater edelfosine resistance than pdk-1(sa709) worms (weaker loss of function allele), whereas pdk-1(mg142) worms (gain of function allele) were as sensitive to edelfosine as wild type worms (Fig. 7A). To verify that akt-1 mutants were resistant to edelfosine, we tested 4 independent akt-1 mutant alleles including 3 loss of function alleles, akt-1(mg306), akt-1(ok525), akt-1(sa573), and a gain of function allele, akt-1(mg144).51-53 The 3 akt-1 loss of function strains led to edelfosine-induced embryonic lethality resistance. However, the akt-1 gain of function strain did not show differences with respect to wild type control embryonic lethality when worms were subjected to edelfosine treatment (Fig. 7A). In summary, these results confirm that loss of function of daf-2, pdk-1 and akt-1 led to resistance against edelfosine-induced embryonic lethality, which indicates a role of the IIS pathway in mediating the effect of edelfosine in C. elegans.
Figure 7.
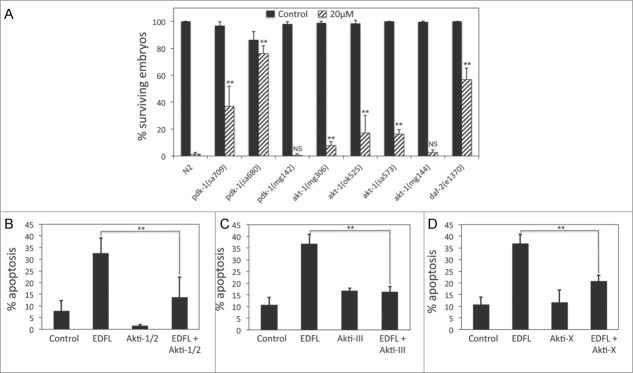
Inhibition of IIS pathway activity leads to edelfosine resistance in C. elegans and in human tumor cells. (A) Percentage of surviving embryos (n ≥ 250 per treatment) laid by L3-L4 N2 control worms as well as pdk-1, akt-1, or daf-2 mutant worms untreated or treated with 20 μM edelfosine. Each bar in the histogram represents the mean values ± SD of at least 3 independent experiments. (**P < 0.01; NS P > 0.05, Student's t test). (B) ZZ-138 cells were preincubated without or with 1 μM Akti-1/2 for 1 h, then incubated in the absence (control) or presence of 10 μM edelfosine for 24 hours and analyzed by flow cytometry to evaluate apoptosis. Data shown are means ± SD of 3 independent experiments (**P < 0.01, Student's t test). (C) Z-138 cells were preincubated without or with 10 μM Akti-III and then incubated in the absence (control) or presence of 10 μM edelfosine for 24 hours, and analyzed by flow cytometry to evaluate apoptosis. Data shown are means ± SD of 3 independent experiments (**P < 0.01, Student's t test). (D) Z-138 cells were preincubated without or with or 5 μM Akti-X for 1 h, and then incubated in the absence (control) or presence of 10 μM edelfosine for 24 hours, and analyzed by flow cytometry to evaluate apoptosis. Data shown are means ± SD of 3 independent experiments (**P < 0.01, Student's t test).
The IIS pathway is well conserved from C. elegans to humans.35 In C. elegans this pathway is crucial in the regulation of many biological responses ranging from growth and development to stress response and longevity. In humans, the IIS pathway serine/threonine protein kinase Akt is deregulated in a large number of cancers.35 Since loss of function of Akt activity in C. elegans led to edelfosine-induced embryonic lethality resistance, we wanted to test whether inhibition of Akt signaling in human tumor cells resulted in cells becoming resistant to edelfosine. To test this, we used the Z-138 mantle cell lymphoma human tumor cell line, which is known to have deregulated Akt activity.15 We treated the tumor culture cells with 3 different Akt inhibitors (Akti-1/2, Akti-III, and Akti-X), which inhibited Akt phosphorylation (Fig. S9) and showed that edelfosine diminished its apoptotic activity when Akt signaling was pharmacologically inhibited (Fig. 7B to D; Fig. S10).
Altogether these data demonstrate that the mechanism of action of edelfosine-induced embryonic lethality in C. elegans or in human tumor cells depends on the IIS pathway. Furthermore, these data are a proof of principle that the mechanisms driving embryonic lethality in C. elegans upon ATL treatment can be extrapolated to human models to understand how ATLs induce selective apoptosis of cancer cells.
Discussion
Edelfosine and other ATLs are of great interest as potential novel anticancer chemotherapy agents.2-4,27 These synthetic lipids selectively recognize and kill cancer cells while sparing healthy cells.6-8 Their selectivity against cancer cells makes ATLs an attractive class of antitumor molecules. However, their mechanism of action including the factors that confer specificity against tumor cells and the molecular pathways that lead to the death of the tumor cells remains to be fully elucidated. In this paper we present a C. elegans paradigm that can facilitate the study of the mechanism of action of ATLs.
Edelfosine and other ATLs selectively kill C. elegans embryos without causing short-term damage to the developing larvae or the adult worm. The selective embryonic lethality of edelfosine in C. elegans resembles the selective effect of edelfosine against cancer cells. What confers the edelfosine selectivity against C. elegans embryos? It has been shown that cholesterol, a fundamental component of cellular membrane microdomains or lipid rafts, plays an important role in the proapoptotic action of edelfosine against tumor cells.7,10,11 In the case of C. elegans, we showed that cholesterol also plays a fundamental role in the edelfosine selectivity against embryos. C. elegans cannot synthesize cholesterol, thus they need to take it from the environment. C. elegans embryos accumulate large amounts of cholesterol via specific LDL-like receptor-mediated endocytosis.46,54 Indeed, visualization of cholesterol in the worm body revealed that the embryo accumulates the largest concentration of cholesterol in the animal.46 The embryonic cholesterol would serve as a reservoir of this essential structural component for the abundant membranes that will subsequently be generated in the embryogenesis rapid cell divisions.46 Edelfosine has a strong affinity for cholesterol,9,42 therefore we propose that edelfosine would interact with the abundant embryonic cholesterol. Edelfosine would accumulate in the embryo until reaching a critical point whereby morphological and structural embryonic alterations would occur, which in turn would lead to developmental arrest and embryonic lethality.
Miltefosine-induced embryonic lethality did not depend on cholesterol as much as edelfosine, perifosine or erucylphosphocholine-induced embryonic lethality. We also observed a lower dependence of miltefosine for cholesterol in human tumor cells, which suggests differences in the mechanism of action of miltefosine with respect to other ATLs.
In addition to the affinity between edelfosine and cholesterol, our data indicate that edelfosine has also a strong affinity to the ganglioside GM-1, a glycolipid that accumulates in lipid rafts and whose concentration in tumor membranes is often altered.55 Interestingly, recent biophysical studies using binary mixed monolayers reported that edelfosine displayed a strong affinity to ganglioside GM-1.56 Thus, ganglioside GM-1 could play a role in the selective recognition of tumor cells by edelfosine and other ATLs.
Our small-scale C. elegans screening demonstrated that, as it is the case for tumor cells, resistance and sensitivity to edelfosine in C. elegans can be genetically regulated. We showed that inhibition of the same signaling pathway in C. elegans and human tumor cells led to resistance to edelfosine. Loss of function of different components of the IIS pathway in C. elegans led to edelfosine-induced embryonic lethality resistance. Moreover, pharmacological inhibition of the key IIS intermediary Akt in tumor culture cells also led to resistance against edelfosine-induced apoptosis. It was recently shown that ATL treatment of mantle cell lymphoma cells displaced Akt as well as other kinases from lipid rafts.15 This ATL-induced lipid raft reorganization altered the PI3K/AKT signaling of the tumor cells and led to their death.15 Therefore, raft-mediated PI3K/Akt signaling appears to play an important role in the ATL mechanism of action of at least some tumor types. Because IIS and Akt signaling are highly conserved in C. elegans and humans,35 the worm model is perfectly suitable to better understand the role of these signaling pathways in the mechanism of ATL chemotherapy.
Another mutant strain that showed resistance to edelfosine in our screening was daf-21(p673). DAF-21 is a member of the Hsp90 family of molecular chaperones57 and associates to lipid rafts.49 Interestingly, pharmacological inhibition of Hsp90 made human leukemic cells more resistant to edelfosine-induced apoptosis.58 Although the mechanism by which inactivation of DAF-21/Hsp90 leads to edelfosine resistance is not fully understood, it appears to be functioning similarly in both C. elegans embryos as well as in human tumor cells.
Other worm mutant strains that promoted resistance to edelfosine involved genes that function in stress response (hsf-1), vesicular trafficking (vps-35), drug uptake (apc-10), and apoptosis (egl-1). Vesicular trafficking and drug uptake had previously been shown to play a central role in ATL-induced cytotoxicity in S. cerevisiae.31 Additionally, worm mutants of vesicular traffic homologs from a edelfosine-induced S. cerevisae toxicity gene deletion screening31 (ZK370.3, hum-1, vps-52, and max-2) were sensitive to edelfosine. Thus, in total, 25% of the worm strains that showed resistance or sensitivity to edelfosine in our screening were mutants of genes involved in vesicular trafficking, what supports the involvement of these cellular pathways in the mechanism of action of ATLs.
A strain with high sensitivity to edelfosine was the yolk receptor mutant rme-2(b1008), a receptor critical for the endocytosis of vitellogenins and cholesterol into the oocyte.54 rme-2(b1008) embryos accumulate considerable less cholesterol, which severely affects their survival.46 Thus, the sensitivity of rme-2(b1008) embryos to edelfosine may be a consequence of the strong affinity of edelfosine to the little cholesterol left in these embryos. rme-2 embryos treated with edelfosine would have no available cholesterol for their new developing embryonic membranes, which would enhance their embryonic lethality.
In summary, using previous knowledge of the mechanism of action of ATLs in tumor cells, we found analogous results in C. elegans. These include the cellular selectivity of ATLs, the ATL dependance on cholesterol, and the similar efficiency of different ATLs in their killing response. Likewise, the results obtained in C. elegans led us to similar results in human tumor cells such as the involvement of the IIS pathway in the ATL mechanism of action or the lower dependence of cholesterol for miltefosine activity.
Because C. elegans embryonic lethality and tumor cell killing induced upon ATL treatment display many important similarities, we propose the use of C. elegans as a bona fide model to study the mechanism of action of ATLs. The simplicity of performing genome-wide screenings in C. elegans combined with its powerful genetic and molecular tools makes C. elegans a simple discovery platform to better understand the mechanism of action of ATLs as novel antitumor chemotherapy agents.
Materials and Methods
Reagents
Edelfosine (R. Berchtold, Biochemisches Labor, Bern, Switzerland); perifosine and erucylphosphocholine (Zentaris, Frankfurt, Germany); and miltefosine (Calbiochem, Cambridge, MA, USA) were prepared as 4 mM stock solutions in distilled water. Methyl-β-cyclodextrin (Sigma, St. Louis, MO, USA) was prepared as 200 mg/ml stock solution in distilled water. Cholesterol (Sigma, St. Louis, MO, USA) was prepared as 10 mg/ml stock solution in 100% ethanol. Ganglioside GM-1 (Avanti Polar Lipids, Inc., Alabaster, AL, USA) was prepared as 5 mg/ml stock solution in 50% ethanol. L-α-Phosphatidylcholine (Sigma, St. Louis, MO) was prepared as 20 mg/ml stock solution in chloroform. Sphingomyelin (Sigma, St. Louis, MO, USA) was prepared as 20 mg/ml stock solution in 100% ethanol. Akt inhibitor III, Akt inhibitor X, Akt inhibitor 1/2 (Calbiochem/Merck, Darmstadt, Germany) were prepared in dimethyl sulfoxide as 10 mM stock solutions.
C. elegans NGM plate preparation
1000 ml of Nematode Growth Medium (NGM) were prepared by mixing 3 g NaCl, 17 g agar, and 2.5 g peptone and then adding 975 ml of distilled water. This solution was autoclaved and allowed to cool in a water bath at 55°C. At this point, 1 ml 1 M CaCl2, 1 ml 5 mg/ml cholesterol in ethanol, 1 ml 1 M MgSO4, and 25 ml 1 M KPO4 buffer were added. The molten NGM agar was dispensed into medium size plates (60 mm diameter).
C. elegans strains and maintenance
Worms were maintained and handled as previously described.59 Unless otherwise stated, all the drugs and reagents tested in C. elegans were added to the molten NGM agar and cast into culture plates. Hardened agar was seeded with Escherichia coli (OP50). All C. elegans experiments were conducted at 20°C.
C. elegans growth assay
Different concentrations of edelfosine were added to the NGM agar. Hardened agar was seeded with Escherichia coli (OP50). Synchonized arrested L1 larvae were placed into seeded NGM plates and worms were photographed at low magnification every 20 hours. Animals were measured for growth using the freehand line tool of the image analysis program ImageJ. The length of 14–69 individual animals was averaged for each measurement.
C. elegans development assay
C. elegans embryos were isolated following standard bleaching protocol using a 1:2 solution of 5 M NaOH:commercial bleach (5% solution of sodium hypochlorite).59 Embryos were allowed to hatch overnight at 20°C in NGM plates without bacterial food. Arrested L1 larvae (around 50 L1 larvae/plate) were transferred to E. coli (OP50) seeded NGM plates with or without edelfosine. After 2.7 d of development at 20°C, worms were examined and the percentage of adults was determined. Adults were recognized by the appearance of a mature vulva and by the presence of eggs in the gonad.
C. elegans progeny determination
Total progeny number was determined by counting the cumulative number of progeny from 5 single hermaphrodites before they reach adulthood, on a daily basis during 4 d at 20°C, point at which hermaphrodites exhausted their sperm and started laying unfertilized oocytes.
C. elegans lifespan assay
Synchronized L4 larvae were transferred to E. coli (OP50) seeded NGM plates with or without drug supplementation. Worms were transferred to fresh plates (to avoid progeny contamination) and scored for dead individuals every other day. To determine whether a worm was dead, we prodded the head of the worm using a platinum wire. Worms that did not respond to prodding were considered dead. Lifespan analyses were conducted at 20°C. Age refers to days following adulthood, and P values were calculated using the log-rank (Mantel-Cox) method. Individuals were excluded from the analysis when their gonad was extruded, or when they desiccated by crawling onto the edge of the housing plate.
C. elegans larval viability assay
L1 larvae were maintained in non-nutritive M9 buffer with or without different concentrations of edelfosine and assessed for viability 5 d after hatching. To determine viability, sequential images of the culture wells containing the worms were captured with a CCD camera 10 seconds apart, and any animal that moved between frames was scored as live. Animals that were motionless between frames were scored as dead. The assay was conducted at 20°C.
C. elegans embryonic lethality assay
Synchronized worms (larvae or adults, depending on the experiment) were transferred to E. coli (OP50) seeded NGM plates, which had been supplemented with the respective ATL. Worms were allowed to lay embryos on the plate for 16–24 hours (in the case of some mutant strains that develop slowly and/or lay embryos at a slower pace, this time was extended up to 48 hours). At this point worms were eliminated from the plates and embryos were allowed to complete embryogenesis and hatch into larvae. In the “ex-utero” embryonic lethality assay, synchronized gravid adult hermaphrodites were allowed to lay embryos for 30 minutes, then 30–50 embryos were transferred to triplicate NGM plates with or without edelfosine. The percentage of embryonic lethality upon ATL treatment was calculated as the percentage of embryos unable to hatch into larvae.
C. elegans 4D Microscopy
Embryos for 4-dimensional microscopy analysis were mounted on a Leica DM6000 microscope equipped with DIC optics as described.60 The 4D microscopy analyses were carried out by recording 30 focal planes of the embryos for 17 hours. As a result, 4-dimensional (4D) movies (3D of the embryo + time) were obtained for each experiment. The SIMI Biocell software (SIMI GmbH) allowed tracing of the cell lineage of the embryos in time and space as well as tracing of the mitoses and apoptoses, as described.61
Cell culture
The human mantle cell lymphoma (MCL) cell line Z-138 was grown in RPMI-1640 culture medium supplemented with 10% heat-inactivated fetal bovine serum, 2 mM L-glutamine, 100 U/ml of penicillin and 100 μg/ml streptomycin at 37°C in humidified 95% air and 5% CO2.
Cellular cholesterol depletion
For cholesterol depletion, 7 × 105 cells were washed in serum free medium 3 times, pretreated with 1 mg/ml or 2.5 mg/ml methyl-β-cyclodextrin for 30 minutes at 37°C in serum-free medium as previously described.62 Cells were then washed 3 times with PBS and resuspended in complete culture medium before edelfosine addition.
Western blot
Cells (4–5 × 106) were lysed with 60 ml lysis buffer (200 mM HEPES, pH 7.5, 10 mM EGTA, 40 mM β-glycerophosphate, 1% (v/v) NP-40, 25 mM MgCl2, 2 mM sodium orthovanadate, 1 mM DTT) supplemented with protease inhibitors (1 mM phenylmethanesulfonyl fluoride, 20 μg/ml aprotinin and 20 μg/ml leupeptin). Proteins (40 μg) were run on SDS-polyacrylamide gels under reducing conditions, transferred to polyvinylidene fluoride membranes (Millipore, Billerica, MA, USA), blocked with 5% (w/v) defatted milk powder in TBST (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 0.1% (v/v) Tween 20) for 1 h at room temperature, and incubated for 1 h at room temperature or overnight at 4°C with specific antibodies: anti-60 kDa Ser473 p-Akt; (Cell Signaling; 1:1000 dilution in TBST with 5% BSA) and anti-60 kDa Akt1/2/3 (Santa Cruz Biotechnology; 1:1000 in TBST with 5% BSA). Antibody reactivity was monitored with horseradish peroxidase-conjugated anti-rabbit or anti-mouse IgG, using an enhanced chemiluminescence detection kit (GE Healthcare, Little Chalfont, UK).
Apoptosis assay
Quantification of apoptotic cells was calculated by flow cytometry as the percentage of cells in the sub-G0/G1 region (hypodiploidy) in cell cycle analysis, as previously described.63
Cytofluorimetric analysis of mitochondrial transmembrane potential and generation of reactive oxygen species
To evaluate mitochondrial transmembrane potential (ΔΨm) disruption and the generation of reactive oxygen species, cells (106/ml) were incubated in phosphate-buffered saline (PBS) with 20 nM 3,3′-dihexyloxacarbocyanine iodide [DiOC6(3)] (green fluorescence) (Molecular Probes, Leiden, The Netherlands) and 2 μM dihydroethidine (red fluorescence after oxidation) (Sigma, St Louis, MO, USA) for 40 min at 37°C, followed by analysis on a FACSCalibur flow cytometer, as previously described.63
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We are thankful to Mayte Montero, Xiao Xu, Alvaro Cuesta-Marbán, Marina Holgado-Madruga, and the members of the Mollinedo lab for critical reading of the manuscript; and to Sara Lima for technical assistance. All the strains were provided by the Caenorhabditis Genetics Center (University of Minnesota, USA), which is funded by NIH Office of Research Infrastructure Programs (P40 OD010440).
Funding
This work was supported by grants from the Spanish Ministerio de Ciencia e Innovación (SAF2011–30518), Spanish Ministerio de Economía y Competitividad (RD12/0036/0065, Red Temática de Investigación Cooperativa en Cáncer, Instituto de Salud Carlos III, cofunded by the Fondo Europeo de Desarrollo Regional of the European Union), European Community's Seventh Framework Program FP7-2007-2013 (grant HEALTH-F2–2011–256986, PANACREAS), and Junta de Castilla y León (CSI052A11–2). AS-B was supported by the CSIC JAE-Doc program.
Supplemental Materials
Supplemental materials for this article can be found on the publisher's website.
References
- 1. Mollinedo F, Fernández-Luna JL, Gajate C, Martín-Martín B, Benito A, Martínez-Dalmau R, Modolell M. Selective induction of apoptosis in cancer cells by the ether lipid ET-18-OCH3 (Edelfosine): molecular structure requirements, cellular uptake, and protection by Bcl-2 and Bcl-X(L). Cancer Res 1997; 57:1320-8; PMID:9102220 [PubMed] [Google Scholar]
- 2. Mollinedo F, Gajate C, Martín-Santamaría S, Gago F. ET-18-OCH3 (edelfosine): a selective antitumour lipid targeting apoptosis through intracellular activation of Fas/CD95 death receptor. Curr Med Chem 2004; 11:3163-84; PMID:15579006; http://dx.doi.org/ 10.2174/0929867043363703 [DOI] [PubMed] [Google Scholar]
- 3. Gajate C, Mollinedo F. Lipid rafts, endoplasmic reticulum and mitochondria in the antitumor action of the alkylphospholipid analog edelfosine. Anticancer Agents Med Chem 2014; 14:509-27; PMID:24628241; http://dx.doi.org/ 10.2174/1871520614666140309222259 [DOI] [PubMed] [Google Scholar]
- 4. Gajate C, Mollinedo F. Biological activities, mechanisms of action and biomedical prospect of the antitumor ether phospholipid ET-18-OCH3 (edelfosine), a proapoptotic agent in tumor cells. Curr. Drug Metab 2002; 3:491-525; PMID:12369895; http://dx.doi.org/ 10.2174/1389200023337225 [DOI] [PubMed] [Google Scholar]
- 5. Jendrossek V, Handrick R. Membrane targeted anticancer drugs: potent inducers of apoptosis and putative radiosensitisers. Curr Med Chem Anticancer Agents 2003; 3:343-53; PMID:12871080; http://dx.doi.org/ 10.2174/1568011033482341 [DOI] [PubMed] [Google Scholar]
- 6. Gajate C, Canto-Jañez E. del Acuña AU, Amat-Guerri F, Geijo E, Santos-Beneit AM, Veldman RJ, Mollinedo F. Intracellular triggering of Fas aggregation and recruitment of apoptotic molecules into Fas-enriched rafts in selective tumor cell apoptosis. J Exp Med 2004; 200:353-65; PMID:15289504; http://dx.doi.org/ 10.1084/jem.20040213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gajate C, Mollinedo F. Edelfosine and perifosine induce selective apoptosis in multiple myeloma by recruitment of death receptors and downstream signaling molecules into lipid rafts. Blood 2007; 109:711-9; PMID:17003375; http://dx.doi.org/ 10.1182/blood-2006-04-016824 [DOI] [PubMed] [Google Scholar]
- 8. Gajate C, Fonteriz RI, Cabaner C, Alvarez-Noves G, Alvarez-Rodriguez Y, Modolell M., Mollinedo F. Intracellular triggering of Fas, independently of FasL, as a new mechanism of antitumor ether lipid-induced apoptosis. Int J Cancer 2000; 85:674-82; PMID:10699948; http://dx.doi.org/ 10.1002/(SICI)1097-0215(20000301)85:5<674::AID-IJC13>3.0.CO;2-Z [DOI] [PubMed] [Google Scholar]
- 9. Ausili A, Torrecillas A, Aranda FJ, Mollinedo F, Gajate C, Corbalán-García S, de Godos A, Gómez-Fernández JC. Edelfosine is incorporated into rafts and alters their organization. J Phys Chem B 2008; 112:11643-54; PMID:18712919; http://dx.doi.org/ 10.1021/jp802165n [DOI] [PubMed] [Google Scholar]
- 10. Mollinedo F, de la Iglesia-Vicente J, Gajate C, Estella-Hermoso de Mendoza A, Villa-Pulgarin JA, Campanero MA, Blanco-Prieto MJ. Lipid raft-targeted therapy in multiple myeloma. Oncogene 2010; 29:3748-57; PMID:20418917; http://dx.doi.org/ 10.1038/onc.2010.131 [DOI] [PubMed] [Google Scholar]
- 11. Gajate C, Mollinedo F. The antitumor ether lipid ET-18-OCH3 induces apoptosis through translocation and capping of Fas/CD95 into membrane rafts in human leukemic cells. Blood 2001; 98:3860-3; PMID:11739199; http://dx.doi.org/ 10.1182/blood.V98.13.3860 [DOI] [PubMed] [Google Scholar]
- 12. Gajate C, Gonzalez-Camacho F, Mollinedo F. Involvement of raft aggregates enriched in Fas/CD95 death-inducing signaling complex in the antileukemic action of edelfosine in Jurkat cells. PLoS One 2009; 4:e5044; PMID:19352436; http://dx.doi.org/ 10.1371/journal.pone.0005044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mollinedo F, Gajate C. Fas/CD95 death receptor and lipid rafts: new targets for apoptosis-directed cancer therapy. Drug Resist Updat 2006; 9:51-73; PMID:16687251; http://dx.doi.org/ 10.1016/j.drup.2006.04.002 [DOI] [PubMed] [Google Scholar]
- 14. Mollinedo F, Gajate C. Lipid rafts and clusters of apoptotic signaling molecule-enriched rafts in cancer therapy. Future Oncol 2010; 6:811-21; PMID:20465392; http://dx.doi.org/ 10.2217/fon.10.34 [DOI] [PubMed] [Google Scholar]
- 15. Reis-Sobreiro M, Roué G, Moros A, Gajate C, de la Iglesia-Vicente J, Colomer D, Mollinedo F. Lipid raft-mediated Akt signaling as a therapeutic target in mantle cell lymphoma. Blood Cancer J 2013; 3:e118; PMID:23727661; http://dx.doi.org/ 10.1038/bcj.2013.15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nieto-Miguel T, Gajate C, González-Camacho F, Mollinedo F. Proapoptotic role of Hsp90 by its interaction with c-Jun N-terminal kinase in lipid rafts in edelfosine-mediated antileukemic therapy. Oncogene 2008; 27:1779-87; PMID:17891170; http://dx.doi.org/ 10.1038/sj.onc.1210816 [DOI] [PubMed] [Google Scholar]
- 17. Gajate C, Matos-da-Silva M, Dakir el-H, Fonteriz RI, Alvarez J, Mollinedo F. Antitumor alkyl-lysophospholipid analog edelfosine induces apoptosis in pancreatic cancer by targeting endoplasmic reticulum. Oncogene 2012; 31:2627-39; PMID:22056873; http://dx.doi.org/ 10.1038/onc.2011.446 [DOI] [PubMed] [Google Scholar]
- 18. Nieto-Miguel T, Fonteriz RI, Vay L, Gajate C, López-Hernández S, Mollinedo F. Endoplasmic reticulum stress in the proapoptotic action of edelfosine in solid tumor cells. Cancer Res 2007; 67:10368-78; PMID:17974980; http://dx.doi.org/ 10.1158/0008-5472.CAN-07-0278 [DOI] [PubMed] [Google Scholar]
- 19. Nieto-Miguel T, Gajate C, Mollinedo F. Differential targets and subcellular localization of antitumor alkyl-lysophospholipid in leukemic versus solid tumor cells. J Biol Chem 2006; 281:14833-40; PMID:16540473; http://dx.doi.org/ 10.1074/jbc.M511251200 [DOI] [PubMed] [Google Scholar]
- 20. Boggs KP, Rock CO, Jackowski S. Lysophosphatidylcholine and 1-O-octadecyl-2-O-methyl-rac-glycero-3-phosphocholine inhibit the CDP-choline pathway of phosphatidylcholine synthesis at the CTP:phosphocholine cytidylyltransferase step. J Biol Chem 1995; 270:7757-64; PMID:7706325; http://dx.doi.org/ 10.1074/jbc.270.19.11612 [DOI] [PubMed] [Google Scholar]
- 21. Baburina I, Jackowski S. Apoptosis triggered by 1-O-octadecyl-2-O-methyl-rac-glycero-3-phosphocholine is prevented by increased expression of CTP:phosphocholine cytidylyltransferase. J Biol Chem 1998; 273:2169-73; PMID:9442058; http://dx.doi.org/ 10.1074/jbc.273.4.2169 [DOI] [PubMed] [Google Scholar]
- 22. Van der Luit AH, Budde M, Ruurs P, Verheij M, van Blitterswijk WJ. Alkyl-lysophospholipid accumulates in lipid rafts and induces apoptosis via raft-dependent endocytosis and inhibition of phosphatidylcholine synthesis. J Biol Chem 2002; 277:39541-47; PMID:12183451; http://dx.doi.org/ 10.1074/jbc.M203176200 [DOI] [PubMed] [Google Scholar]
- 23. Unger C, Peukert M, Sindermann H, Hilgard P, Nagel G, Eibl H. Hexadecylphosphocholine in the topical treatment of skin metastases in breast cancer patients. Cancer Treat Rev 1990; 17:243-46; PMID:2272039; http://dx.doi.org/ 10.1016/0305-7372(90)90054-J [DOI] [PubMed] [Google Scholar]
- 24. Leonard R, Hardy J, van Tienhoven G, Houston S, Simmonds P, David M, Mansi J. Randomized, double-blind, placebo-controlled, multicenter trial of 6% miltefosine solution, a topical chemotherapy in cutaneous metastases from breast cancer. J Clin Oncol 2001; 19:4150-9; PMID:11689583 [DOI] [PubMed] [Google Scholar]
- 25. Terwogt JM, Mandjes IA, Sindermann H, Beijnen JH, Ten Bokkel Huinink WW. Phase II trial of topically applied miltefosine solution in patients with skin-metastasized breast cancer. Br J Cancer 1999; 79:1158-61; PMID:10098751; http://dx.doi.org/ 10.1038/sj.bjc.6690184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dummer R, Krasovec M, Röger J, Sindermann H, Burg G. Topical administration of hexadecylphosphocholine in patients with cutaneous lymphomas: results of a phase I/II study. J Am Acad Dermatol 1993; 29:963-70; PMID:8245262; http://dx.doi.org/ 10.1016/0190-9622(93)70275-X [DOI] [PubMed] [Google Scholar]
- 27. Mollinedo F. Antitumour ether lipids: proapoptotic agents with multiple therapeutic indications. Expert Opin Ther Patents 2007; 17:385-405; http://dx.doi.org/ 10.1517/13543776.17.4.385 [DOI] [Google Scholar]
- 28. Fensterle J, Aicher B, Seipelt I, Teifel M, Engel J. Current view on the mechanism of action of perifosine in cancer. Anticancer Agents Med Chem 2014; 14:629-35; PMID:24628236; http://dx.doi.org/ 10.2174/1871520614666140309225912 [DOI] [PubMed] [Google Scholar]
- 29. Van Blitterswijk WJ, Verheij M. Anticancer mechanisms and clinical application of alkylphospholipids. Biochim Biophys Acta 2013; 1831:663-74; PMID:23137567; http://dx.doi.org/ 10.1016/j.bbalip.2012.10.008 [DOI] [PubMed] [Google Scholar]
- 30. Verheij M, Moolenaar WH, Blitterswijk van WJ. Combining anti-tumor alkyl-phospholipid analogs and radiotherapy: rationale and clinical outlook. Anticancer Agents Med Chem 2014; 14:618-28; PMID:24628238; http://dx.doi.org/ 10.2174/1871520614666140309224145 [DOI] [PubMed] [Google Scholar]
- 31. Cuesta-Marbán Á, Botet J, Czyz O, Cacharro LM, Gajate C, Hornillos V, Delgado J, Zhang H, Amat-Guerri F, Acuña AU, et al. Drug uptake, lipid rafts, and vesicle trafficking modulate resistance to an anticancer lysophosphatidylcholine analogue in yeast. J Biol Chem 2013; 288:8405-18; http://dx.doi.org/ 10.1074/jbc.M112.425769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Czyz O, Bitew T, Cuesta-Marbán A, McMaster CR, Mollinedo F, Zaremberg V. Alteration of plasma membrane organization by an anticancer lysophosphatidylcholine analogue induces intracellular acidification and internalization of plasma membrane transporters in yeast. J Biol Chem 2013; 288:8419-32; PMID:23344949; http://dx.doi.org/ 10.1074/jbc.M112.425744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sulston JE, Schierenberg E, White JG, Thomson JN. The embryonic cell lineage of the nematode Caenorhabditis elegans. Dev Biol 1983; 100:64-119; PMID:6684600; http://dx.doi.org/ 10.1016/0012-1606(83)90201-4 [DOI] [PubMed] [Google Scholar]
- 34. Kirienko NV, Mani K, Fay DS. Cancer models in Caenorhabditis elegans. Dev Dyn 2010; 239:1413-48; PMID:20175192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Shaw R, Dillin A. PPTR-1 counterAkts insulin signaling. Cell 2009; 136:816-8; PMID:19269361; http://dx.doi.org/ 10.1016/j.cell.2009.02.029 [DOI] [PubMed] [Google Scholar]
- 36. Lettre G, Hengartner MO. Developmental apoptosis in C. elegans: a complex CEDnario. Nat Rev Mol Cell Biol 2006; 7:97-108; PMID:16493416; http://dx.doi.org/ 10.1038/nrm1836 [DOI] [PubMed] [Google Scholar]
- 37. Sternberg PW, Han M. Genetics of RAS signaling in C. elegans. Trends Genet 1998; 14:466-72; PMID:9825675; http://dx.doi.org/ 10.1016/S0168-9525(98)01592-3 [DOI] [PubMed] [Google Scholar]
- 38. Poulin G, Nandakumar R, Ahringer J. Genome-wide RNAi screens in Caenorhabditis elegans: impact on cancer research. Oncogene 2004; 23:8340-5; PMID:15517014; http://dx.doi.org/ 10.1038/sj.onc.1208010 [DOI] [PubMed] [Google Scholar]
- 39. Greenwald I. LIN-12/Notch signaling: lessons from worms and flies. Genes Dev 1998; 12:1751-62; PMID:9637676; http://dx.doi.org/ 10.1101/gad.12.12.1751 [DOI] [PubMed] [Google Scholar]
- 40. Mollinedo F, de la Iglesia-Vicente J, Gajate C, Estella-Hermoso de Mendoza A, Villa-Pulgarin JA, de Frias M, Roué G, Gil J, Colomer D, Campanero MA, et al. In vitro and in vivo selective antitumor activity of Edelfosine against mantle cell lymphoma and chronic lymphocytic leukemia involving lipid rafts. Clin Cancer Res 2010; 16:2046-54; PMID:20233887; http://dx.doi.org/ 10.1158/1078-0432.CCR-09-2456 [DOI] [PubMed] [Google Scholar]
- 41. Hughes SE, Evason K, Xiong C, Kornfeld K. Genetic and pharmacological factors that influence reproductive aging in nematodes. PLoS Genet 2007; 3:e25; PMID:17305431; http://dx.doi.org/ 10.1371/journal.pgen.0030025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Busto JV, Del Canto-Jañez E, Goñi FM, Mollinedo F, Alonso A. Combination of the anti-tumour cell ether lipid edelfosine with sterols abolishes haemolytic side effects of the drug. J Chem Biol 2008; 1:89-94; PMID:19568801; http://dx.doi.org/ 10.1007/s12154-008-0009-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kurzchalia TV, Ward S. Why do worms need cholesterol? Nat Cell Biol 2003; 5:684-688; PMID:12894170; http://dx.doi.org/ 10.1038/ncb0803-684 [DOI] [PubMed] [Google Scholar]
- 44. Hieb WF, Rothstein M. Sterol requirement for reproduction of a free-living nematode. Science 1968; 160:778-80; PMID:4869093; http://dx.doi.org/ 10.1126/science.160.3829.778 [DOI] [PubMed] [Google Scholar]
- 45. Brenner S. The genetics of Caenorhabditis elegans. Genetics 1974; 77:71-94; PMID:4366476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Matyash V, Geier C, Henske A, Mukherjee S, Hirsh D, Thiele C, Grant B, Maxfield FR, Kurzchalia TV. Distribution and transport of cholesterol in Caenorhabditis elegans. Mol Biol Cell 2001; 12:1725-36; PMID:11408580; http://dx.doi.org/ 10.1091/mbc.12.6.1725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Frand AR, Russel S, Ruvkun G. Functional genomic analysis of C. elegans molting. PLoS Biol 2005; 3:e312; PMID:16122351; http://dx.doi.org/ 10.1371/journal.pbio.0030312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Morley JF, Morimoto RI. Regulation of longevity in Caenorhabditis elegans by heat shock factor and molecular chaperones. Mol Biol Cell 2004; 15:657-64; PMID:14668486; http://dx.doi.org/ 10.1091/mbc.E03-07-0532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Rao W, Isaac RE, Keen JN. An analysis of the Caenorhabditis elegans lipid raft proteome using geLC-MS/MS. J Proteomics 2011; 74:242-53; PMID:21070894; http://dx.doi.org/ 10.1016/j.jprot.2010.11.001 [DOI] [PubMed] [Google Scholar]
- 50. Paradis S, Ailion M, Toker A, Thomas JH, Ruvkun G. A PDK1 homolog is necessary and sufficient to transduce AGE-1 PI3 kinase signals that regulate diapause in Caenorhabditis elegans. Genes Dev 1999; 13:1438-52; PMID:10364160; http://dx.doi.org/ 10.1101/gad.13.11.1438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Paradis S, Ruvkun G. Caenorhabditis elegans Akt/PKB transduces insulin receptor-like signals from AGE-1 PI3 kinase to the DAF-16 transcription factor. Genes Dev 1998; 12:2488-98; PMID:9716402; http://dx.doi.org/ 10.1101/gad.12.16.2488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hu PJ, Xu J, Ruvkun G. Two membrane-associated tyrosine phosphatase homologs potentiate C. elegans AKT-1/PKB signaling. PLoS Genet 2006; 2:e99; PMID:16839187; http://dx.doi.org/ 10.1371/journal.pgen.0020099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Chen C-S, Bellier A, Kao C-Y, Yang Y-L, Chen H-D, Los FCO, Aroian RV. WWP-1 is a novel modulator of the DAF-2 insulin-like signaling network involved in pore-forming toxin cellular defenses in Caenorhabditis elegans. PLoS One 2010; 5:e9494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Grant B, Hirsh D. Receptor-mediated endocytosis in the Caenorhabditis elegans oocyte. Mol Biol Cell 1999; 10:4311-4326; PMID:10588660; http://dx.doi.org/ 10.1091/mbc.10.12.4311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Yates AJ, Thompson DK, Boesel CP, Albrightson C, Hart RW. Lipid composition of human neural tumors. J Lipid Res 1979; 20:428-36; PMID:458265 [PubMed] [Google Scholar]
- 56. Hac-Wydro K, Dynarowicz-Łatka P. The relationship between the concentration of ganglioside GM1 and antitumor activity of edelfosine-the Langmuir monolayer study. Colloids Surf B Biointerfaces 2010; 81:385-8; PMID:20692132; http://dx.doi.org/ 10.1016/j.colsurfb.2010.07.026 [DOI] [PubMed] [Google Scholar]
- 57. Gaiser AM, Kaiser CJO, Haslbeck V, Richter K. Downregulation of the Hsp90 system causes defects in muscle cells of Caenorhabditis elegans. PLoS One 2011; 6:e25485; PMID:21980476; http://dx.doi.org/ 10.1371/journal.pone.0025485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Nieto-Miguel T, Gajate C, González-Camacho F, Mollinedo F. Proapoptotic role of Hsp90 by its interaction with c-Jun N-terminal kinase in lipid rafts in edelfosine-mediated antileukemic therapy. Oncogene 2008; 27:1779-87; PMID:17891170; http://dx.doi.org/ 10.1038/sj.onc.1210816 [DOI] [PubMed] [Google Scholar]
- 59. Stiernagle T. Maintenance of C. elegans. WormBook 2006; 11:1-11; PMID:18050451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Schnabel R, Hutter H, Moerman D, Schnabel H. Assessing normal embryogenesis in Caenorhabditis elegans using a 4D microscope: variability of development and regional specification. Dev Biol 1997; 184:234-65; PMID:9133433; http://dx.doi.org/ 10.1006/dbio.1997.8509 [DOI] [PubMed] [Google Scholar]
- 61. Nieto C, Almendinger J, Gysi S, Gómez-Orte E, Kaech A, Hengartner MO, Schnabel R, Moreno S, Cabello J. ccz-1 mediates the digestion of apoptotic corpses in C. elegans. J Cell Sci 2010; 123:2001-7; PMID:20519582; http://dx.doi.org/ 10.1242/jcs.062331 [DOI] [PubMed] [Google Scholar]
- 62. Gajate C, Gonzalez-Camacho F, Mollinedo F. Lipid raft connection between extrinsic and intrinsic apoptotic pathways. Biochem Biophys Res Commun 2009; 380:780-4; PMID:19338752; http://dx.doi.org/ 10.1016/j.bbrc.2009.01.147 [DOI] [PubMed] [Google Scholar]
- 63. Gajate C, Santos-Beneit AM, Macho A, Lazaro MD, Hernandez-De Rojas A, Modolell M, Muñoz E, Mollinedo F. Involvement of mitochondria and caspase-3 in ET-18-OCH3-induced apoptosis of human leukemic cells. Int J Cancer 2000; 86:208-18; PMID:10738248; http://dx.doi.org/ 10.1002/(SICI)1097-0215(20000415)86:2<208::AID-IJC10>3.0.CO;2-E [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.