

Abstract
Genome stability is ensured by multiple surveillance mechanisms that monitor the duplication, segregation, and integrity of the genome throughout the cell cycle. Depletion of components of the spliceosome, a macromolecular machine essential for mRNA maturation and gene expression, has been associated with increased DNA damage and cell cycle defects. However, the specific role for the spliceosome in these processes has remained elusive, as different cell cycle defects have been reported depending on the specific spliceosome subunit depleted. Through a detailed cell cycle analysis after spliceosome depletion, we demonstrate that the spliceosome is required for progression through multiple phases of the cell cycle. Strikingly, the specific cell cycle phenotype observed after spliceosome depletion correlates with the extent of depletion. Partial depletion of a core spliceosome component results in defects at later stages of the cell cycle (G2 and mitosis), whereas a more complete depletion of the same component elicits an early cell cycle arrest in G1. We propose a quantitative model in which different functional dosages of the spliceosome are required for different cell cycle transitions.
Keywords: cell cycle, DNA damage, mitosis, mRNA splicing, spliceosome
Introduction
The mechanism of heredity relies on the accurate conservation of the genome throughout generations, at both the organismal and cellular levels. At the cellular level, the genome is transmitted through the faithful replication of genomic DNA during S phase, followed by accurate partition of the sister chromosomes during mitosis. Failures in any of these mechanisms compromise genomic integrity and result in genomic instability and aneuploidy, a common feature of cancer cells.1,2
Multiple surveillance mechanisms termed checkpoints monitor errors and pause cell cycle progression to allow time for error correction.3 These cell cycle checkpoints include the spindle checkpoint, whose role is to prevent premature chromosome segregation during mitosis,4 and multiple DNA damage checkpoints that monitor DNA integrity.1,5 Several genome-wide small interfering RNA (siRNA) screens designed to identify genes with roles in genome maintenance and cell cycle progression have reported a role for spliceosome components in these processes.6-12 Mutations in multiple spliceosome subunits have been found in leukemia, myeloid neoplasms, and some solid tumors,13,14 suggesting that spliceosome defects might promote tumorigenesis perhaps through its roles in genome maintenance and cell division.
The spliceosome is a highly dynamic ribonucleoprotein (RNP) complex required for the generation of mature mRNAs by removal of introns during a process termed splicing.15 In higher eukaryotes, splicing is required for the expression of most genes. Furthermore, it has been estimated that over 95% of human genes can be alternatively spliced, resulting in increased proteomic diversity through the generation of multiple mature mRNAs and eventually proteins from a single pre-mRNA.16 The process of splicing involves 2 transesterification reactions that first generate a free 5′ exon which is then ligated to a 3′ exon, leading to excision of the intervening intron.17,18 During the splicing reaction, spliceosomes are assembled by recruitment of spliceosome subcomplexes onto pre-mRNAs in a stepwise manner, where they recognize the 2 intron-exon boundaries and catalyze the splicing reactions.17,18 In humans, the major spliceosome consists of 5 uridine-rich small nuclear RNAs (snRNAs), termed U1, U2, U4, U5, and U6, and over 300 different proteins.18 These snRNAs form a scaffold for assembly of small-nuclear RNPs (snRNPs), the major building blocks of the spliceosome. During the splicing process, spliceosomes form by the stepwise recruitment of snRNPs onto the pre-mRNA and this assembly is in part directed by the substrate pre-mRNA.17 Different snRNPs and accessory proteins are recruited or excluded at different steps of the splicing process, resulting in a very dynamic composition of the spliceosome machine and the generation of extensive rearrangements during different stages of splicing.17,18
A connection between the spliceosome and cell cycle progression has been found in many organisms including budding yeast,19-24 fission yeast,25-27 Drosophila,9,28 chicken,29 mouse,30 and human cells.6,11,12,29,31,32 In human cells, depletion of different spliceosome components with siRNAs results in multiple cell cycle defects, with most siRNAs analyzed eliciting mitotic defects6,11,12,31 although accumulation of cells in S phase32 has also been observed. The mitotic defects observed after spliceosome depletion have been linked to splicing defects affecting the chromosome cohesion protein sororin11,12 and Apc2,11 a subunit of the anaphase-promoting complex/cyclosome (APC/C) required for mitotic progression.
In contrast, inactivation of the splicing factor ASF/SF2 in chicken cells29 results in genomic instability, formation of RNA-DNA loops (R-loops), and arrest in the G2 phase of the cell cycle in a splicing-independent manner. Similarly, mouse cells with the spliceosome component PLRG1 deleted show an increase in DNA damage with no evidence of major splicing defects.30 However, unlike the G2 arrest observed after ASF/SF2 depletion,29 PLRG1 deficiency causes an earlier cell cycle arrest, as cells cannot progress through S phase.30
Hence, a connection between spliceosome depletion and cell cycle defects has been observed after depletion of many different spliceosome subunits. However, the specific cell cycle phenotype observed varies depending on the spliceosome subunit targeted and the type of cells used. Here, we report that the spliceosome is required for progression through multiple phases of the cell cycle in human cells. Spliceosome depletion results in arrests in G1, S, G2, and mitosis. More importantly, we show evidence of a correlation between spliceosome dosage and the specific cell cycle phase affected. That is, partial spliceosome depletion leads to defects in later stages of the cell cycle (mitosis and G2), while a more complete depletion results in early cell cycle arrest (G1). Our results support a global requirement for the spliceosome throughout the cell cycle, likely due to its role in the expression of multiple cell cycle proteins.
Results
Depletion of spliceosome components induces cell cycle delays
We recently reported a genome-wide siRNA screen designed to identify regulators of the response to mitotic arrest elicited by taxol.10 Taxol arrests cells in mitosis by disrupting microtubule dynamics and triggering activation of the spindle checkpoint, a surveillance mechanism that monitors chromosome attachment to the spindle during mitosis.4 Once the cells are arrested in mitosis, 2 fates are possible: a cell can escape the mitotic arrest and exit mitosis abnormally (adaptation) or it undergoes apoptosis.33 Our screen revealed that siRNAs against several spliceosome components resulted in increased cell survival in the presence of taxol.10 The spliceosome components uncovered in our screen included: SNRPB (SmB/B'), one of 7 core structural spliceosome proteins that directly assemble on the U-rich snRNAs;34 P14 (SF3B14), a protein that directly contacts the adenosine at the branch point and carries out the first transesterification reaction during splicing;35 as well as the spliceosome associated components CWC2236 and CRNKL1.37
To understand how depletion of these spliceosome components increased cell survival in taxol, we transfected HeLa cells with a pool of 4 siRNAs designed against each of the indicated spliceosome components. At 24 h after transfection, taxol was added at a final concentration of 220 nM. Samples were analyzed by flow cytometry after 42 h in taxol (Fig. 1A). Under these conditions, the majority of mock-depleted cells had already undergone apoptosis after the prolonged mitotic arrest,10 as evidenced by the shift in DNA content to subG1 levels. In contrast, cells transfected with pooled siRNAs targeting CRNKL1 (siCRNKL1), SNRPB (siSNRPB), or P14 (siP14) had a marked reduction in subG1 cells and clear peaks of live cells with a 4C DNA content (G2/M). These results confirmed that depletion of several spliceosome subunits from HeLa cells decreased apoptosis and increased cell survival.
Figure 1.

Spliceosome depletion induces cell cycle defects. (A) HeLa cells were transfected with the indicated siRNA pools (4 siRNAs per pool). Taxol (200 nM) was added at 24 h after transfection. Samples were collected after 42 h in taxol. DNA content histograms obtained by flow cytometry using propidium iodide (PI) staining are shown. A sample histogram of an asynchronously growing culture is shown as comparison (left). (B) Cell cycle analysis after spliceosome depletion. Cells were transfected with the individual indicated siRNAs, grown asynchronously, collected 48 h after transfection and analyzed by flow cytometry. The DNA content histograms obtained by PI staining are shown.
In addition to a decrease in apoptosis and presence of cells in G2/M, cells transfected with the siSNRPB pool also had a small population of cells in G1 even after 42 h in taxol (Fig. 1A). These results suggested that SNRPB depletion elicited a cell cycle delay in G1. To directly test whether depletion of different spliceosome components resulted in cell cycle defects in the absence of taxol, we transfected HeLa cells with a control siRNA (siControl) or individual siRNAs targeting the different spliceosome components, and analyzed the cell cycle distribution of asynchronously growing populations with flow cytometry at 48 h after transfection (Fig. 1B). Interestingly, at least 2 siRNAs per target gene produced abnormal cell cycle profiles. Two main phenotypes were observed: cells transfected with siCRNKL1–3, siCRNKL1–4, and all 4 siRNAs against CWC22 and siP14–3 showed an increase in S phase cells, while siP14–1, siSNRPB-3, and siSNRPB-4 caused an increase in the number of cells in G2. These results indicate that spliceosome depletion results in cell cycle delays in S and G2 phases. Cell cycle delays in phases prior to mitosis can explain the increased survival observed in taxol by preventing mitotic entry and therefore taxol-induced mitotic arrest and apoptosis.
Spliceosome depletion results in multiple cell cycle phenotypes
To further characterize the cell cycle phenotypes caused by spliceosome depletion in more detail, we next synchronized HeLa cells at the G1/S boundary by thymidine block and then released them into medium containing taxol to allow for progression through a single cell cycle (Fig. 2). Cells transfected with siControl progressed synchronously through S phase and reached G2/M by 9 h after release. In contrast, cells transfected with siCRNKL1–3 or siCRNKL1–4 arrested in S phase (Fig. 2A). Strikingly, cells transfected with siRNAs against SNRPB showed different cell cycle phenotypes, depending on the specific siRNA used (Fig. 2B). A majority of the cells transfected with siSNRPB-1 were not able to enter S phase and remained arrested at the G1/S boundary, while most of the cells transfected with siSNRPB-3 progressed to G2/M, albeit with a delay. Furthermore, quantification of mitotic cells by flow cytometry using the anti-MPM2 antibody, which recognized phospho-epitopes present only in mitotic cells, revealed that cells depleted of SNRPB had a significant delay in mitotic entry. Only 1.3% of cells transfected with siSNRPB-1 and 2.8% of cells transfected with siSNRPB-3 reached mitosis by 12 h after release into taxol, compared to 15.2% in siControl-treated cells (Fig. 2C-D). Consistent with a delay of siSNRPB treated cells to enter mitosis, Western blot analysis of total lysates from the same time course revealed less robust cyclin B1 accumulation in these cells (Fig. 2E). Consistent with an earlier study, depletion of SNRPB reduced sororin protein levels (Fig. 2F), presumably due to inefficient splicing of sororin mRNA. Taken together, these results indicate that depletion of different spliceosome components can lead to cell cycle arrest/delays at the G1, S, and G2 phases of the cell cycle.
Figure 2.

Spliceosome depletion induces interphase delays. (A-B) HeLa cells transfected with the indicated siRNAs were synchronized in G1 by thymidine block and subsequently released into medium containing taxol. Samples were taken at the indicated time points after release and subjected to cell cycle profile analysis by flow cytometry using propidium iodide (PI) and anti-MPM2 staining. Overlaid DNA content histograms are shown. (C) Quantification of mitotic cells (MPM2+) for the experiment in (B). (D) Representative flow cytometry profiles of cells stained with PI and MPM2 at 12 h after release. The percentage of mitotic cells (MPM2+) is indicated. (E) Western blots of total lysates of cells in (B) blotted with the indicated antibodies. (F) Western blots of total lysates of cells in (B) at the time of release (t = 0 h) blotted with the indicated antibodies.
Spliceosome depletion increases DNA damage and elicits a G2 arrest
A role for spliceosome components in preventing DNA damage has been reported.7,29 In the presence of DNA damage, the DNA damage checkpoint is triggered and delays cell cycle progression in G2 to allow time to repair the damage.5 To test whether the G2 arrest we observed after SNRPB depletion was due to DNA damage, we first quantified γH2AX, a marker of DNA double-strand breaks, by immunostaining (Fig. 3A-C). Compared to siControl-treated cells, SNRPB depletion resulted in an increase in DNA damage in terms of both the percentage of cells with γH2AX foci (Fig. 3B) and the overall intensity of γH2AX signal (Fig. 3C). Next, we tested whether the G2 arrest observed in cells transfected with siSNRPB-3 was due to activation of the DNA damage checkpoint by co-transfecting cells with siRNAs targeting components of this checkpoint (Fig. 3D-E). We focused on the kinases Chk1, Chk2, ATR, and ATM, since they are essential for the DNA damage checkpoint response.5 Cells were co-transfected with siControl or siSNRPB-3 and siRNAs against the indicated DNA damage checkpoint kinases. At 24 h after transfection, taxol was added for another 15 h, and the percentage of mitotic cells was quantified by flow cytometry. SNRPB depletion using siSNRPB-3 resulted in a strong G2 arrest, with only 17.2% of the cells entering mitosis, compared to 74.8% in siControl-treated cells (Fig. 3E). Co-transfection with siRNAs against Chk1, Chk2, or ATM partially alleviated the G2 arrest and increased the percentage of cells in mitosis, while siRNAs against ATR did not have any effect. Combination of siChk1 and siChk2 siRNAs, together with siSNRPB-3, had additive effects, increasing the percentage of mitotic cells to 41.4%. Together, these results indicate that the G2 arrest observed in cells transfected with siSNRPB-3 is at least partially due to an increase in DNA damage and subsequent activation of the DNA damage checkpoint. Interestingly, the checkpoint response elicited by SNRPB depletion requires Chk1, Chk2, and ATM, but not ATR, although we cannot rule out the trivial possibility that this is due to incomplete ATR depletion.
Figure 3.

Spliceosome depletion results in increased DNA damage and checkpoint-dependent G2 arrest. (A) Analysis of DNA damage by immunostaining. HeLa cells were transfected with the indicated siRNAs. Cells were fixed at 48 h after transfection and stained with antibodies against γ-H2AX and DAPI. Sample micrographs are shown. (B) Quantification of the percentage of γ-H2AX-positive cells in (A). (C) Quantification of nuclear γ-H2AX intensity in (A). Each dot represents one nucleus. (D) HeLa cells were transfected with Control (siControl) or siSNRPB-3 siRNA together with siRNAs against the indicated DNA damage checkpoint components. Western blots of total lysates blotted with the indicated antibodies are shown. (E) Quantification of mitotic entry. HeLa cells transfected with the indicated combinations of siRNAs were incubated with taxol at 24 h after siRNA transfection. Samples were collected after 15 h in taxol, and the cell cycle profile was analyzed by flow cytometry using Propidium Iodide (PI) and anti-MPM2 staining. The percentage of mitotic cells (MPM2+) is indicated.
Spliceosome depletion also causes mitotic defects
Mitotic defects after depletion of the spliceosome components SON,31 SNW1,11 and MFAP112 have been reported. To test whether SNRPB depletion also caused mitotic defects, we performed live cell imaging of HeLa cells stably expressing H2B-GFP, and analyzed their progression through mitosis (Fig. 4). Because depletion of SNRPB using siRNAs at a final concentration of 5 nM resulted in interphase arrest in G1 and G2 (Fig. 2B-D), we decided to reduce the siRNA concentration to achieve a level of partial SNRPB depletion that allowed the cells to progress past G2 and into mitosis. Transfection with 1 nM siSNRPB-1 for 24 h resulted in 88% of the cells entering mitosis within the time of our time lapse analysis (30 h), albeit with a slight delay compared to siControl-transfected cells (Fig. 4A). Analysis of mitotic progression after transfection with either siSNRPB-1 or siSNRPB-3 at a final concentration of 1 nM resulted in an increase in mitotic defects, including multipolar mitoses, misaligned chromosomes, and lagging chromosomes (Fig. 4B-C). These results are in agreement with and extend previously reported roles for other spliceosome components in mitosis.11,12,31
Figure 4.

Spliceosome depletion also results in mitotic defects. (A) HeLa H2B-GFP cells were transfected with the indicated siRNAs (1 nM), and live cell imaging was performed starting at 24 h after siRNA transfection. Cumulative frequency of cells entering mitosis during the time lapse is shown. (B) Quantification of the mitotic phenotypes observed in (A). (C) Representative micrographs of the time lapse imaging in (A). Nuclear envelope breakdown (NEBD) is set as time 0 h. Arrow points to a misaligned chromosome. Arrowhead marks a lagging chromosome.
Different degrees of SNRPB depletion produce distinct cell cycle phenotypes
Our analyses thus far indicate that depletion of different spliceosome components can result in diverse cell cycle phenotypes, including arrest in G1, S, or G2 (Fig. 1–2), increased DNA damage (Fig. 3), and mitotic defects (Fig. 4). Interestingly, depletion of a single spliceosome component, SNRPB, using 2 different siRNAs also results in different cell cycle phenotypes: siSNRPB-1 transfection causes predominantly a G1 arrest while siSNRPB-3 transfection results predominantly in a G2 arrest (Fig. 2B). More importantly, reduction of siSNRPB-1 concentration relieves the G1 arrest, allowing the cells to progress into mitosis (Fig. 4A). These results suggested that the multiple cell cycle phenotypes could be due to the specific level of knockdown achieved by the different siRNAs.
To further test this hypothesis, we compared the phenotypes and levels of SNRPB depletion obtained by transfection of HeLa cells with 3 different siRNAs against SNRPB (Fig. 5A-B). HeLa cells were transfected with siControl or one of the 3 SNRPB siRNAs at a final concentration of 5 nM in triplicate samples. Taxol was added to one sample 24 h after the siRNA transfection, and the sample was collected after 15 h in taxol. The other 2 samples were grown asynchronously (Log) for 29 h and collected for cell cycle analysis. As expected, siControl-treated cells showed a typical cell cycle distribution in asynchronous culture and were arrested in mitosis in the presence of taxol, with 78% of the cells in mitosis (Fig. 5A). Interestingly, the 3 siRNAs against SNRBP produced different cell cycle phenotypes. Cells transfected with siSNRPB-1 and siSNRPB-2 had a strong G1 arrest, since many cells were still in G1 even after 15 h in taxol. In contrast, siSNRPB-3 caused primarily a G2 arrest that could be observed both in asynchronous and taxol-treated cultures.
Figure 5.

Different degrees of spliceosome depletion cause different cell cycle phenotypes. (A) Cell cycle analysis of HeLa cells transfected with the indicated siRNAs (5 nM). Top panel: Overlaid DNA content histograms obtained by flow cytometry using PI staining in the absence (Log) or presence of taxol (Tax)(15 h). Bottom panel: Quantification of mitotic cells in the taxol samples by PI and anti-MPM2 staining. Percentage of mitotic cells is indicated. (B) Western blots of total lysates from HeLa cells transfected with the indicated siRNAs at the indicated concentrations. Samples were taken at 48 h after transfection and blotted with the indicated antibodies. (C-D) HeLa cells were transfected with the indicated siRNAs at the indicated concentrations. Samples were taken at 48 h after transfection and analyzed by Western blotting (C) or flow cytometry using PI (D).
We then compared the degree of SNRPB depletion by siSNRPB-1 and siSNRPB-3. Lysates of HeLa cells transfected with different concentrations of siSNRPB-1 or siSNRPB-3 were analyzed by Western blotting. While both siRNAs effectively reduced the levels of SNRPB protein, siSNRPB-1 depleted SNRPB more completely than siSNRPB-3 did (Fig. 5B). Thus, the efficiency of SNRPB depletion correlated with the severity of the cell cycle arrest. Cells treated with siSNRPB-1 had less residual SNRPB protein and underwent the earliest cell cycle arrest (G1). Cells transfected with siSNRPB-3 had more residual SNRBP and were arrested at a later stage in the cell cycle (G2). These results indicated a correlation between SNRPB depletion and cell cycle progression.
Because of its central role in mRNA splicing, we reasoned that variations in the level of spliceosome function could lead to defects in the expression of cell cycle proteins that would in turn result in cell cycle progression defects. Consistent with a role for the spliceosome in gene expression, the levels of several cell cycle proteins, including Apc2, Mad2, BubR1, and γ-tubulin, were reduced after SNRPB depletion. More importantly, the level of the decrease for these cell cycle proteins correlated with the level of SNRPB depletion. Together, these results suggest that spliceosome depletion results in cell cycle defects due to decreased expression of multiple cell cycle proteins. The nature of the cell cycle defect observed after depletion of spliceosome components depends on the severity of the depletion, and is likely due to the specific type of cell cycle proteins affected and the level of their reduction.
To further confirm this notion, we next tested if the correlation between SNRPB depletion and cell cycle phenotype could be recapitulated using a single siRNA. HeLa cells were transfected with different concentrations of siSNRPB-1. Samples were collected 48 h after transfection, and analyzed with Western blotting and flow cytometry. Because the levels of residual SNRPB did not vary much when cells were transfected with 1–20 nM of siSNRPB-1 (Fig. 5B), we used a much wider concentration range, from 1 pM to 5 nM. Remarkably, the different levels of SNRPB depletion correlated with the cell cycle phenotype. While high concentrations of siSNRPB-1 (1–5 nM) produced a strong G1 arrest (Fig. 5A and D), lower siSNRPB-1 concentrations (1–40 pM) resulted in a G2 arrest (Fig. 5D). These results demonstrate that different degrees of depletion of a single spliceosome component (SNRPB) can produce distinct cell cycle phenotypes.
Discussion
Through a detailed cell cycle analysis of human cells depleted of several spliceosome components, we have defined a global role for the spliceosome at multiple stages of the cell cycle. Furthermore, by analyzing cell cycle progression in the presence of different levels of the core spliceosome component SNRPB, we identified a correlation between the spliceosome level and the specific cell cycle stage at which the cells undergo arrest. These results are consistent with a dosage-dependent model in which the extent of progression through the cell cycle is dependent on the level of spliceosome activity (Fig. 6). High spliceosome levels support progression through multiple phases of the cell cycle, while low spliceosome levels result in an early arrest.
Figure 6.
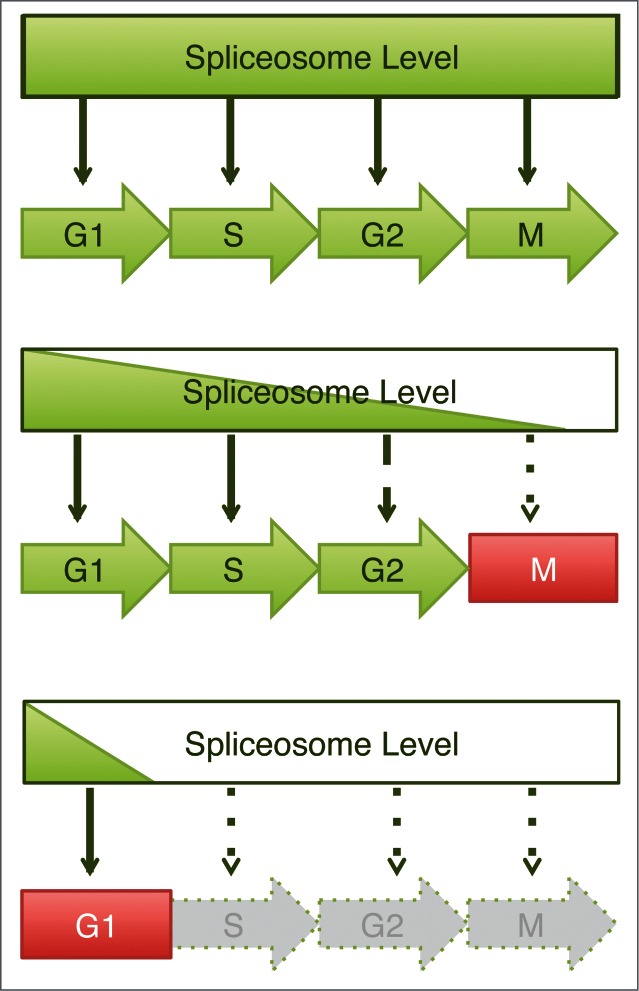
Graded requirement for the spliceosome during the cell cycle. Schematic drawing of a dosage model to explain the varying cell cycle phenotypes caused by spliceosome inactivation. In this model, spliceosome is required for pre-mRNA splicing and subsequent expression of multiple proteins at all stages of the cell cycle. Depletion of spliceosome components results in cell cycle defects whose severity depends upon the efficiency of spliceosome depletion. Mild spliceosome depletion results in defects in the later stages of the cell cycle (e.g. mitosis or G2), whereas severe spliceosome depletion elicits a block in G1.
Previous studies have indicated that depletion of individual spliceosome components causes splicing defects in only a small subset of mRNAs, including the chromosome cohesion protector sororin and the APC/C subunit Apc2.11,12 Two interpretations were suggested, either the specific spliceosome components depleted are preferentially required for splicing of these mitotic pre-mRNAs or the sororin and Apc2 pre-mRNAs are particularly difficult to splice and require high spliceosome levels. Our results show that depletion of the core spliceosome SNRPB reduced the protein levels of sororin (Fig. 2F) and Apc2 (Fig. 5C, E). However, we also observed reduction in the levels of several other mitotic and cell cycle proteins. Although it is possible that specific pre-mRNAs are affected first, as suggested for sororin and Apc2,11,12 the cell cycle phenotypes observed here are more likely a result of the reduction of multiple cell cycle proteins.
Our results show that depletion of spliceosome proteins to different levels results in defects in different cell cycle phases, with mitosis being the most sensitive phase, and G1 being the least sensitive one. Why does progression through different cell cycle stages require different levels of spliceosome function? Several scenarios could explain this graded response to spliceosome depletion. First, variations in spliceosome function during different phases of the cell cycle might result in a need for increased spliceosome levels during phases when splicing activity is diminished. This idea is supported by evidence showing that splicing activity in mitotic extracts is reduced, compared to that of interphase extracts, due to activation of the spliceosome inhibitor SRp38 during mitosis.38 In addition, spliceosome assembly and maturation occurs at the Cajal bodies, nuclear bodies whose size and number change during the different phases of the cell cycle and are disassembled during mitosis.39,40 Thus reduced spliceosome activity during mitosis due to inhibition and reduced assembly/maturation might explain a requirement for higher spliceosome dosage at this stage. Higher spliceosome levels during mitosis might compensate for the repression and maintain appropriate levels of splicing of genes transcribed and translated in mitosis, such as cyclin B1.41
A related scenario can be envisioned in which the different requirements for spliceosome dosage are due to cell cycle-regulated changes in spliceosome abundance. Recently, a proteome study reported a striking decrease in the abundance of more than 50 spliceosome proteins at the G1/S transition.42 This sharp decrease might explain why late cell cycle phases are more sensitive to spliceosome depletion compared to G1. Further depletion of spliceosome components during these phases could readily compromise the already reduced spliceosome levels.
Lastly, it is also possible that the different spliceosome dosage requirements during the different cell cycle phases are due to splicing-independent roles for the spliceosome. A direct role for the spliceosome component PRP19 during in vitro spindle assembly in Xenopus extracts has been reported.43 This seems to be a splicing-independent role, since spindle assembly in this system does not require transcription and is not inhibited by the splicing inhibitor spliceostatin A. Similarly, a splicing-independent role for the spliceosome-associated protein ASF/SF2 in the prevention of DNA damage has been reported.29 Depletion of ASF/SF2 results in increased DNA damage and G2 arrest due to the formation of DNA-RNA hybrids (R-loops). Formation of R-loops in ASF/SF2-depleted cells can be reversed by overexpression of RNaseH, an enzyme that specifically cleaves RNA in DNA-RNA hybrids, suggesting that the role of ASF/SF2 in R-loop prevention is independent of its role in splicing. Here, we have shown that SNRPB depletion also increases DNA damage and elicits a G2 arrest (Fig. 3). Depletion of the DNA damage checkpoint kinases Chk1, Chk2, and ATM partially alleviates the G2 arrest, indicating that the cell cycle arrest in G2 is, at least in part, due to the increase in DNA damage. However, the fact that inhibition of the DNA damage checkpoint only rescues the G2 arrest in about 50% of the cells indicates the concomitant presence of additional cell cycle defects that also contribute to the G2 arrest.
We hypothesize that the specific cell cycle phenotypes observed after spliceosome depletion might be the result of multiple splicing-related and splicing-independent defects, which lead to complex and diverse phenotypic outcomes. Understanding the specific contributions of the different spliceosome activities to the cell cycle and how the cell cycle in turn regulates the spliceosome are important questions that need to be addressed. Further understanding of the link between the spliceosome and cell division, may ultimately contribute to our understanding of proliferative diseases, such as cancer.
Materials and Methods
Mammalian cell culture and reagents
HeLa Tet-On (Clontech) cells were grown in Dulbecco's modified Eagle's medium (Life Technologies) supplemented with 10% fetal bovine serum (Life Technologies), 10 mM L-glutamine, and penicillin/streptomycin (Life Technologies). siRNA transfections were performed using Lipofectamine RNAiMAX (Life Technologies) according to instructions from the manufacturer. For arresting cells in mitosis, cells were treated with 220 nM taxol (Sigma) for 14–16 h or as indicated. For cell cycle synchronization, cells were cultured in medium containing 2.5 mM thymidine (Sigma) for 14 h and released into fresh medium with taxol for the desired times.
Antibodies and immunoblotting
Cells were lysed in 2 × loading buffer. Lysates were separated by SDS-PAGE, transferred to nitrocellulose membranes, and blotted with the indicated antibodies. Antibodies against Apc2, Mad2, Mad1, and BubR1 have been previously described.44-46 The following antibodies were purchased from commercial sources: anti-SNRPB (Sigma), anti-γ-tubulin (Sigma), anti-actin (Millipore) and anti-γ-H2AX (Millipore).
Flow cytometry
Cells resuspended in PBS were fixed in cold 70% ethanol, washed once in PBS, and permeabilized in PBS containing 0.25% Triton-X for 5 min. Anti-MPM2 antibody (Millipore) was diluted 1:400 in PBS containing 3% BSA (Sigma), added to the permeabilized cells, and incubated at room temperature for 3 h. After washing with PBS containing 3% BSA, cells were resuspended in the same buffer containing anti-mouse Alexa-488 secondary antibody (Invitrogen; 1:200) and incubated for 30 min in the dark. Cells were washed in PBS and resuspended in PBS supplemented with 0.1% Triton-X, 200 µg/ml DNase-free RNase A (QIAGEN), and 2 µg/ml propidium iodide (PI) (Sigma). Samples were processed using a FACScalibur flow cytometer (BD Biosciences). Data were analyzed using the FlowJo software.
Immunofluorescence
Cells for immunostaining were grown on chamber slides (LabTek), fixed using cold methanol (−20°C), blocked in PBS containing 3% BSA and 0.2% Triton-X, and mounted using ProLong with DAPI (Invitrogen) after incubation with the appropriate primary and secondary antibodies. The antibodies used were: anti-γ-H2AX (1:250, Millipore) and Alexa secondary antibodies (1:500, Life Technologies). Samples were imaged using a DeltaVision system (Applied Precision) taking z-stacks at 0.2 µm. Images were deconvolved using the manufacturer algorithm and projected using the Sum method. For γ-H2AX staining quantification, nuclei were segmented in the deconvolved and projected images in the DAPI channel using ImageJ and the Integrated Density of γ-H2AX for every nuclei was measured.
Live cell microscopy
HeLa cells stably expressing H2B-GFP were cultured on 4- or 8-well chambered coverslips (LabTek) and transfected with a control siRNA or the indicated siRNAs at 5 nM. Cells were imaged using a DeltaVision system (Applied Precision) fitted with a Xenon light source, CoolSnap HQ2 camera, a humidified environmental chamber at 37°C and 5% CO2. Images were acquired using a 40× objective at 2–10 min intervals for 12–24 h. Three 5-μm z-stacks per timepoint were acquired and projected by the maximum intensity method. The images were further processed in ImageJ to create time-lapse movies. All images in each movie were subjected to the same brightness/contrast adjustment.
Disclosure of Potential Conflicts of Interest
HY is an Investigator with the Howard Hughes Medical Institute.
Acknowledgments
We thank members of the Yu lab for helpful discussions.
Funding
LADM was supported in part by a Research Supplement to Promote Diversity in the Health Sciences (R01CA125269). This work was also supported by the Welch Foundation (I-1441) and the Cancer Prevention and Research Institute of Texas (RP110465-P3 and RP120717-P2).
References
- 1.Aguilera A, García-Muse T. Causes of genome instability. Annu Rev Genet 2013; 47:1-32; PMID:23909437; http://dx.doi.org/ 10.1146/annurev-genet-111212-133232 [DOI] [PubMed] [Google Scholar]
- 2.Ricke RM, van Deursen JM. Aneuploidy in health, disease, and aging. J Cell Biol 2013; 201:11-21; PMID:23547028; http://dx.doi.org/ 10.1083/jcb.201301061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hartwell LH, Weinert TA. Checkpoints: controls that ensure the order of cell cycle events. Science 1989; 246:629-34; PMID:2683079; http://dx.doi.org/ 10.1126/science.2683079 [DOI] [PubMed] [Google Scholar]
- 4.Musacchio A, Salmon ED. The spindle-assembly checkpoint in space and time. Nat Rev Mol Cell Biol 2007; 8:379-93; PMID:17426725; http://dx.doi.org/ 10.1038/nrm2163 [DOI] [PubMed] [Google Scholar]
- 5.Kousholt AN, Menzel T, Sørensen CS. Pathways for genome integrity in G2 phase of the cell cycle. Biomolecules 2012; 2:579-607; PMID:24970150; http://dx.doi.org/ 10.3390/biom2040579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kittler R, Putz G, Pelletier L, Poser I, Heninger A-K, Drechsel D, Fischer S, Konstantinova I, Habermann B, Grabner H, et al.. An endoribonuclease-prepared siRNA screen in human cells identifies genes essential for cell division. Nature 2004; 432:1036-40; PMID:15616564; http://dx.doi.org/ 10.1038/nature03159 [DOI] [PubMed] [Google Scholar]
- 7.Paulsen RD, Soni DV, Wollman R, Hahn AT, Yee M-C, Guan A, Hesley JA, Miller SC, Cromwell EF, Solow-Cordero DE, et al.. A genome-wide siRNA screen reveals diverse cellular processes and pathways that mediate genome stability. Mol Cell 2009; 35:228-39; PMID:19647519; http://dx.doi.org/ 10.1016/j.molcel.2009.06.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ceron J, Rual J-F, Chandra A, Dupuy D, Vidal M, van den Heuvel S. Large-scale RNAi screens identify novel genes that interact with the C. elegans retinoblastoma pathway as well as splicing-related components with synMuv B activity. BMC Dev Biol 2007; 7:30; PMID:17417969; http://dx.doi.org/ 10.1186/1471-213X-7-30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Somma MP, Ceprani F, Bucciarelli E, Naim V, De Arcangelis V, Piergentili R, Palena A, Ciapponi L, Giansanti MG, Pellacani C, et al.. Identification of drosophila mitotic genes by combining co-expression analysis and RNA interference. PLoS Genet 2008; 4:e1000126; PMID:18797514; http://dx.doi.org/ 10.1371/journal.pgen.1000126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Díaz-Martínez LA, Karamysheva ZN, Warrington R, Li B, Wei S, Xie X-J, Roth MG, Yu H. Genome-wide siRNA screen reveals coupling between mitotic apoptosis and adaptation. EMBO J 2014; 33:1960-76; PMID:25024437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.van der Lelij P, Stocsits RR, Ladurner R, Petzold G, Kreidl E, Koch B, Schmitz J, Neumann B, Ellenberg J, Peters J-M. SNW1 enables sister chromatid cohesion by mediating the splicing of sororin and APC2 pre-mRNAs. EMBO J 2014; 33:2643-58; PMID:25257309; http://dx.doi.org/ 10.15252/embj.201488202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sundaramoorthy S, Vázquez-Novelle MD, Lekomtsev S, Howell M, Petronczki M. Functional genomics identifies a requirement of pre-mRNA splicing factors for sister chromatid cohesion. EMBO J 2014; 33:2623-42; PMID:25257310; http://dx.doi.org/ 10.15252/embj.201488244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Padgett RA. New connections between splicing and human disease. Trends Genet 2012; 28:147-54; PMID:22397991; http://dx.doi.org/ 10.1016/j.tig.2012.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Scott LM, Rebel VI. Acquired mutations that affect pre-mRNA splicing in hematologic malignancies and solid tumors. J Natl Cancer Inst 2013; 105:1540-9; PMID:24052622; http://dx.doi.org/ 10.1093/jnci/djt257 [DOI] [PubMed] [Google Scholar]
- 15.Sharp PA. The discovery of split genes and RNA splicing. Trends Bioch Sci 2005; 30:279-81; PMID:15950867; http://dx.doi.org/ 10.1016/j.tibs.2005.04.002 [DOI] [PubMed] [Google Scholar]
- 16.Nilsen TW, Graveley BR. Expansion of the eukaryotic proteome by alternative splicing. Nature 2010; 463:457-63; PMID:20110989; http://dx.doi.org/ 10.1038/nature08909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wahl MC, Will CL, LUhrmann R. The spliceosome: design principles of a dynamic RNP machine. Cell 2009; 136:701-18; PMID:19239890; http://dx.doi.org/ 10.1016/j.cell.2009.02.009 [DOI] [PubMed] [Google Scholar]
- 18.Hoskins AA, Moore MJ. The spliceosome: a flexible, reversible macromolecular machine. Trends Bioch Sci 2012; 37:179-88; PMID:22480731; http://dx.doi.org/ 10.1016/j.tibs.2012.02.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ben-Yehuda S, Russell CS, Dix I, Beggs JD, Kupiec M. Extensive genetic interactions between PRP8 and PRP17/CDC40, two yeast genes involved in pre-mRNA splicing and cell cycle progression. Genetics 2000; 154:61-71; PMID:10628969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ben-Yehuda S, Dix I, Russell CS, McGarvey M, Beggs JD, Kupiec M. Genetic and physical interactions between factors involved in both cell cycle progression and pre-mRNA splicing in Saccharomyces cerevisiae. Genetics 2000; 156:1503-17; PMID:11102353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Russell CS, Ben-Yehuda S, Dix I, Kupiec M, Beggs JD. Functional analyses of interacting factors involved in both pre-mRNA splicing and cell cycle progression in Saccharomyces cerevisiae. RNA 2000; 6:1565-72; PMID:11105756; http://dx.doi.org/ 10.1017/S1355838200000984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Burns CG, Ohi R, Mehta S, O'Toole ET, Winey M, Clark TA, Sugnet CW, Ares M, Gould KL. Removal of a single alpha-tubulin gene intron suppresses cell cycle arrest phenotypes of splicing factor mutations in Saccharomyces cerevisiae. Mol Cell Biol 2002; 22:801-15; PMID:11784857; http://dx.doi.org/ 10.1128/MCB.22.3.801-815.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dahan O, Kupiec M. Mutations in genes of Saccharomyces cerevisiae encoding pre‐mRNA splicing factors cause cell cycle arrest through activation of the spindle checkpoint. Nucleic Acids Res 2002; 30:4361-70; PMID:12384582; http://dx.doi.org/ 10.1093/nar/gkf563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kaplan Y, Kupiec M. A role for the yeast cell cycle/splicing factor Cdc40 in the G1/S transition. Curr Genet 2007; 51:123-40; PMID:17171376; http://dx.doi.org/ 10.1007/s00294-006-0113-y [DOI] [PubMed] [Google Scholar]
- 25.Bishop DT, McDonald WH, gould KL, Forsburg SL. Isolation of an essential Schizosaccharomyces pombe gene, prp31(+), that links splicing and meiosis. Nucl Acids Res 2000; 28:2214-20; PMID:10871341; http://dx.doi.org/ 10.1093/nar/28.11.2214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ochotorena IL, Hirata D, Kominami K, Potashkin J, Sahin F, Wentz-Hunter K, gould KL, Sato K, Yoshida Y, Vardy L, et al.. Conserved Wat1/Pop3 WD-repeat protein of fission yeast secures genome stability through microtubule integrity and may be involved in mRNA maturation. J Cell Sci 2001; 114:2911-20; PMID:11686295 [DOI] [PubMed] [Google Scholar]
- 27.Lundgren K, Allan S, Urushiyama S, Tani T, Ohshima Y, Frendewey D, Beach D. A connection between pre-mRNA splicing and the cell cycle in fission yeast: cdc28+ is allelic with prp8+ and encodes an RNA-dependent ATPase/helicase. Mol Biol Cell 1996; 7:1083-94; PMID:8862522; http://dx.doi.org/ 10.1091/mbc.7.7.1083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Andersen DS, Tapon N. Drosophila MFAP1 is required for pre-mRNA processing and G2/M progression. J Biol Chem 2008; 283:31256-67; PMID:18765666; http://dx.doi.org/ 10.1074/jbc.M803512200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li X, Manley JL. Inactivation of the SR protein splicing factor ASF/SF2 results in genomic instability. Cell 2005; 122:365-78; PMID:16096057; http://dx.doi.org/ 10.1016/j.cell.2005.06.008 [DOI] [PubMed] [Google Scholar]
- 30.Kleinridders A, Pogoda HM, Irlenbusch S, Smyth N, Koncz C, Hammerschmidt M, Bruning JC. PLRG1 is an essential regulator of cell proliferation and apoptosis during vertebrate development and tissue homeostasis. Mol Cell Biol 2009; 29:3173-85; PMID:19307306; http://dx.doi.org/ 10.1128/MCB.01807-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huen MSY, Sy SMH, Leung KM, Ching Y-P, Tipoe GL, Man C, Dong S, Chen J. SON is a spliceosome-associated factor required for mitotic progression. Cell Cycle (Georgetown, Tex) 2010; 9:2679-85; PMID:20581448; http://dx.doi.org/ 10.4161/cc.9.13.12151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li Z, Pützer BM. Spliceosomal protein E regulates neoplastic cell growth by modulating expression of cyclin E/CDK2 and G2/M checkpoint proteins. J Cell Mol Med 2008; 12:2427-38; PMID:18208561; http://dx.doi.org/ 10.1111/j.1582-4934.2008.00244.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gascoigne KE, Taylor SS. How do anti-mitotic drugs kill cancer cells? J Cell Sci 2009; 122:2579-85; PMID:19625502; http://dx.doi.org/ 10.1242/jcs.039719 [DOI] [PubMed] [Google Scholar]
- 34.Leung AKW, Nagai K, Li J. Structure of the spliceosomal U4 snRNP core domain and its implication for snRNP biogenesis. Nature 2011; 473:536-9; PMID:21516107; http://dx.doi.org/ 10.1038/nature09956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schellenberg MJ, Dul EL, MacMillan AM. Structural model of the p14/SF3b155-branch duplex complex. RNA 2010; 17:155-65; PMID:21062891; http://dx.doi.org/ 10.1261/rna.2224411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yeh T-C, Liu H-L, Chung C-S, Wu N-Y, Liu Y-C, Cheng S-C. Splicing factor Cwc22 is required for the function of Prp2 and for the spliceosome to escape from a futile pathway. Mol Cell Biol 2011; 31:43-53; PMID:20956557; http://dx.doi.org/ 10.1128/MCB.00801-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chung S, Zhou Z, Huddleston KA, Harrison DA, Reed R, Coleman TA, Rymond BC. Crooked neck is a component of the human spliceosome and implicated in the splicing process. Biochim Biophys Acta 2002; 1576:287-97; PMID:12084575; http://dx.doi.org/ 10.1016/S0167-4781(02)00368-8 [DOI] [PubMed] [Google Scholar]
- 38.Shin C, Manley JL. The SR protein SRp38 represses splicing in M phase cells. Cell 2002; 111:407-17; PMID:12419250; http://dx.doi.org/ 10.1016/S0092-8674(02)01038-3 [DOI] [PubMed] [Google Scholar]
- 39.Morris GE. The Cajal body. Biochim Biophys Acta 2008; 1783:2108-15; PMID:18755223; http://dx.doi.org/ 10.1016/j.bbamcr.2008.07.016 [DOI] [PubMed] [Google Scholar]
- 40.Matera AG, Wang Z. A day in the life of the spliceosome. Nat Rev Mol Cell Biol 2014; 15:108-21; PMID:24452469; http://dx.doi.org/ 10.1038/nrm3742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mena AL, Lam EWF, Chatterjee S. Sustained Spindle-Assembly Checkpoint Response Requires De Novo Transcription and Translation of Cyclin B1. PLoS One 2010; 5:e13037; PMID:20927403; http://dx.doi.org/ 10.1371/journal.pone.0013037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lane KR, Yu Y, Lackey PE, Chen X, Marzluff WF, Cook JG. Cell cycle-regulated protein abundance changes in synchronously proliferating HeLa cells include regulation of pre-mRNA splicing proteins. PLoS One 2013; 8:e58456; PMID:23520512; http://dx.doi.org/ 10.1371/journal.pone.0058456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hofmann JC, Tegha-Dunghu J, Dräger S, Will CL, LUhrmann R, Gruss OJ. The Prp19 Complex Directly Functions in Mitotic Spindle Assembly. PLoS One 2013; 8:e74851; PMID:24069358; http://dx.doi.org/ 10.1371/journal.pone.0074851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fang G, Yu H, Kirschner MW. The checkpoint protein MAD2 and the mitotic regulator CDC20 form a ternary complex with the anaphase-promoting complex to control anaphase initiation. Genes Dev 1998; 12:1871-83; PMID:9637688; http://dx.doi.org/ 10.1101/gad.12.12.1871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tang Z, Bharadwaj R, Li B, Yu H. Mad2-Independent inhibition of APCCdc20 by the mitotic checkpoint protein BubR1. Dev Cell 2001; 1:227-37; PMID:11702782; http://dx.doi.org/ 10.1016/S1534-5807(01)00019-3 [DOI] [PubMed] [Google Scholar]
- 46.Kim S, Sun H, Tomchick DR, Yu H, Luo X. Structure of human Mad1 C-terminal domain reveals its involvement in kinetochore targeting. Proc Natl Acad Sci 2012; 109:6549-54; http://dx.doi.org/ 10.1073/pnas.1118210109 [DOI] [PMC free article] [PubMed] [Google Scholar]