

Abstract
Intrahepatic cholangiocarcinoma (ICC) is an aggressive cancer associated with the bile ducts within the liver. These tumors are characterized by frequent gain-of-function mutations in the isocitrate dehydrogenase 1 and 2 (IDH1 and IDH2) genes—that are also common in subsets of neural, haematopoietic and bone tumors, but rare or absent in the other types of gastrointestinal malignancy. Mutant IDH acts through a novel mechanism of oncogenesis, producing high levels of the metabolite 2-hydroxyglutarate, which interferes with the function of α-ketoglutarate-dependent enzymes that regulate diverse cellular processes including histone demethylation and DNA modification. Recently, we used in vitro stem cell systems and genetically engineered mouse models (GEMMs) to demonstrate that mutant IDH promotes ICC formation by blocking hepatocyte differentiation and increasing pools of hepatic progenitors that are susceptible to additional oncogenic hits leading to ICC. We found that silencing of HNF4A—encoding a master transcriptional regulator of hepatocyte identity and quiescence—was critical to mutant IDH-mediated inhibition of liver differentiation. In line with these findings, human ICC with IDH mutations are characterized by a hepatic progenitor cell transcriptional signature suggesting that they are a distinct ICC subtype as compared to IDH wild type tumors. The role of mutant IDH in controlling hepatic differentiation state suggests the potential of newly developed inhibitors of the mutant enzyme as a form of differentiation therapy in a solid tumor.
Keywords: cholangiocarcinoma, IDH1, IDH2, Isocitrate Dehydrogenase, mouse models
Introduction
Biliary tract cancers (BTCs) are a group of epithelial malignancies with shared histopathologic features and include intrahepatic cholangiocarcinoma (ICC) and extrahepatic cholangiocarcinoma (ECC)—cancers of the bile ducts within and outside the liver, respectively, and gall bladder cancer (GBC).1,2 The BTC categorization has been useful to guide the clinical approach for diagnostic work-up and surgical resection in localized disease. However, these tumors are also treated identically in the metastatic setting as they were thought to share a common biology and cell of origin. This view has now been irrevocably changed with the publication of a series of genetic studies that show that the subgroups of BTC have highly distinct mutational profiles. Most notably, recent findings by Borger, et al.3 and corroborated by other groups,4-8 demonstrated that mutations in IDH1 and IDH2 were common in ICC (22–36% of cases) but rare or absent in other hepatobiliary cancers (ECC, GBC, and hepatocellular carcinoma) and in additional gastrointestinal tumor types (e.g. pancreatic cancer, gastric cancer, and colon cancer) (Fig. 1).
Figure 1.
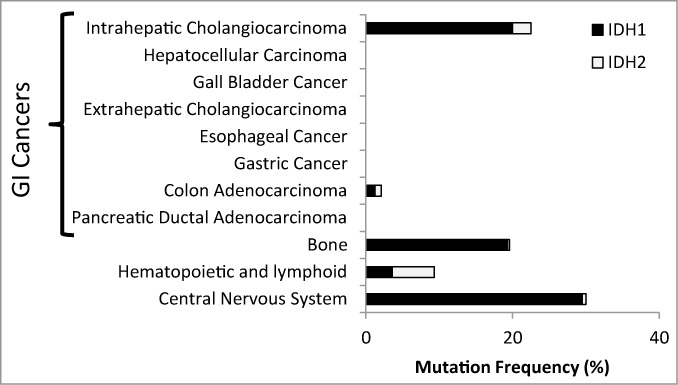
ICC is unique among gastrointestinal (GI) malignancies in harboring a high frequency of IDH mutations. The chart shows the frequency of each tumor type exhibiting a gain-of-function hotspot mutation in either IDH1 or IDH2.3 (http://www.sanger.ac.uk/cosmic) Major GI tumor types are shown as are other tissues where tumors with IDH mutations are common.
IDH1 and IDH2 encode metabolic enzymes localized to the cytoplasm and peroxisome (IDH1) or to the mitochondria (IDH2), whose normal functions are to catalyze a reversible reaction converting isocitrate to α-ketoglutarate (αKG) coupled with the production of reduced nicotinamide adenine dinucleotide phosphate (NADPH). The products of this reaction contribute to lipid biosynthesis, redox balance, energy metabolism, and the supply of αKG as an essential co-substrate for a diverse group of dioxygenase enzymes.9 Cancer-associated hotspot IDH1/IDH2 mutations result in a gain-of-function activity, where αKG is converted to 2-hydroxyglutarate (2-HG) with the consumption of NADPH.10,11 2-HG is not used in any biochemical pathways and accumulates to high levels within the cell.10 The tumor promoting effect of mutant IDH1 and IDH2 is thought to be due to the action of 2-HG as an ‘oncometabolite’ that interferes with the function of the αKG-dependent dioxygenases, which include regulators of DNA and histone demethylation, as well as multiple other nuclear and cytoplasmic processes.12-19 IDH mutant tumors consequently exhibit widespread epigenetic alterations.6,13,16 Changes in redox state due to depletion of NAPDH may also contribute to tumorigenesis in IDH mutant cells.20
The remarkable tissue-specificity of IDH1/IDH2 mutations in ICC was puzzling since they had never before been identified at a high frequency in epithelial malignancies and since the role of these mutations in tumorigenesis was poorly understood. Recently, we developed model systems that have shed light on the mechanisms by which mutant IDH promotes ICC formation as discussed below.21 Deciphering mutant IDH function has added significance as specific pharmacologic inhibitors of the mutant enzyme have entered clinical trials, and understanding the mechanisms underlying this oncogenic pathway will be a critical step toward informing optimal approaches for deploying these drugs, defining relevant biomarkers of response, and predicting mechanisms of drug resistance that may arise.
Role of IDH Mutations in ICC Pathogenesis
Although both ICC and ECC have traditionally been thought to arise through the malignant transformation of bile duct epithelium, several lines of evidence suggest that the pathogenesis of ICC may be more complex. Embryologic and lineage tracing studies have established that during development, extrahepatic bile ducts arise directly from the ventral foregut mesoderm, while intrahepatic bile ducts arise from a common progenitor cell for hepatocytes, referred to as the hepatoblast (HB).22 In the normal adult liver, cell turnover is very low and is accomplished by replication of existing cholangiocytes and hepatocytes. Following injury, intrahepatic bile duct epithelium, or cholangiocytes, may be replaced by a number of potential sources: (1) cell cycle reentry of neighboring cholangiocytes; (2) de-differentiation of hepatocytes into adult bipotential liver progenitor cells (referred to as oval cells), which then replace the damaged cholangiocytes; or (3) activation of resident liver progenitor cells, which may be present as a rare population in the liver, but can expand and differentiate into cholangiocytes (Fig. 2A).23 Likewise, injured hepatocytes can be replaced by neighboring hepatocytes, oval cells or cholangiocytes that de-differentiate into oval cells.23 Although the precise mechanisms of liver regeneration remain controversial, recent lineage-tracing studies24,25 suggest that perhaps any of these sources may be called upon, depending on the injury context. This remarkable cellular plasticity within the liver offers the possibility that ICC can arise from any of these cell types, a notion that is further supported by the appearance of tumors with mixed hepatocellular carcinoma (HCC)/ICC histopathology (Fig. 2B).26,27 The demonstration that transgenic mouse models in which genetic alterations—combined AKT/Notch activation or PTEN/p53 inactivation—targeted to the hepatocytes result in an ICC phenotype provides direct experimental evidence of the potential of hepatocytes to give rise to ICC.28,29 Similarly, HB cells engineered to express various oncogenes can give rise to either HCC or ICC following subcutaneous implantation, depending on the identity of the oncogene.30-33
Figure 2.

Cellular plasticity in the liver. (A) Under normal conditions, the liver is largely quiescent and its low rate of cell turnover is maintained by proliferation of differentiated bile duct cells (cholangiocytes) and hepatocytes. The liver also exhibits extensive capacity to regenerate following damage. Depending on the nature of the injury, cholangiocytes or hepatocytes can be replaced by their neighboring non-injured counterparts, or by oval cells—existing as rare endogenous progenitors or generated by de-differentiation of either cholangioctyes or hepatocytes. (B) The major types of adult liver cancer are ICC and HCC, which show histologic and immunophenotypic resemblance to the normal cholangiocytes and hepatocytes, respectively. Experimental studies indicate that differentiated liver cells can give rise to both tumor types directly or though an oval cell intermediate. Mixed HCC/ICC tumors with histopathologic features of both tumors may be associated with oval cell expansion. (C) left panel, Impact of mutant IDH on differentiation of bipotential hepatoblast (HB) cells in vitro. Expression of mutant IDH in HBs leads to production of 2HG which blocks hepatocyte differentiation through suppression of HNF4α via an unknown mechanism. Right panel, IDH acts in the adult liver to block the differentiation of hepatic progenitors. These liver progenitors are sensitized to transformation by additional oncogenic hits, and can progress through graded premalignant biliary lesions leading to ICC.
Given this striking cellular plasticity in the liver, it is notable that IDH1/IDH2 mutations are present in >20 % of ICC tumors but have not been observed in hepatocellular carcinomas3 (http://www.sanger.ac.uk/cosmic). In leukemogenesis, mutant IDH is thought to act as an early pathogenic event to disrupt haematopoietic stem cell differentiation.13 Therefore, we hypothesized that mutant IDH may be acting in an analogous fashion in the liver progenitor cell to drive ICC. To test the potential function of mutant IDH in this context, we expressed various mutant IDH alleles in liver progenitor (HB) cells and assessed the resulting impact on cell differentiation programs in vitro. Mutant IDH had no observable effect on the morphology or proliferation rates of HB cells under basal conditions, nor did it affect the ability of these cells to undergo bile duct differentiation as measured by tubule formation and the upregulation of biliary markers. By contrast, IDH mutant HBs exhibited a pronounced block in hepatocyte differentiation. While control cells form hepatocyte spheres, strongly upregulate an extensive program of hepatocyte markers and undergo proliferative arrest, IDH mutant HB cells failed to induce these hepatocyte genes and continued to grow in monolayer, maintaining their stem cell phenotype. This was due to the production of 2HG since it was phenocopied in wild type HB cells treated with cell-permeable 2HG esters, and since a pharmacologic inhibitor of the mutant enzyme rescued the ability of IDH mutant HBs to undergo hepatocyte differentiation.
Global gene expression profiling and gene set enrichment analysis (GSEA) revealed that mutant IDH strongly suppressed a program regulated by hepatocyte nuclear factor (HNF) 4α, a transcription factor that is a key component of a regulatory network directing the development of hepatocytes from liver progenitors.22 Accordingly, in control cells, HNF4α was potently induced upon hepatocyte differentiation of control HB cells, but remained at basal levels in mutant IDH HBs in a 2HG-dependent manner. The Hnf4a promoter was also devoid of the histone mark associated with active transcription, tri-methyl histone 3 lysine 4 (H3K4Me3), consistent with the observed transcriptional silencing of this locus in IDH mutant-expressing cells. The functional significance of this HNF4α regulation was validated in genetic epistasis experiments where knock-down of HNF4α in wild-type HBs potently suppressed hepatocyte differentiation while ectopic expression of HNF4α in IDH mutant HBs effectively restored differentiation. Thus, mutant IDH inhibits hepatocyte fate decisions in liver progenitor cells through the production of 2HG and transcriptional suppression of HNF4α as a key downstream target.
To extend our studies in vivo, we generated a genetically engineered mouse model (GEMM) that expresses mutant IDH in adult hepatocytes using a doxycycline (Dox)-inducible system. After treating these mice with Dox for 1 month, no detectable alterations in hepatocyte differentiation or proliferation were detected. In retrospect, this result should not have been surprising based on our prior in vitro studies. Indeed, while expression of mutant IDH in multipotent HB cells blocked hepatocyte differentiation in vitro, mutant IDH had little effect if we induced its expression late in the hepatocyte differentiation process. As the normal adult liver lacks a significant progenitor cell population, but such a population is activated following injury to the organ, we hypothesized that mutant IDH may act analogously in vivo to override hepatocyte differentiation from a progenitor cell state arising in the setting of hepatic injury. To address this question, we utilized the liver toxin 3,5-diethoxycarbonyl-1,4-dihydrocollidine (DDC) which damages hepatocytes and activates oval cells in the adult liver. Our IDH mutant GEMMs were exposed to Dox to induce transgene expression and then transiently treated with DDC for 5 d. After 3 weeks of recovery from DDC treatment, the hepatocytes in wild-type mice had returned to quiescence, while the IDH mutant hepatocytes continued to proliferate and expressed dramatically lower levels of HNF4α as well as a large set of hepatocyte markers, indicative of a failure to restore normal hepatocyte identity. In a related GEMM expressing mutant IDH in the bile duct and hepatic progenitor cells, there were again no gross effects on the normal liver. However, as the mice were aged >1 year, we observed accumulation of oval cells throughout the liver lobules, which was not seen in wild type controls. These observations indicate that mutant IDH acts in the adult liver to block the differentiation of hepatic progenitors—activated in response to injury or spontaneously during aging—specifically impairing hepatocyte lineage progression.
Progenitor cells are thought to be more prone to oncogenic transformation as compared to differentiated cells, possibly due to their dynamic and accessible chromatin states being favorable for activation of mitogenic programs. Therefore, although mutant IDH did not cause tumorigenesis directly, we predicted that the expanded pools of progenitor cells might result in sensitization to other oncogenic driver mutations. To test this possibility, we studied the interactions between liver-targeted activating mutations in IDH2 and KRAS, a genetic lesion also found in human ICC.6,7 Crosses of our IDH mutant GEMM with a knock-in KRASG12D model,34 revealed pronounced oncogenic cooperation. The compound mutant mice exhibited dramatic oval cell expansion and multistage ICC pathogenesis, with tumors progressing from graded premalignant biliary lesions and culminating in metastatic ICC. By contrast, KRASG12D expression alone failed to induce oval cells and only resulted in HCCs and mixed HCC/ICC tumors with long latency—while combined p53 deletion accelerated KRASG12D-driven tumors, it did not result in oval cell expansion nor did most animals exhibit the range of precursor lesions. Collectively, the data are consistent with a model whereby mutant IDH subverts the hepatocyte differentiation/quiescence program in proliferating hepatocytes or bipotential progenitors, creating a persistent pre-neoplastic state primed for transformation by additional oncogenic mutations such as activated KRAS (Fig. 2C).
In parallel studies we investigated potential roles of HNF4α silencing in ICC biology. Notably, genetic ablation of HNF4α in the adult mouse liver results in loss of hepatocyte differentiation and quiescence, and expansion of oval cells.35 Following treatment with the carcinogen DEN, the HNF4α-deficient cells are predisposed to developing into ICC. These findings indicate that HNF4α is a regulator of oval cell function and an ICC tumor suppressor. Although further investigation is needed to establish the direct targets of mutant IDH and 2HG contributing to ICC pathogenesis in vivo, these studies are consistent with a central role of HNF4α silencing in the process.
The ability of mutant IDH to thwart liver progenitor cell differentiation and the differing tumor phenotypes of the KRAS-IDH and KRAS-p53 models together suggest that IDH mutations may define a distinct subtype of ICC in humans. In this regard, examination of a set of more than 100 human ICCs36 revealed that IDH mutant tumors strongly express a liver progenitor cell gene signature when compared to IDH wild-type tumors. This is of clinical relevance as allele-specific enzymatic inhibitors of mutant IDH37-39 are currently in clinical trials. Such inhibitors have resulted in rapid and dramatic complete responses in several patients with refractory acute myelogenous leukemia (AML).40 The proposed mechanism for this response, which has been corroborated in animal models of IDH mutant leukemia, relates to the ability of these inhibitors to induce differentiation of leukemic blasts into terminally-differentiated myeloid cells.19,37,41,42 While it is not yet known whether mutant IDH activity is required for tumor maintenance in ICC harboring these mutations, the presence of this progenitor cell signature may indicate the potential for such “differentiation therapy” in these tumors as well. The progenitor cell state of IDH mutant ICC may also result in a distinct set of targetable signaling dependencies in addition to the function of the mutant enzyme. Thus, understanding the biology of this genetically-defined ICC subtype offers the potential of identifying multiple new patient-specific therapies.
Allele Frequencies Vary Widely Among IDH Mutant Cancers
Mutations in IDH1 or IDH2 have now been identified at high frequencies in a wide spectrum of seemingly unrelated neoplasias, including acute myelogenous leukemias (AML), angioimmunoblastic T-cell lymphomas (AITL), myelodysplastic syndrome (MDS), low grade and secondary gliomas, chondrosarcomas, and cholangiocarcinomas.9 It appears that like we have observed in ICC, the role played by mutant IDH in haematopoietic cancers and sarcomas relates to a capacity to override stem/progenitor cell differentiation.15,41-43 While all hotspot IDH1 and IDH2 mutations confer neomorphic enzymatic activity resulting in markedly elevated levels of 2HG, it is notable that these different diseases have greatly variable frequencies of the specific IDH mutant alleles. The most obvious of these examples is the relatively high incidence of IDH2 R140Q allele in AML and MDS, while this mutant allele has not yet been identified in any solid tumor (Fig. 3). By contrast, the IDH2 R172K allele is relatively common in biliary tract cancers and hematopoeitic malignancies but extremely rare in central nervous system and bone tumors (Fig. 3). Another striking example of this specificity is the prevalence of IDH1 R132H mutants in glioma (>90 %) and complete absence of such mutations in ICC. Reciprocally, IDH1 R132C mutations are the most frequent IDH mutations in ICC and rare in glioma. Both IDH1 R132H and R132C mutations involve a conversion of a CpG dinucleotide to TpG on opposite strands of the IDH R132 codon, which likely results from a spontaneous deamination event (Fig. 4A). These observations imply that rather than reflecting differences in mutagenic mechanisms between tissues, the distinct spectrum of mutant alleles may be due to functional differences in the resulting mutant enzymes.
Figure 3.
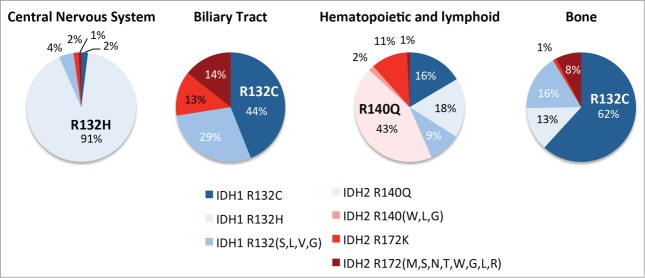
Mutant IDH allele frequency varies widely across different cancers. The relative frequency of the different mutant IDH1 or IDH2 alleles in the indicated cancer subtypes are shown. The most common alleles for each tumor type are labeled on the individual pie charts. Less common variants of IDH1 and IDH2 are grouped together.
Figure 4.
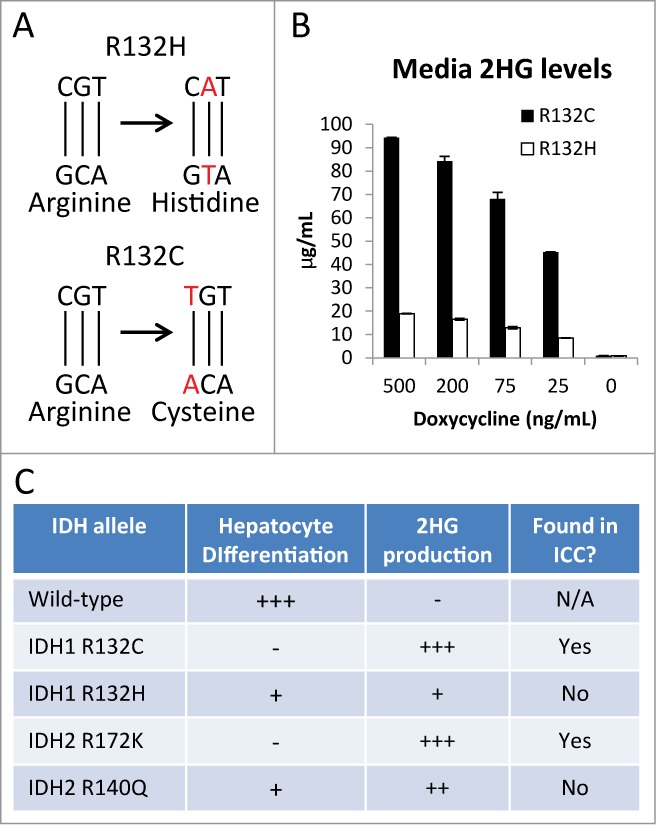
Functional significance of different IDH mutant alleles. (A) The R132H mutation appears to occur as a result of a spontaneous deamination of a CpG dinucleotide of the reverse (bottom) strand, yielding a TpG dinucleotide. Similarly, the R132C mutation appears to occur from the same spontaneous deamination event of a CpG to a TpG dinucleotide on the forward strand in the same codon. (B) HB cells were engineered to express different IDH alleles under a doxycycline-inducible promoter. At all levels of expression, the R132C-expressing HBs produced ∼5 fold higher concentrations of 2HG compared with the R132C-expressing HB cells. (C) Table describing the phenotypes of HB cells expressing the indicated alleles.
While the mechanisms underlying these frequency variations remains unclear, our recent work suggests at least one possible explanation. Using a doxycycline-inducible expression system in liver progenitor HB cells, we titrated ectopic expression of mutant IDH1 R132C and IDH1 R132H to levels indistinguishable from endogenous wild-type IDH1 and measured the 2HG produced by these mutant IDH-expressing HBs. Strikingly, the level of 2HG produced by IDH1 R132C, the most common mutant allele in ICC, was 5-fold greater than that produced by the IDH1 R132H mutant enzyme at every level of expression (Fig. 4B). Moreover, this difference in 2HG production was functionally significant as the IDH1 R132H allele was less potent in blocking hepatocyte differentiation—as assessed by morphologic changes, hepatocyte gene induction, and proliferative arrest—a critical step toward the development of ICC (Fig. 4C). Similarly, the IDH2 R172K mutant allele, which is found in ICC, produced 2-fold more 2HG than the IDH2 R140Q allele—which has not yet been identified in ICC— and was more effective in preventing in vitro differentiation.21 Thus, the IDH1 and IDH2 mutant alleles associated with ICC result in production of highest levels of 2HG and this correlates with the extent of impairment in hepatocyte differentiation. Therefore, we propose that the key targets of 2HG involved in the pathogenesis of IDH mutant ICC may require a relatively high level of 2HG production for their regulation, which cannot be generated by the IDH1 R132H allele and/or that such high levels of 2HG production may be toxic in different cell types.
An additional question is whether there are differences in the effect of IDH1 and IDH2 mutations on tumorigenesis. The common production of 2HG as a mechanism to bypass differentiation and the mutual exclusivity of IDH1/IDH2 mutations suggest that their oncogenic programs are similar. However, beyond their apparently overlapping functions driving cancer formation, mutations in IDH1 and IDH2 are likely to have distinct collateral effects in cell physiology relating to the specific subcellular localization and contribution to metabolic pathways of the wild type enzymes. For example IDH1 mutant cells could have defects in lipid biosynthesis through the impairment of the reductive glutaminolysis pathway and increased dependency on oxidative phosphorylation,44,45 while IDH2 mutant cells could have a compromise in mitochondrial redox balance.46 Although such differences are speculative at present, it seems likely that there exist alterations in cellular states characteristic of either mutant isoform that may influence the acquisition of additional oncogenic lesions required for tumor progression as well as response to certain therapeutic interventions.
Conclusion
We have demonstrated that mutant IDH subverts hepatocyte differentiation and results in the expansion of liver progenitors primed for transformation by additional oncogenic insults. Moreover, our data indicate that IDH mutations define a distinct subtype of ICC, characterized by a liver progenitor gene signature. Key questions include the need to resolve the critical immediate targets of mutant IDH/2HG that contribute to the differentiation block and, most importantly, to determine whether mutant IDH is a good target in these tumors once they have developed. As clinical trials are currently underway using mutant specific inhibitors, additional insights into the mechanism of action of these inhibitors will be needed to enable full interpretation of the results from these initial trials and to help inform future therapeutic strategies.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by grants from the NIH (R01CA136567–02 and P50CA1270003) and TargetCancer Foundation to N.B. N.B. holds the Gallagher Endowed Chair in Gastrointestinal Cancer Research at Massachusetts General Hospital and is supported by a V Foundation Translational Award, TargetCancer Foundation, and grants from the NCI. S.K.S. is the recipient of an American Cancer Society Postdoctoral Fellowship (PF-13–294–01-TBG). C.A.P is the recipient of a CIHR postdoctoral fellowship.
References
- 1. Hezel AF, Deshpande V, Zhu AX. Genetics of biliary tract cancers and emerging targeted therapies. J Clin Oncol 2010; 28:3531-40; PMID:20547994; http://dx.doi.org/10.1200/JCO.2009.27.4787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Razumilava N, Gores GJ. Cholangiocarcinoma. Lancet; 383:2168-79; PMID:24581682; http://dx.doi.org/ 10.1016/S0140-6736(13)61903-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Borger DR, Tanabe KK, Fan KC, Lopez HU, Fantin VR, Straley KS, Schenkein DP, Hezel AF, Ancukiewicz M, Liebman HM, et al. . Frequent Mutation of Isocitrate Dehydrogenase (IDH)1 and IDH2 in Cholangiocarcinoma Identified Through Broad-Based Tumor Genotyping. Oncol 2012; 17:72-9; http://dx.doi.org/ 10.1634/theoncologist.2011-0386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chan-on W, Nairismagi M-L, Ong CK, Lim WK, Dima S, Pairojkul C, Lim KH, McPherson JR, Cutcutache I, Heng HL, et al. . Exome sequencing identifies distinct mutational patterns in liver fluke-related and non-infection-related bile duct cancers. Nat Genet 2013; 45:1474-8; PMID:24185513; http://dx.doi.org/ 10.1038/ng.2806 [DOI] [PubMed] [Google Scholar]
- 5. Jiao Y, Pawlik TM, Anders RA, Selaru FM, Streppel MM, Lucas DJ, Niknafs N, Guthrie VB, Maitra A, Argani P, et al. . Exome sequencing identifies frequent inactivating mutations in BAP1, ARID1A and PBRM1 in intrahepatic cholangiocarcinomas. Nat Genet 2013; 45:1470-3; PMID:24185509; http://dx.doi.org/ 10.1038/ng.2813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wang P, Dong Q, Zhang C, Kuan PF, Liu Y, Jeck WR, Andersen JB, Jiang W, Savich GL, Tan TX, et al. . Mutations in isocitrate dehydrogenase 1 and 2 occur frequently in intrahepatic cholangiocarcinomas and share hypermethylation targets with glioblastomas. Oncogene 2013; 32:3091-100; PMID:22824796; http://dx.doi.org/ 10.1038/onc.2012.315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Voss JS, Holtegaard LM, Kerr SE, Fritcher EGB, Roberts LR, Gores GJ, Zhang J, Highsmith WE, Halling KC, Kipp BR. Molecular profiling of cholangiocarcinoma shows potential for targeted therapy treatment decisions. Hum Pathol 2013; 44:1216-22; PMID:23391413; http://dx.doi.org/ 10.1016/j.humpath.2012.11.006 [DOI] [PubMed] [Google Scholar]
- 8. Kipp BR, Voss JS, Kerr SE, Barr Fritcher EG, Graham RP, Zhang L, Highsmith WE, Zhang J, Roberts LR, Gores GJ, et al. . Isocitrate dehydrogenase 1 and 2 mutations in cholangiocarcinoma. Hum Pathol 2012; 43:1552-8; PMID:22503487; http://dx.doi.org/ 10.1016/j.humpath.2011.12.007 [DOI] [PubMed] [Google Scholar]
- 9. Losman J-A, Kaelin WG. What a difference a hydroxyl makes: mutant IDH, (R)-2-hydroxyglutarate, and cancer. Gene Dev 2013; 27:836-52; PMID:23630074; http://dx.doi.org/ 10.1101/gad.217406.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, Fantin VR, Jang HG, Jin S, Keenan MC, et al. . Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009; 462:739-44; PMID:19935646; http://dx.doi.org/ 10.1038/nature08617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ward PS, Patel J, Wise DR, Abdel-Wahab O, Bennett BD, Coller HA, Cross JR, Fantin VR, Hedvat CV, Perl AE, et al. . The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting α-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 2010; 17:225-34; PMID:20171147; http://dx.doi.org/ 10.1016/j.ccr.2010.01.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim S-H, Ito S, Yang C, Wang P, Xiao M-T, et al. . Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell 2011; 19:17-30; PMID:21251613; http://dx.doi.org/ 10.1016/j.ccr.2010.12.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, Li Y, Bhagwat N, Vasanthakumar A, Fernandez HF, et al. . Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 2010; 18:553-67; PMID:21130701; http://dx.doi.org/ 10.1016/j.ccr.2010.11.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chowdhury R, Yeoh KK, Tian YM, Hillringhaus L, Bagg EA, Rose NR, Leung IKH, Li XS, Woon ECY, Yang M, et al. . The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lu C, Ward PS, Kapoor GS, Rohle D, Turcan S, Abdel-Wahab O, Edwards CR, Khanin R, Figueroa ME, Melnick A, et al. . IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 2012; 483:474-8; PMID:22343901; http://dx.doi.org/ 10.1038/nature10860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Turcan S, Rohle D, Goenka A, Walsh LA, Fang F, Yilmaz E, Campos C, Fabius AWM, Lu C, Ward PS, et al. . IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 2012; 483:479-83; PMID:22343889; http://dx.doi.org/ 10.1038/nature10866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sasaki M, Knobbe CB, Munger JC, Lind EF, Brenner D, Brustle A, Harris IS, Holmes R, Wakeham A, Haight J, et al. . IDH1(R132H) mutation increases murine haematopoietic progenitors and alters epigenetics. Nature 2012; 488:656-9; PMID:22763442; http://dx.doi.org/ 10.1038/nature11323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Koivunen P, Lee S, Duncan CG, Lopez G, Lu G, Ramkissoon S, Losman JA, Joensuu P, Bergmann U, Gross S, et al. . Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked to EGLN activation. Nature 2012; 483:484-8; PMID:22343896; http://dx.doi.org/ 10.1038/nature10898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Losman J-A, Looper RE, Koivunen P, Lee S, Schneider RK, McMahon C, Cowley GS, Root DE, Ebert BL, Kaelin WG. (R)-2-hydroxyglutarate is sufficient to promote leukemogenesis and its effects are reversible. Science 2013; 339:1621-5; PMID:23393090; http://dx.doi.org/ 10.1126/science.1231677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sasaki M, Knobbe CB, Itsumi M, Elia AJ, Harris IS, Chio IIC, Cairns RA, McCracken S, Wakeham A, Haight J, et al. . D-2-hydroxyglutarate produced by mutant IDH1 perturbs collagen maturation and basement membrane function. Gene Dev 2012; 26:2038-49; PMID:22925884; http://dx.doi.org/ 10.1101/gad.198200.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Saha SK, Parachoniak CA, Ghanta KS, Fitamant J, Ross KN, Najem MS, Gurumurthy S, Akbay EA, Sia D, Cornella H, et al. . Mutant IDH inhibits HNF-4alpha to block hepatocyte differentiation and promote biliary cancer. Nature 2014; advance online publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Si-Tayeb K, Lemaigre FP, Duncan SA. Organogenesis and development of the liver. Dev Cell 2010; 18:175-89; PMID:20159590; http://dx.doi.org/ 10.1016/j.devcel.2010.01.011 [DOI] [PubMed] [Google Scholar]
- 23. Michalopoulos GK. Principles of liver regeneration and growth homeostasis. Comprehensive Physiology 2013; 3:485-513; PMID:23720294; http://dx.doi.org/10.1002/cphy.c120014 [DOI] [PubMed] [Google Scholar]
- 24. Yanger K, Zong Y, Maggs LR, Shapira SN, Maddipati R, Aiello NM, Thung SN, Wells RG, Greenbaum LE, Stanger BZ. Robust cellular reprogramming occurs spontaneously during liver regeneration. Gene Dev 2013; 27:719-24; PMID:23520387; http://dx.doi.org/ 10.1101/gad.207803.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tarlow BD, Finegold MJ, Grompe M. Clonal tracing of Sox9+ liver progenitors in mouse oval cell injury. Hepatol 2014; 60:278-89; http://dx.doi.org/ 10.1002/hep.27084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Garancini M, Goffredo P, Pagni F, Romano F, Roman S, Sosa JA, Giardini V. Combined hepatocellular-cholangiocarcinoma: A population-level analysis of an uncommon primary liver tumor. Liver Transplant 2014; 20:952-9; http://dx.doi.org/ 10.1002/lt.23897 [DOI] [PubMed] [Google Scholar]
- 27. Singh S, Chakraborty S, Bonthu N, Radio S, Hussain S, Sasson A. Combined hepatocellular cholangiocarcinoma: a case report and review of literature. Dig Dis Sci 2013; 58:2114-23; PMID:23397471; http://dx.doi.org/ 10.1007/s10620-013-2585-1 [DOI] [PubMed] [Google Scholar]
- 28. Fan B, Malato Y, Calvisi DF, Naqvi S, Razumilava N, Ribback S, Gores GJ, Dombrowski F, Evert M, Chen X, et al. . Cholangiocarcinomas can originate from hepatocytes in mice. J Clin Investi 2012; 122:2911-5; http://dx.doi.org/ 10.1172/JCI63212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Xue W, Chen S, Yin H, Tammela T, Papagiannakopoulos T, Joshi NS, Cai W, Yang G, Bronson R, Crowley DG, et al. . CRISPR-mediated direct mutation of cancer genes in the mouse liver. Nature 2014; advance online publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zender L, Xue W, Cordón-Cardo C, Hannon GJ, Lucito R, Powers S, Flemming P, Spector MS, Lowe SW. Generation and analysis of genetically defined liver carcinomas derived from bipotential liver progenitors. Cold Spring Harb Sym 2005; 70:251-61; http://dx.doi.org/ 10.1101/sqb.2005.70.059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zender L, Spector MS, Xue W, Flemming P, Cordon-Cardo C, Silke J, Fan S-T, Luk JM, Wigler M, Hannon GJ, et al. . Identification and validation of oncogenes in liver cancer using an integrative oncogenomic approach. Cell 2006; 125:1253-67; PMID:16814713; http://dx.doi.org/ 10.1016/j.cell.2006.05.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Saborowski A, Saborowski M, Davare MA, Druker BJ, Klimstra DS, Lowe SW. Mouse model of intrahepatic cholangiocarcinoma validates FIG–ROS as a potent fusion oncogene and therapeutic target. Proc Natl Acad Sci 2013; 110:19513-8; PMID:24154728; http://dx.doi.org/ 10.1073/pnas.1311707110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Holczbauer Á, Factor VM, Andersen JB, Marquardt JU, Kleiner DE, Raggi C, Kitade M, Seo D, Akita H, Durkin ME, et al. . Modeling pathogenesis of primary liver cancer in lineage-specific mouse cell types. Gastroenterol 2013; 145:221-31; http://dx.doi.org/ 10.1053/j.gastro.2013.03.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. O'Dell MR, Li Huang J, Whitney-Miller CL, Deshpande V, Rothberg P, Grose V, Rossi RM, Zhu AX, Land H, Bardeesy N, et al. . KrasG12D and p53 mutation cause primary intrahepatic cholangiocarcinoma. Cancer Res 2012; 72:1557-67; PMID:22266220; http://dx.doi.org/ 10.1158/0008-5472.CAN-11-3596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Walesky C, Edwards G, Borude P, Gunewardena S, O'Neil M, Yoo B, Apte U. Hepatocyte nuclear factor 4 alpha deletion promotes diethylnitrosamine-induced hepatocellular carcinoma in rodents. Hepatol 2013; 57:2480-90; http://dx.doi.org/ 10.1002/hep.26251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sia D, Hoshida Y, Villanueva A, Roayaie S, Ferrer J, Tabak B, Peix J, Sole M, Tovar V, Alsinet C, et al. . Integrative molecular analysis of intrahepatic cholangiocarcinoma reveals 2 classes that have different outcomes. Gastroen 2013; 144:829-40; http://dx.doi.org/ 10.1053/j.gastro.2013.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang F, Travins J, DeLaBarre B, Penard-Lacronique V, Schalm S, Hansen E, Straley K, Kernytsky A, Liu W, Gliser C, et al. . Targeted inhibition of mutant IDH2 in leukemia cells induces cellular differentiation. Science 2013; 340:622-6; PMID:23558173; http://dx.doi.org/ 10.1126/science.1234769 [DOI] [PubMed] [Google Scholar]
- 38. Rohle D, Popovici-Muller J, Palaskas N, Turcan S, Grommes C, Campos C, Tsoi J, Clark O, Oldrini B, Komisopoulou E, et al. . An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science 2013; 340:626-30; PMID:23558169; http://dx.doi.org/ 10.1126/science.1236062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Davis MI, Gross S, Shen M, Straley KS, Pragani R, Lea WA, Popovici-Muller J, DeLaBarre B, Artin E, Thorne N, et al. . Biochemical, cellular, and biophysical characterization of a potent inhibitor of mutant isocitrate dehydrogenase IDH1. J Biol Chem 2014; 289:13717-25; PMID:24668804; http://dx.doi.org/ 10.1074/jbc.M113.511030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Stein E. Clinical safety and activity in a phase I trial of AG-221, a first in class, potent inhibitor of the IDH2-mutant protein, in patients with IDH2 mutant positive advanced hematologic malignancies [abstract]. Proc Ann Meeting AACR 2014:CT103. [Google Scholar]
- 41. Chen C, Liu Y, Lu C, Cross JR, Morris JP, Shroff AS, Ward PS, Bradner JE, Thompson C, Lowe SW. Cancer-associated IDH2 mutants drive an acute myeloid leukemia that is susceptible to Brd4 inhibition. Gene Dev 2013; 27:1974-85; PMID:24065765; http://dx.doi.org/ 10.1101/gad.226613.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kats Lev M, Reschke M, Taulli R, Pozdnyakova O, Burgess K, Bhargava P, Straley K, Karnik R, Meissner A, Small D, et al. . Proto-oncogenic role of mutant IDH2 in leukemia initiation and maintenance. Cell Stem Cell 2014; 14:329-41; PMID:24440599; http://dx.doi.org/ 10.1016/j.stem.2013.12.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lu C, Venneti S, Akalin A, Fang F, Ward PS, DeMatteo RG, Intlekofer AM, Chen C, Ye J, Hameed M, et al. . Induction of sarcomas by mutant IDH2. Gene Dev 2013; 27:1986-98; PMID:24065766; http://dx.doi.org/ 10.1101/gad.226753.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Reitman ZJ, Duncan CG, Poteet E, Winters A, Yan L-J, Gooden DM, Spasojevic I, Boros LG, Yang S-H, Yan H. Cancer-associated isocitrate ehydrogenase 1 (IDH1) R132H mutation and d-2-hydroxyglutarate stimulate glutamine metabolism under hypoxia. J Biol Chem 2014; 289:23318-28; PMID:24986863; http://dx.doi.org/ 10.1074/jbc.M114.575183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Grassian AR, Parker SJ, Davidson SM, Divakaruni AS, Green CR, Zhang X, Slocum KL, Pu M, Lin F, Vickers C, et al. . IDH1 mutations alter citric acid cycle metabolism and increase dependence on oxidative mitochondrial metabolism. Cancer Res 2014; 74:3317-31; PMID:24755473; http://dx.doi.org/ 10.1158/0008-5472.CAN-14-0772-T [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lewis Caroline A, Parker Seth J, Fiske Brian P, McCloskey D, Gui Dan Y, Green Courtney R, Vokes Natalie I, Feist Adam M, Vander Heiden Matthew G, Metallo Christian M. Tracing compartmentalized NADPH metabolism in the cytosol and mitochondria of mammalian cells. Mol Cell 2014; 55:253-63; PMID:24882210; http://dx.doi.org/ 10.1016/j.molcel.2014.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]