

Abstract
Up to 80% of human cancers, in particular solid tumors, contain cells with abnormal chromosomal numbers, or aneuploidy, which is often linked with marked chromosomal instability. Whereas in some tumors the aneuploidy occurs by missegregation of one or a few chromosomes, aneuploidy can also arise during proliferation of inherently unstable tetraploid cells generated by whole genome doubling from diploid cells. Recent findings from cancer genome sequencing projects suggest that nearly 40% of tumors underwent whole genome doubling at some point of tumorigenesis, yet its contribution to cancer phenotypes and benefits for malignant growth remain unclear. Here, we investigated the consequences of a whole genome doubling in both cancerous and non-transformed p53 positive human cells. SNP array analysis and multicolor karyotyping revealed that induced whole-genome doubling led to variable aneuploidy. We found that chromosomal instability (CIN) is a frequent, but not a default outcome of whole genome doubling. The CIN phenotypes were accompanied by increased tolerance to mitotic errors that was mediated by suppression of the p53 signaling. Additionally, the expression of pro-apoptotic factors, such as iASPP and cIAP2, was downregulated. Furthermore, we found that whole genome doubling promotes resistance to a broad spectrum of chemotherapeutic drugs and stimulates anchorage-independent growth even in non-transformed p53-positive human cells. Taken together, whole genome doubling provides multifaceted benefits for malignant growth. Our findings provide new insight why genome-doubling promotes tumorigenesis and correlates with poor survival in cancer.
Keywords: aneuploidy, cancer, CIN, drug resistance, p53, tetraploidy, whole genome doubling
Introduction
Many malignant tumors contain cells with aberrant chromosome numbers. These changes vary from aneuploidy (imbalanced chromosome numbers) of one or multiple chromosomes, to numbers approaching triploidy or tetraploidy.1 Aneuploidy is often accompanied by chromosomal instability (CIN), which manifests as ongoing gains and losses of chromosomes during mitosis2 CIN contributes to tumor heterogeneity and is associated with increased resistance to drug treatment and poor patient prognosis.3 Studies revealed that CIN might be triggered by mutations in genes that control chromosome segregation.4-6 Whole-genome doubling that leads to tetraploidy provides another route by which aneuploidy can arise in tumors independently of mutations in mitotic genes.7,8 In this model, tetraploid cells formed by cytokinesis failure, endoreduplication or cell-cell fusion divide in a highly unstable manner due to supernumerary centrosomes and increased chromosome numbers. The compromised maintenance of genomic stability of tetraploids facilitates CIN and tumorigenesis. Indeed, injection of p53-deficient tetraploid cells into nude mice triggers tumor formation, whereas isogenic diploid cells show no effect.9 Similarly, tetraploids arising from mouse ovarian surface embryonic cells develop aneuploidy, CIN and promote tumorigenesis when injected into mice.10
Remarkably, tetraploid cells were found in early stages of several tumors and in total tetraploidy was documented in 37% of cancers.11 This frequent occurrence suggests that passage through tetraploidy provides advantages that facilitate tumor growth. However, experimentally generated tetraploid cells often fail to propagate, as these cells arrest in a p53-dependent manner immediately after whole genome doubling or after the first tetraploid mitosis, which is often severely erroneous.9,12-15 This further impairs the proliferation after tetraploidization, as chromosome missegregation of even a single chromosome triggers a p53-dependent arrest.14,16,17 Also, aneuploid cells arising due to chromosome missegregation suffer from proliferation delays and other physiological defects.18,19 So far, only little is known how human cells survive tetraploidy and what the long-term effects of whole genome doubling are. Previous data suggest that tetraploidy increases CIN to generate karyotypic heterogeneity, while also providing tolerance to potentially deleterious genetic changes.20,21 However, little experimental data is available to document the long-term effects of whole genome doubling in human cells.
Results
Whole genome doubling triggers aneuploidy and chromosome instability
To determine the long-term consequences of tetraploidization in human cells, we analyzed the fate of tetraploids generated from 2 p53-positive cell lines, HCT116 and hTERT-RPE1. HCT116 is a near-diploid cell line derived from human hereditary non-polyposis colon cancer and is chromosomally stable with characteristic microsatellite instability.2,22 hTERT-RPE1 (hereafter RPE1) is a chromosomally stable diploid retinal pigment epithelium cell line immortalized by constitutive expression of human telomerase. Both cell lines stably express H2B-GFP for visualization of chromatin by fluorescence microscopy. Tetraploid cells were generated by inhibition of the actomyosin ring with the actin depolymerizing drug cytochalasin D (DCD), which lead to cytokinesis failure in approximately 60% of cells.9,14 600 DCD-treated cells were plated by limiting dilution into 96-well plates and allowed to expand for 6 weeks (Fig. 1A). All surviving cell populations were analyzed by flow cytometry. This analysis revealed near-tetraploid DNA content in 8 out of the 64 surviving HCT116-derived cell lines and in 7 out of 58 RPE1-derived cell lines (Fig. S1A), hereafter referred to as PostTetraploid (PT) cell lines (HPT – derived from HCT116, RPT – derived from RPE1). To minimize the effect of further evolution, all PTs underwent minimal passages before the analysis and were compared at the same “early” passage, unless otherwise stated. The posttetraploid cell lines showed only a mild proliferation delay and the duration of mitosis as well as the robustness of mitotic checkpoint activation were comparable to that of controls (Fig. S1B-G).
Figure 1.

Posttetraploid progenies (PTs) are chromosomally unstable. (A) Schematic depiction of the experimental strategy. (B, C) SNP array profiles of HCT116, RPE1 and their posttetraploid derivatives. Copy numbers are indicated by colors. The log R represents the copy number; B-allele frequency (BAF) indicates the allele composition: BAF of 0 or 1 represents genotype of AA / A- / BB / B-, respectively; BAF of 0.5 represents AB. (D) Multicolor FISH karyotyping of 2 cells from the HPT2 cell line (number of chromosomes was 72 and 79, respectively). Note the difference in copy number of chromosomes 1, 3, 4, 6, 7, 8, 9, 10, 11, 12, 13, 15, 16, 17, 18 and 20.
To determine the copy number changes after tetraploidization, we performed SNP array analysis in 6 HPTs, 3 RPTs cell lines and the respective parental cells. Recurrent chromosome copy number changes were observed in 3 out of 9 analyzed PTs (HPT1, HPT6 and RPT3; Fig. 1B, C, and Fig. S2A, B). SNP arrays analyze a pool of cells, which only allows detection of common changes that are shared by a majority of cells. To detect chromosomal changes in individual cells, we used multicolor fluorescence in situ hybridization (mFISH) karyotyping of HCT116, HPT1 and HPT2 cell lines. This analysis revealed heterogeneity within the PT populations, but not in parental HCT116: all 12 clonal cell lines originating from individual HCT116 cells remained chromosomally stable even after 80 generations, thus excluding that the emergence of karyotypic diversity is a consequence of clonal outgrowth of cells with preexisting karyotypic heterogeneity or an intrinsic characteristic of the parental line (Fig. 1D, and Fig. S2C, S3A-E). Moreover, the PTs also contained more chromosomal rearrangements than the diploids, although the difference was not statistically significant (Fig. S3A-E).
Since all PTs arose from a single cell, we hypothesized that the karyotypic heterogeneity indicates chromosomal instability arising after tetraploidization. We used fluorescence in situ hybridization (FISH) utilizing the chromosome enumeration probes to compare “early” karyotypes with karyotypes after additional 36 passages. The distribution of the chromosome copy numbers remained nearly identical in early and late HCT116 cells, whereas in HPT1 and HPT2 the numbers of chromosomes differed markedly (Fig. 2A and Fig. S4A). Chromosomal instability was also identified in RPT3 after 12 passages; in contrast, RPT1 cell line did not show significant changes in the FISH signal (Fig. S4B). Additionally, FISH analysis revealed a loss of the signal of chromosome 7 in 2 out of 4 analyzed posttetraploid cell lines (HPT1 and RPT3) that was present in both early and late passages. Changes in the number or structure of chromosome 7 are common in human cancers: trisomy of chromosome 7 is among the most frequently observed aberrations in cancers of the large intestine, while a loss of part or all of one copy of chromosome 7 is common in leukemia and lymphoma.23 Taken together, transient tetraploidization can generate aneuploid and chromosomally unstable progeny even in non-transformed p53-proficient parental cell lines.
Figure 2.
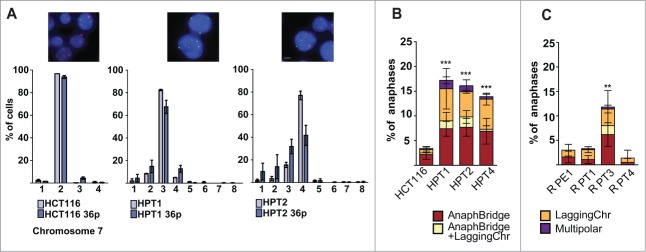
Posttetraploid cells display chromosomal instability and an increased frequency of abnormal mitosis. (A) Fluorescence in situ hybridization (FISH) against centromeric regions of HCT116 and HCT116-derived PTs. Comparison of chromosome number distribution for chromosome 7 in early passages and 36 passages later; mean and SEM of 2 independent FISH experiments. Chromosome 7 – red, chromosome 1- green, DNA was counterstained with DAPI (objective 63x, bar: 10 μm). Percentage of abnormal mitoses evaluated in fixed images of HCT116 (B) and RPE1 (C) and their respective posttetraploid derivatives; mean and SD of 3 independent experiments. AnaphBridge – cells that contain an anaphase bridge; LaggingChr – cells containing a lagging chromosome; AnaphBridge-LaggingChr – cells containing both an anaphase bridge and a lagging chromosome; Multipolar – cells that underwent multipolar anaphase.
Mitotic errors frequently occur in posttetraploid cell lines
To further characterize the chromosomal instability in the posttetraploid progeny, we imaged fixed cells and found that 15.8%, 15.0% and 13.8% of anaphases displayed segregation aberrancies in HPT1, HPT2 and HPT4, respectively, whereas only 3.7% of HCT116 underwent erroneous anaphase (Fig. 2B). The frequency of both anaphase bridges as well as the presence of lagging or unattached chromosomes was increased, suggesting that the frequency of both pre-mitotic and mitotic errors was elevated. Among the 3 RPT cell lines, only one displayed increased frequency of abnormal mitoses: 11.6% in RPT3 in comparison to 3.0% in RPE1, 3.1% in RPT1 and 1.4% in RPT4 (Fig. 2C). The multipolar mitoses were rare in PTs; the vast majority of cells segregated their chromosomes in a bipolar manner (Fig. S5A, B). In addition, a detailed analysis by imaging fixed cells stained with centrosome- and centriole-recognizing antibodies revealed no significant increase in the numbers of centrosomes and centrioles in PTs compared to the respective parental cell lines (Fig. S5C-G). This indicates that multipolarity alone cannot explain the high frequency of chromosome segregation errors in PTs.
The frequency of chromosome segregation errors could be elevated simply as a consequence of the increased chromosome numbers under the assumption that the error frequency correlates to chromosome number. However, normalization of the frequency of mitotic errors to the median chromosome number of each cell line revealed that the frequency of abnormal mitoses increases more than expected according to chromosome number in CIN+ PTs (Fig. S4C). Specifically, the mitotic error frequency increased 3.7–4.3 fold (from 3.7% to 13.8–15.8%), whereas modal chromosome numbers increased 1.7–1.8 fold (from 44 to 75–78) in HPTs. Similarly, the increase in chromosome numbers was 1.7 fold in RPT3, whereas the increase in mitotic errors reached 3.9 fold. Thus, the high frequency of mitotic errors in CIN+ PTs does not result from a simple linear increase of mitotic errors due to the higher numbers of chromosomes. Taken together, whole genome doubling supports the emergence of a CIN+ phenotype even in p53-positive non-cancerous cells, but it is not a default consequence.
Posttetraploid cells show increased tolerance to mitotic errors
The efficient proliferation of PT cell lines despite their aneuploidy and CIN suggests that the cells became more tolerant to mitotic errors. We analyzed the fate of parental diploid and tetraploid cells, as well as PTs after abnormal mitosis by long-term live cell imaging. The analysis revealed that 34.2% of diploid and 54.1% of newly formed tetraploid HCT116 cells that missegregated chromosomes arrested for at least 48 h or died in the subsequent interphase (Fig. 3A). In contrast, only 11.0% of HPT1 and 9.8% of HPT2 cells arrested after chromosome missegregation (Fig. 3A, for fate analysis of individual cells see Fig. S6). Thus, whole genome doubling increases cell tolerance to errors in chromosome segregation.
Figure 3.
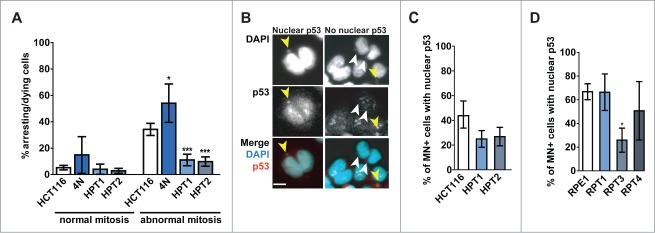
Posttetraploid cells are tolerant to mitotic errors. (A) Frequency of cell cycle arrest/cell death after bipolar mitosis with no apparent defects (normal mitosis) and with visible chromosome segregation defects (abnormal mitosis). Mean of 4 independent experiments and SD is plotted. Unpaired Student t-test was used to test for statistical significance. (B) Examples of p53 accumulation in the nuclei and micronuclei of the micronucleated cells. Yellow and white arrowheads indicate the micronuclei with and without p53 enrichment, respectively. p53-red, DNA was counterstained with DAPI, bar: 10 μm. (C and D) Accumulation of p53 in the nuclei of cells forming micronuclei (MN+) in HCT116, RPE1 and their respective posttetraploid derivatives (panels C, D, respectively). Mean of 4 independent experiments and SEM are plotted.
The p53 pathway is deregulated in posttetraploid cells
We hypothesized that the tolerance to abnormal mitosis in PTs is due to changes in activation of the p53 pathway. To test this, we performed a micronucleation assay which allows visualization of missegregated lagging chromosome as a micronucleus in the daughter cells, combined with immunostaining with an antibody against p53. Whereas nearly 42.0% of HCT116 cells with micronuclei accumulated nuclear p53, only 25.0% of HPT1 and 26.9% of HPT2 cells showed nuclear p53 accumulation when a micronucleus was present in the cell (Fig. 3B, C). A similar experiment in RPE1 and its posttetraploid derivatives revealed that 66.4% and 50.7% of RPT1 and RPT4 cells that underwent missegregation accumulated nuclear p53, which is similar to parental RPE1 (66.8%). In contrast, we observed that only 25.9% of RPT3 cells with a micronucleus accumulated p53 in the nucleus (Fig. 3C, D). Thus, whereas chromosomally stable PTs accumulated nuclear p53 as the parental cell line, the p53 signaling upon chromosome missegregation is attenuated in the CIN+ posttetraploids (compare Figs. 2C and 3B-D).
The activation of the p53 pathway upon chromosome missegregation occurs possibly via phosphorylation of p53 on serine 15 or by activation of the p38/MAPK pathway.14,16,17 Analysis of the p53 and p38 protein levels revealed no significant changes in parental and PT cell lines during unperturbed growth (Fig. S7A, B). Next, we induced chromosome missegregation by treatment with 20 μM VS83, an inhibitor of the kinesin Eg5 that results in the formation of mitotic cells with monopolar spindles. Subsequent wash out of VS83 ensures bipolar spindle formation and progress through mitosis; however, the mitosis is highly erroneous.16 The frequency and type of errors upon treatment with VS83 was similar in both posttetraploid and parental cell lines (Fig. S7C, D). Markedly, we observed that p53 was not stabilized in HPTs and in RPT3 upon VS83 treatment, whereas the levels of p53 increased in HCT116 and RPE1, RPT1 and RPT4 (Fig. 4A, C). This finding is in agreement with the diminished nuclear accumulation of p53 that we identified in the CIN+ clones (Fig. 3C, D).
Figure 4.

The p53 pathway is deregulated in posttetraploids. (A, B) Changes in abundance in p53, p38 and p21 and phosphorylation of p53 on Ser15 (p53-p) and phosphorylation of p38-p on Threonine180 and Tyrosine 182 (p38-p) in HCT116, RPE1 and respective posttetraploid cell lines with and without VS83 treatment. Four independent experiments were performed, an example of immunoblotting is shown; a Ponceau S stain was used as a loading control. (C, D) Quantification of the response to the missegregation triggered by release from VS83 treatment. The relative signal levels are presented as fold change of treated-to-untreated cells. Mean of at least 3 independent experiments with SD is shown, * marks statistically significant difference (P < 0.05). (E) Heat map of transcriptional fold changes of 91 significantly altered p53 interactors in posttetraploid cell lines (normalized to the respective parental cell lines).
Next, we analyzed transcriptome changes in 2 HPTs (HPT1 and HPT2) and 3 RPTs (RPT1, 3 and 4). We focused on 388 previously identified upstream and downstream interactors of p53 (see Material and Methods). Markedly, 23% of the analyzed genes were significantly deregulated with respect to the parental lines with a fold change of 1.5 in at least one of the HPTs or RPTs, but only 6 factors were upregulated in all PTs: LDHA (lactate dehydrogenese A), DGKA (diacylglycerol kinase), HIF1A (hypoxia induced factor 1), 2 inhibitors of apoptosis iASPP (inhibitory member of the ASPP family, encoded by PPP1R13L) and BIRC3 (Baculoviral IAP Repeat Containing 3), transcriptional factor ETS1 and transforming growth factor TGFA (Fig. 4E, and Table S1). Two genes were downregulated in all PTs: DUSP5, an inhibitor that negatively regulates members of the mitogen-activated protein (MAP) kinase superfamily (MAPK/ERK, SAPK/JNK, p38) and MST1 (macrophage signaling growth factor), a member of the MSP-RON signaling that plays a role in malignant invasive growth. We found 2 genes that were upregulated specifically in CIN+ cells: FOXO1 and NDRG1 that are both involved in response to oxidative and metabolic stress. Taken together, whole genome doubling promotes upregulation of factors promoting cell survival upon stress (HIF1, FOXO1) and alters expression of factors that inhibit apoptosis and MAP kinases (iASPP, BIRC3, DUSP5).
Posttetraploid cell lines acquire multidrug resistance and transform in vitro
To test whether whole genome doubling promotes increased resistance to cancer treatments, we compared the sensitivity of PTs with their respective parental cell lines to a broad range of anti-cancer agents. Effects of compounds on cell proliferation were determined using measurement of intracellular ATP content as an indirect readout of cell number.24 We profiled 17 different anti-cancer agents at a wide concentration range over 9 points from 32 μM to 0.32 nm on the 2 parental cell lines and in HPT1 and 2, RPT1, 3 and 4 in 3 independent experiments. The inhibitory potency of the compounds was expressed as pIC50 (-10logIC50) values. Comparison of the relative drug sensitivity of the parental cells with that of the PTs revealed a general multidrug resistant phenotype in all analyzed posttetraploid lines derived from the non-transformed hTERT-RPE1 cell line (Table S2, Fig. 5A) as well as in the 3 posttetraploid lines derived from the colon cancer line HCT116 (Table S2, Fig. 5B). All PTs showed significant resistance to the topoisomerase II inhibitors daunorubicin, doxorubicin and etoposide (Tables S2 and S3; Fig. 5A, B). In addition, RPTs were significantly resistant to the pyrimidine antagonist 5-fluoracil, the inhibitor of the p53-MDM2 interaction nutlin3a and the growth factor receptor kinase inhibitor pelitinib. HPT cell lines showed significant resistance to the DNA crosslinker cisplatin, the microtubule-targeting agents docetaxel and paclitaxel, and the inhibitor of histone deacytelases vorinostat (Tables S2 and S3). Interestingly, HPTs showed increased sensitivity to the purine antagonist 6-mercaptopurine (Fig. 5A). Thus, whole genome doubling provides a general protection against drug treatment in both non-transformed and cancer cells. This marked feature might explain why whole genome doubling correlates with a poor prognosis and resistance to therapy in some cancer types.21
Figure 5.

Posttetraploid cell lines are resistant to a broad spectrum of chemotherapeutic drugs and transform in vitro. (A) Dose-response curves of compounds showing different sensitivities in proliferation assays with the hTERT-RPE1 cell line and the posttetraploid cell lines RPT1, RPT3 and RPT4. (B) Dose-response curves of compounds showing different sensitivities in proliferation assays with the HCT116 cell line and the posttetraploid cell lines HPT1, HPT2 and HPT4. The posttetraploids lines are resistant to a broad spectrum of anti-cancerous drugs; except HPT1, 2 and 4 that are relatively more sensitive to 6-mercaptopurine. Fitted curve for 2 replicates from one or 2 independent experiments is plotted. Note that no fitted curve was determined for HPT1 upon etoposide treatment. See Material and Methods for details. (C) Phase contrast images of anchorage-independent colony growth in soft agar.
Finally, we determined the impact of tetraploidization on transformation capacity by assaying the anchorage independent growth of the posttetraploid progeny in soft agar (Fig. 5C). Since HCT116 is a cell line derived from colorectal cancer and therefore proficient in anchorage independent growth, we tested RPE1, a primary p53–positive immortalized cell line and its derivatives. The diploid RPE1 showed no anchorage-independent growth even after initiation/promotion treatment, where cells were exposed to the mutagen 7,12-dimethylbenz[a]anthracene (DMBA) alone or followed by exposure to the tumor promoter 12-O-tetradecanoylphorbol-13-acetate (TPA). In contrast, RPT cell lines efficiently formed colonies in soft agar even in absence of any treatment, indicating that the selected surviving populations became transformed in vitro (Fig. 5C). This result was observed in 2 independent experiments in all 3 posttetraploid cells lines derived from RPE1. Thus, whole genome doubling promotes in vitro transformation even in p53-positive non-cancerous cell lines.
Discussion
Whole genome doubling (WGD) is considered to contribute to eukaryotic evolution by facilitating adaptation while simultaneously buffering the possible effects of deleterious mutations.25 Recent findings support a similar role for WGD in fostering tumor genome evolution in mammalian cells, however, it remains unclear how exactly tetraploidy benefits malignant growth. The cellular fates of tetraploids were previously analyzed in populations isolated by serial FACS sorting.20,26 These cells were found to be chromosomally stable likely because the serial sorting to cellular DNA content eliminated aneuploid cells that arise from tetraploids.26 In another study, spontaneously arising tetraploid cells were isolated from the cancer cell line HCT116.21 This approach can only be used in cancer cell lines as spontaneous tetraploidization is rare in non-transformed human cells; an additional drawback is that the mechanism of spontaneous tetraploidization and possible underlying mutations remain enigmatic. Here, we isolated clonal cell lines from both cancerous and non-transformed cells that arose from survivors of induced cytokinesis failure. We found that all PTs harbored aneuploid karyotypes, but showed marked differences in the degree of CIN (Figs. 1, 2). Our findings demonstrate that tetraploidy leads to aneuploidy and chromosomal instability even in p53-positive non-cancerous cell lines, but also establish that a bona fide CIN+ phenotype is not a default outcome of whole genome doubling.
What causes the chromosomal instability in posttetraploid cell lines? CIN after whole genome doubling is often explained by a doubling of the centrosome number, however, the centrosome numbers in HPT and RPT cell lines were nearly normal (Fig. S5), in line with recent finding that multipolar mitoses are highly detrimental in human cells and thus an early loss of extra centrosomes is necessary to ensure survival.26,27 The fact the centrosome numbers and multipolarity are not responsible for the CIN phenotype in PTs is best illustrated by the comparison of the RPT cell lines. Although both RPT1 and RPT3 show similar distribution of centrosome numbers, RPT1 is chromosomally stable, while RPT3 is chromosomally unstable and highly aneuploid (Fig. S5E and Fig. 2C). This implies that the chromosome composition of the cells may determine whether they are CIN+ or not. For example, imbalanced gene copy numbers due to aneuploidy might affect the functionality of protein complexes required for spindle functions or for the spindle assembly checkpoint.28 Alternatively, microtubule dynamics might be altered due to changes in expression of microtubule associate proteins and motors, thus interfering with the stability of microtubule-kinetochore attachments. This hypothesis is in line with the previous findings that microtubule dynamics are often altered in CIN+ cancer cell lines and affect the frequency of errors during mitosis.29,30
Tetraploidy as well as chromosome segregation errors activate p53 pathway, thereby driving cells into irreversible arrest.14,16,17 Accordingly, TP53 mutations and p53 pathway alterations are frequently found in CIN tumors.31 Importantly, we found that the CIN+ posttetraploids were able to overcome the p53 activation as the levels of p53 and its nuclear accumulation were diminished upon chromosome missegregation (Figs. 3 and 4A). Additionally, analysis of transcriptome changes of the p53 interactors found 11 factors to be similarly deregulated in all PTs (Fig. 4D); all of the identified factors positively affect cell survival. Two genes were downregulated in all PTs: DUSP5, a direct target of p53 and an inhibitor of the mitogen-activated protein (MAP) kinases32 and MST1 (macrophage signaling growth factor), a member of the MSP-RON signaling; expression of MST1 is repressed in many types of human cancer.33 Notably, expression of apoptotic inhibitors iASPP and BIRC3 (cIAP2) was upregulated in all PTs. Both iASPP and cIAP2 are upregulated in many cancers and facilitates their survival.34-36 TGFα, a growth factor, which activates signaling pathways for proliferation, differentiation and development and has been associated with many types of cancers,37 is upregulated in all posttetraploids, similar to another pro-proliferative factor Ets1 that controls the expression of cytokines and chemokines.38 Another upregulated transcription factor, Hif-1α, is required for the response to hypoxia as well as for regulation of apoptosis.39 PTs also upregulate expression of a downstream target of Hif-1α, the lactate dehydrogenase A (LDHA) that catalyzes the conversion of pyruvate to lactate. LDHA is a key factor of anaerobic glycolysis and instrumental for the switch from oxidative phosphorylation to increased glycolysis, so called Warburg effect that is typical for malignant cells.40
Two p53 interactors were overexpressed only in CIN+ cells: FOXO1, the main target of insulin signaling and a transcription factor that regulates metabolic homeostasis in response to oxidative stress,41 and NDRG1 that has a poorly characterized function in stress response.42 The upregulation of stress-response factors in CIN+ PTs suggests that chromosomal instability imposes an ongoing metabolic and oxidative stress on human cells that might arise in response to the disruption of the protein homeostasis due to ongoing changes in chromosome content.43
Aneuploidy and CIN in tumors are often associated with increased resistance to drug treatment and therefore poor prognosis for cancer patients.44,45 We found that the posttetraploid cell lines gained resistance to a broad spectrum of small inhibitors that are used in chemotherapy. This multidrug resistance profile was found in both cells derived from a cancer cell line HCT116 as well as in posttetraploids originating from RPE1, a non-transformed and p53-positive cell line (Fig. 5). Several lines of evidence have been advanced recently demonstrating that aneuploid and tetraploid cancer cells confer resistance to some drugs.20,46,47 Why whole genome doubling provides increased multidrug resistance remains enigmatic and should be analyzed in future. Finally, we found that whole genome doubling promotes anchorage-independent growth, and hence in vitro transformation even in p53-positive cells. This ability was independent of the CIN+ phenotype. We propose that the expression changes that allow efficient proliferation despite abnormal karyotype by inhibiting apoptosis and stimulating pro-proliferative pathways contribute to the multidrug resistance and to in vitro transformation of human cells.
Taken together, tetraploidization benefits uncontrolled growth in both cancerous and non-cancerous cells. The molecular mechanisms underlying these effects remain to be addressed in the future. Identification of pathways that promote tetraploidy and its survival will be essential not only to understand the mechanisms leading to tumor formation but also for the development of novel strategies to prevent acquired multidrug resistance during cancer treatment.
Materials and Methods
Generation and culturing of posttetraploid cell lines
HCT116 H2B-GFP and RPE1 H2B-GFP (a gift from Dr. Steven Taylor, The University of Manchester, UK) were treated with 0.75 μM of the actomyosin inhibitor dihydrocytochalasin D (DCD, Sigma) for 18 h. The cells were then washed, placed into a drug-free medium and subcloned by limiting dilution in 96-well plates (0.5 cell per well). Tetraploid RPE1 H2B-GFP cells were grown on plates coated with gelatin (Merck). After clone expansion, cells were harvested for flow cytometry to measure the DNA content. All cell lines were cultured in Dulbecco's modified medium (Gibco), supplemented with 10% fetal calf serum (Gibco), 50 IU/ml penicillin, and 50 μg/ml streptomycin (Gibco) at 37°C with 5% CO2.
Immunoblotting
Total cell lysate was separated by SDS-PAGE and transferred to a polyvinylidene fluoride membrane (Roche) or nitrocellulose membrane (Whatman) as previously described. Following antibodies were used: anti-p53 antibody (1:100,Santa Cruz Biotechnology, Inc.), anti-pSer15-p53 antibody (1:500, Santa Cruz Biotechnology, Inc.), anti- p21 antibody (1:1000, Cell Signaling), anti-p38 antibody (1:1000, Cell signaling), anti-pThr180/Tyr182-p38 antibody (1:200, Cell signaling). Immunoblot quantification was performed using Image J software.
Live cell microscopy
Long term live cell data were recorded on an inverted Zeiss Observer.Z1 microscope (Visitron Systems) equipped with a humidified chamber (EMBLEM) at 37°C, 40% humidity and in the atmosphere of 5% CO2 using CoolSNAP HQ2 camera (Photometrics), Plan Neofluar 20x or 10x air objective NA 1.0, epifluorescent X-Cite 120 Series lamp (EXFO), using GFP filter and differential interference contrast (DIC) in DMEM. Imaging of fixed cells was carried out on Marianas SDC™ system (inverted Zeiss Observer.Z1 microscope, Plan Apochromat 63x magnification oil objective or 20x magnification air objective, equipped with spinning disc head (Yokogawa) and a CoolSNAP-HQ2 and CoolSNAP-EZ CCD Photometrics cameras (Intelligent Imaging Innovations, Inc.).
Chromosome spreads
The cells were treated with 50 ng/ml microtubule-depolymerizing drug colchicine (Serva) for 4.5 h, collected and pelleted using table-top centrifuge, swollen in 75 mM KCl in a 37°C water bath for 15 minutes, fixed with Carnoy solution (75% methanol and 25% acetic acid) and spread on a wet glass slide with a glass Pasteur pipette. The slides were dried at 42°C and stained with Giemsa dye (Fluka).
Fluorescence in situ hybridization (FISH)
FISH was carried out using satellite enumeration probes against centromeric regions of specific chromosomes (1, 3, 7, and 12) conjugated either to a red or a green fluorophore according to manufacturer's protocol (Cytocell, UK). DNA was counterstained with DAPI, and the cover slips were mounted on slides using antifade solution (Cytocell, UK).
Whole chromosome multicolor FISH (mFISH karyotyping)
Multicolor FISH was performed as previously described with a DNA probe mixture (24XCyte Human Multicolor FISH Probe Kit, MetaSystems, Altlussheim, Germany). The analysis was carried out using Adobe Photoshop (Adobe Systems, San Jose, USA) for visual inspection of the images; statistical analysis was performed using MS Excel (Microsoft) and Prism. Aberration ratio was calculated as number of derivative chromosomes normalized to a total number of chromosomes identified in analyzed cell spread.
SNP array profiling
Human CytoSNP-12 bead chip arrays (Illumina) were used for detection of copy number aberrations (CNAs) in clonal aneuploid and diploid cell lines. Array hybridization was performed according to the manufacturer's recommendations. CNAs were identified using Nexus software (version 7.5.1) with standard settings. To identify unique CNAs in clonal cell lines, we used the Nexus call coordinates and removed all calls of the same type with a reciprocal overlap of at least 60%. All profiles were manually checked.
Analysis of mitotic abnormalities
The cells were grown in the glass-bottom 96-black well plates and fixed with 100% methanol for 10 min at -20°C. DNA was stained with SYTOX Green Nucleic Acid dye (Molecular Probes, Invitrogen) with added RNAse. The imaging was carried out on Visitron Systems microscope.
Micronucleation test followed by anti-p53 immunostaining
The cells were seeded in the glass-bottomed 96-black well plates 48 h prior the experiment and then treated with DCD for 18 h. Only cells that became binucleated were scored. Cells were fixed with 100% MeOH and stained with DAPI (Carl Roth). Anti-p53 antibody (1:500, Cell Signaling) was used. The acquisition and analysis were performed using Slidebook 5 software with 3I microscope, 20x magnification objective. p53 status in the nuclei was determined by automated measurement of median intensities of p53 in the nucleus normalized by median intensity of p53 in cytoplasm.
Transcriptome data processing and analysis
Total RNA was extracted using RNeasy Mini Kit (QIAGEN). For the HPT cell lines microarray data preprocessing, normalization and analysis was conducted as described previously.48 For next generation RNA sequencing of the RPTs, TruSeq RNA library preparation and Illumina HiSeq2500 sequencing with 25 million 100bp single reads per library were performed by the Max Planck-Genome-Center Cologne, Germany (http://mpgc.mpipz.mpg.de/home/). Subsequently to adapter removal with cutadapt, reads were mapped to the human genome (hg19) using TopHat (v2.0.10) with the following parameters: “tophat2 -g1 -G”. RefSeq information in the GTF file was downloaded from the UCSC genome browser. featureCounts (v1.4.3) was used to generate the count matrix with the same GTF file as for the alignment and the following parameters: “-t exon -g gene_id”. Normalization and differential expression analysis of the count matrix data was performed using the R/Bioconductor package DESeq2. For differential expression analysis, PTs were compared to the parental diploid cell line. Processed and normalized RNA sequencing data (RPE1 derived cell lines) or microarray data (HCT116 derived cell lines) were analyzed by QIAGEN's Ingenuity® Pathway Analysis and visualized with R. p53 physical and genetic interactors were identified in the IPA knowledge base.
Cell proliferation assay
The compounds were obtained from commercial suppliers and dissolved in 100% DMSO. Cells were dispensed in a 384-well plate at 400 cells per well. After 24 h, 5 μl of compound dilution was added and plates were further incubated for another 72 h, after which 25 μl of ATPlite 1Step™ (PerkinElmer, Groningen, The Netherlands) solution was added to each well. Luminescence was recorded on an Envision™ multimode reader (PerkinElmer, Waltham, MA, USA). IC50s were fitted by non-linear regression using XLfit™5. A two-tailed Student's t-test was performed to determine whether differences in sensitivity (ΔpIC50) were statistically significant (i.e., P < 0.05).
Anchorage independent growth assay
Cells were treated with ethanol as a vehicle or DMBA (2μM or 4 μM) for 3 days. Subsequently cells were either seeded on soft agar or treated with 100 ng/ml TPA or with DMSO as a vehicle control for next 10 days before seeding on soft agar.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Aline Sewo Pires de Campos for excellent technical assistance and Neysan Donnelly and Verena Passerini for comments to the manuscript.
Funding
This work was supported by the Max Planck Society and by Center for Integrated Protein Science Munich (CIPSM) to ZS. KS is supported by Marie Curie Action: PloidyNet.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- 1.Storchova Z, Kuffer C. The consequences of tetraploidy and aneuploidy. J Cell Sci 2008; 121:3859-66; PMID:19020304; http://dx.doi.org/ 10.1242/jcs.039537 [DOI] [PubMed] [Google Scholar]
- 2.Lengauer C, Kinzler KW, Vogelstein B. Genetic instability in colorectal cancers. Nature 1997; 386:623-7; PMID:9121588; http://dx.doi.org/ 10.1038/386623a0 [DOI] [PubMed] [Google Scholar]
- 3.Burrell RA, McGranahan N, Bartek J, Swanton C. The causes and consequences of genetic heterogeneity in cancer evolution. Nature 2013; 501:338-45; PMID:24048066; http://dx.doi.org/ 10.1038/nature12625 [DOI] [PubMed] [Google Scholar]
- 4.Sotillo R, Hernando E, Diaz-Rodriguez E, Teruya-Feldstein J, Cordon-Cardo C, Lowe SW, Benezra R. Mad2 overexpression promotes aneuploidy and tumorigenesis in mice. Cancer Cell 2007; 11:9-23; PMID:17189715; http://dx.doi.org/ 10.1016/j.ccr.2006.10.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weaver BA, Silk AD, Montagna C, Verdier-Pinard P, Cleveland DW. Aneuploidy acts both oncogenically and as a tumor suppressor. Cancer Cell 2007; 11:25-36; PMID:17189716; http://dx.doi.org/ 10.1016/j.ccr.2006.12.003 [DOI] [PubMed] [Google Scholar]
- 6.Ricke RM, Jeganathan KB, van Deursen JM. Bub1 overexpression induces aneuploidy and tumor formation through Aurora B kinase hyperactivation. J Cell Biol 2011; 193:1049-64; PMID:21646403; http://dx.doi.org/ 10.1083/jcb.201012035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shackney SE, Smith CA, Miller BW, Burholt DR, Murtha K, Giles HR, Ketterer DM, Pollice AA. Model for the genetic evolution of human solid tumors. Cancer Res 1989:3344-54; PMID:2720687 [PubMed] [Google Scholar]
- 8.Storchova Z, Pellman D. From polyploidy to aneuploidy, genome instability and cancer. Nat Rev Mol Cell Biol 2004; 5:45-54; PMID:14708009; http://dx.doi.org/ 10.1038/nrm1276 [DOI] [PubMed] [Google Scholar]
- 9.Fujiwara T, Bandi M, Nitta M, Ivanova EV, Bronson RT, Pellman D. Cytokinesis failure generating tetraploids promotes tumorigenesis in p53-null cells. Nature 2005; 437:1043-7; PMID:16222300 [DOI] [PubMed] [Google Scholar]
- 10.Lv L, Zhang T, Yi Q, Huang Y, Wang Z, Hou H, Guo Z, Cooke HJ, Shi Q. Tetraploid cells from cytokinesis failure induce aneuploidy and spontaneous transformation of mouse ovarian surface epithelial cells. Cell Cycle 2012; 11:2864-75; PMID:22801546; http://dx.doi.org/ 10.4161/cc.21196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zack TI, Schumacher SE, Carter SL, Cherniack AD, Saksena G, Tabak B, Lawrence MS, Zhsng CZ, Wala J, Mermel CH, et al. Pan-cancer patterns of somatic copy number alteration. Nat Genet 2013; 45:1134-40; PMID:24071852; http://dx.doi.org/ 10.1038/ng.2760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lanni JS, Jacks T. Characterization of the p53-dependent postmitotic checkpoint following spindle disruption. Mol Cell Biol 1998; 18:1055-64; PMID:9448003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Andreassen PR, Lohez OD, Lacroix FB, Margolis RL. Tetraploid state induces p53-dependent arrest of nontransformed mammalian cells in G1. Mol Biol Cell 2001; 12:1315-28; PMID:11359924; http://dx.doi.org/ 10.1091/mbc.12.5.1315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kuffer C, Kuznetsova A, Storchová Z. Abnormal mitosis triggers p53-dependent cell cycle arrest in human tetraploid cells. Chromosoma 2013:1-14; PMID:23192763 [DOI] [PubMed] [Google Scholar]
- 15.Ganem NJ, Cornils H, Chiu SY, O'Rourke KP, Arnaud J, Yimlamai D, Théry M, Camargo FD, Pellman D. Cytokinesis failure triggers hippo tumor suppressor pathway activation. Cell 2014; 158:833-48; PMID:25126788; http://dx.doi.org/ 10.1016/j.cell.2014.06.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thompson SL, Compton DA. Proliferation of aneuploid human cells is limited by a p53-dependent mechanism. J Cell Biol 2010; 188:369-81; PMID:20123995; http://dx.doi.org/ 10.1083/jcb.200905057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li M, Fang X, Baker DJ, Guo L, Gao X, Wei Z, Han S, van Deursen JM, Zhang P. The ATM-p53 pathway suppresses aneuploidy-induced tumorigenesis. Proc Natl Acad Sci U S A 2011; 107:14188-93; PMID:20663956; http://dx.doi.org/ 10.1073/pnas.1005960107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Williams BR, Prabhu VR, Hunter KE, Glazier CM, Whittaker CA, Housman DE, Amon A. Aneuploidy affects proliferation and spontaneous immortalization in mammalian cells. Science 2008; 322:703-9; PMID:18974345; http://dx.doi.org/ 10.1126/science.1160058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stingele S, Stoehr G, Peplowska K, Cox J, Mann M, Storchova Z. Global analysis of genome, transcriptome and proteome reveals the response to aneuploidy in human cells. Mol Sys Biol 2012; 8:608; PMID:22968442; http://dx.doi.org/ 10.1038/msb.2012.40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Castedo M, Coquelle A, Vivet S, Vitale I, Kauffmann A, Dessen P, Pequignot MO, Casares N, Valent A, Mouhamad S, et al. Apoptosis regulation in tetraploid cancer cells. EMBO J 2006; 25:2584-95; PMID:16675948; http://dx.doi.org/ 10.1038/sj.emboj.7601127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dewhurst SM, McGranahan N, Burrell RA, Rowan AJ, Gronroos E, Endesfelder D, Joshi T, Mouradov D, Gibbs P, Ward RL, et al. Tolerance of whole-genome doubling propagates chromosomal instability and accelerates cancer genome evolution. Cancer Dis 2014; 4:175-85; PMID:24436049; http://dx.doi.org/ 10.1158/2159-8290.CD-13-0285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Masramon L, Ribas M, Cifuentes P, Arribas R, García F, Egozcue J, Peinado MA, Miró R. Cytogenetic characterization of two colon cell lines by using conventional G-banding, comparative genomic hybridization, and whole chromosome painting. Cancer Genet Cytogenet 2000; 121:17-21; PMID:10958935; http://dx.doi.org/ 10.1016/S0165-4608(00)00219-3 [DOI] [PubMed] [Google Scholar]
- 23.Honda H, Nagamachi A, Inaba T. -7/7q- syndrome in myeloid-lineage hematopoietic malignancies: attempts to understand this complex disease entity. Oncogene 2014; 0. [DOI] [PubMed] [Google Scholar]
- 24.Uitdehaag JC, de Roos JA, van Doornmalen AM, Prinsen MB, de Man J, Tanizawa Y, Kawase Y, Yoshino K, Buijsman RC, Zaman GJ. Comparison of the cancer gene targeting and biochemical selectivities of all targeted kinase inhibitors approved for clinical use. PloS One 2014; 9:e92146; PMID:24651269; http://dx.doi.org/ 10.1371/journal.pone.0092146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Semon M, Wolfe KH. Consequences of genome duplication. Curr Opin Genet Dev 2007; 17:505-12; PMID:18006297; http://dx.doi.org/ 10.1016/j.gde.2007.09.007 [DOI] [PubMed] [Google Scholar]
- 26.Ganem NJ, Godinho SA, Pellman D. A mechanism linking extra centrosomes to chromosomal instability. Nature 2009; 460:278-82; PMID:19506557; http://dx.doi.org/ 10.1038/nature08136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Krzywicka-Racka A, Sluder G. Repeated cleavage failure does not establish centrosome amplification in untransformed human cells. J Cell Biol 2011; 194:199-207; PMID:21788368; http://dx.doi.org/ 10.1083/jcb.201101073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhu J, Pavelka N, Bradford WD, Rancati G, Li R. Karyotypic determinants of chromosome instability in aneuploid budding yeast. PLoS Genet 2012; 8:e1002719; PMID:22615582; http://dx.doi.org/ 10.1371/journal.pgen.1002719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bakhoum SF, Genovese G, Compton DA. Deviant kinetochore microtubule dynamics underlie chromosomal instability. Curr Biol 2009; 19:1937-42; PMID:19879145; http://dx.doi.org/ 10.1016/j.cub.2009.09.055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ertych N, Stolz A, Stenzinger A, Weichert W, Kaulfuss S, Burfeind P, Aigner A, Wordeman L, Bastians H. Increased microtubule assembly rates influence chromosomal instability in colorectal cancer cells. Nat Cell Biol 2014; 16:779-91; PMID:24976383; http://dx.doi.org/ 10.1038/ncb2994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang B, Wang J, Wang X, Zhu J, Liu Q, Shi Z, Chambers MC, Zimmerman LJ, Shaddox KF, Kim S, et al. Proteogenomic characterization of human colon and rectal cancer. Nature 2014; 513:382-7; PMID:25043054; http://dx.doi.org/ 10.1038/nature13438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ueda K, Arakawa H, Nakamura Y. Dual-specificity phosphatase 5 (DUSP5) as a direct transcriptional target of tumor suppressor p53. Oncogene 2003; 22:5586-91; PMID:12944906; http://dx.doi.org/ 10.1038/sj.onc.1206845 [DOI] [PubMed] [Google Scholar]
- 33.Zalcenstein A, Weisz L, Stambolsky P, Bar J, Rotter V, Oren M. Repression of the MSP//MST-1 gene contributes to the antiapoptotic gain of function of mutant p53. Oncogene 2005; 25:359-69. [DOI] [PubMed] [Google Scholar]
- 34.Bergamaschi D, Samuels Y, O'Neil NJ, Trigiante G, Crook T, Hsieh J-K, O'Connor DJ, Zhong S, Campargue I, Tomlinson ML, et al. iASPP oncoprotein is a key inhibitor of p53 conserved from worm to human. Nat Genet 2003; 33:162-7; PMID:12524540 [DOI] [PubMed] [Google Scholar]
- 35.Bertrand MJM, Milutinovic S, Dickson KM, Ho WC, Boudreault A, Durkin J, Gillard JW, Jaquith JB, Morris SJ, Barker PA. cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol Cell 2008; 30:689-700; PMID:18570872 [DOI] [PubMed] [Google Scholar]
- 36.LaCasse EC, Mahoney DJ, Cheung HH, Plenchette S, Baird S, Korneluk RG. IAP-targeted therapies for cancer. Oncogene 2008; 27:6252-75; PMID:18931692 [DOI] [PubMed] [Google Scholar]
- 37.Sandgren EP, Luetteke NC, Palmiter RD, Brinster RL, Lee DC. Overexpression of TGFα in transgenic mice: induction of epithelial hyperplasia, pancreatic metaplasia, and carcinoma of the breast. Cell 1990; 61:1121-35; PMID:1693546 [DOI] [PubMed] [Google Scholar]
- 38.Pei H, Li C, Adereth Y, Hsu T, Watson DK, Li R. Caspase-1 is a direct target gene of ETS1 and plays a role in ETS1-induced apoptosis. Cancer Res 2005; 65:7205-13; PMID:16103071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kilic M, Kasperczyk H, Fulda S, Debatin KM. Role of hypoxia inducible factor-1 alpha in modulation of apoptosis resistance. Oncogene 2006; 26:2027-38; PMID:17043658 [DOI] [PubMed] [Google Scholar]
- 40.Le A, Cooper CR, Gouw AM, Dinavahi R, Maitra A, Deck LM, Royer RE, Vander Jagt DL, Semenza GL, Dang CV. Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc Natl Acad Sci 2010; 107:2037-42; PMID:20133848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tothova Z, Kollipara R, Huntly BJ, Lee BH, Castrillon DH, Cullen DE, McDowell EP, Lazo-Kallanian S, Williams IR, Sears C, et al. FoxOs are critical mediators of hematopoietic stem cell resistance to physiologic oxidative stress. Cell 2007; 128:325-39; PMID:17254970 [DOI] [PubMed] [Google Scholar]
- 42.Melotte V, Qu X, Ongenaert M, van Criekinge W, de Bruïne AP, Baldwin HS, van Engeland M. The N-myc downstream regulated gene (NDRG) family: diverse functions, multiple applications. FASEB J 2010; 24:4153-66; PMID:20667976 [DOI] [PubMed] [Google Scholar]
- 43.Donnelly N, Storchova Z. Causes and consequences of protein folding stress in aneuploid cells. Cell Cycle 2015; 14:495-501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Carter SL, Eklund AC, Kohane IS, Harris LN, Szallasi Z. A signature of chromosomal instability inferred from gene expression profiles predicts clinical outcome in multiple human cancers. Nat Genet 2006; 38:1043-8; PMID:16921376 [DOI] [PubMed] [Google Scholar]
- 45.McGranahan N, Burrell RA, Endesfelder D, Novelli MR, Swanton C. Cancer chromosomal instability: therapeutic and diagnostic challenges. EMBO Rep 2012; 13:528-38; PMID:22595889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Duesberg P, Stindl R, Hehlmann R. Origin of multidrug resistance in cells with and without multidrug resistance genes: chromosome reassortments catalyzed by aneuploidy. Proc Natl Acad Sci U S A 2001; 98:11283-8; PMID:11553793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee AJ, Endesfelder D, Rowan AJ, Walther A, Birkbak NJ, Futreal PA, Downward J, Szallasi Z, Tomlinson IP, Howell M, et al. Chromosomal instability confers intrinsic multidrug resistance. Cancer Res 2011; 71:1858-70; PMID:21363922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Durrbaum M, Kuznetsova AY, Passerini V, Stingele S, Stoehr G, Storchova Z. Unique features of the transcriptional response to model aneuploidy in human cells. BMC Genomics 2014; 15:139; PMID:24548329 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.