

Through multiple steps of invasion and metastasis cancer cells can escape from the primary tumor, and colonize at distant sites.1 These processes are triggered by mutations though not all of them pose the same risk. Evolution of invasion and metastasis, in particular in colon cancer, has been studied in experimental models, however, the mechanism that triggers the process is still not clear and the available mice models of colon cancer are far from being satisfactory.2 To create an experimental mouse model of invasive colon cancer, one needs to address the question what are the major players and the driver mutations inducing the invasion? One of the early events of metastasis is assumed to be epithelial to mesenchymal transition (EMT), during which the tumor cells change their phenotype, lose adhesion contacts with the neighboring cells and gain ability to migrate.1
In normal cells the EMT program is silenced. What are candidate pathways suspected to contribute to the switch toward EMT phenotype? Although evidences on molecular regulators of EMT are documented in multiple studies, the most important EMT regulators and their possible synergistic interactions have to be elucidated.3 The question to be addressed is what genetic alterations will remove the inhibitory barrier? Since single transgenic mouse mutants currently described never developed distant metastasis, we hypothesized that reactivation of EMT program requires perturbing the activity of several genes that can interact in a synergistic manner. Testing experimentally all possible synergistic combinations of EMT regulators is costly and time-consuming. Therefore, we designed a computational systems biology approach for determining the synergistic interactions between molecular players triggering the switch to EMT.4
We first collected information from the literature and pathway databases to reconstruct a comprehensive map of signaling processes reported to regulate EMT.5 The map depicts the mechanisms of EMT regulation by p53, Notch, AKT, Wnt, TGFbeta signaling and microRNAs. The core EMT program is represented by EMT transcription factors SNAIL, SLUG, TWIST, ZEB1 and ZEB2 and their partially overlapping sets of downstream target genes executing the EMT program. Important regulators of differentiation, apoptosis and proliferation are included into the map to depict regulatory circuits between these processes and EMT.
How to figure out what are the main inducers of the EMT program out of hundreds players on the signaling map? Structural analysis and stepwise simplification of the map allowed identifying the core regulatory mechanisms and highlighting the role of the key players.6 It has been concluded that the balance between the positive (Notch and Wnt, at the transcriptional level) and the negative (p53, p63 and p73 mediated by microRNAs at the level of translation) effectors of EMT determines the EMT phenotype (Fig. 1). The reduced EMT regulation network has been used for evaluating the effect of double mutants, which led to the prediction that the simultaneous activation of Notch and inactivation of p53 genes is capable to trigger the EMT phenotype. Based on the network analysis we were able to suggest the mechanistic model of their synergy (Fig. 1).
Figure 1.
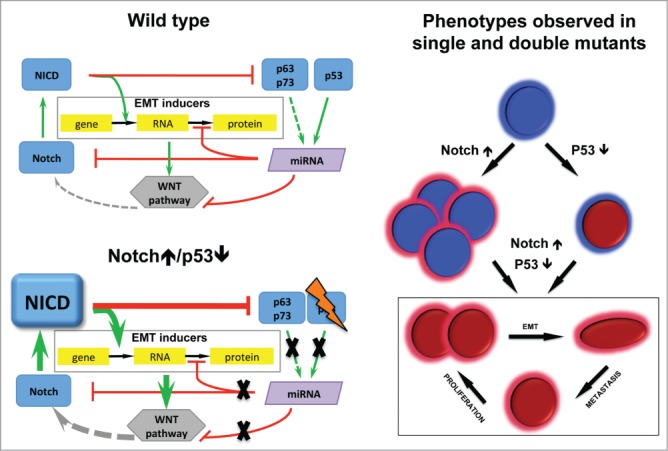
Mechanism of EMT induction derived from signaling network analysis validated in the mouse model. In Wild type EMT program is inhibited at the level of Notch and EMT genes by microRNAs activated by p53 family. Notch↑/p53↓ results in EMT genes induction, stabilised by WNT pathway. Phenotypes observed in single and double mutants: Notch↑ proliferative adenomas; p53↓ vulnerable to cancer; Notch↑/p53↓ proliferate, EMT, metastasize.
This hypothesis has been validated in transgenic mouse model expressing a constitutively active Notch1 receptor in a p53-deleted background, specifically in the digestive epithelium. Linking green fluorescent protein (GFP) expression to the Notch1 receptor activation allows lineage tracing of epithelial tumor cells during cancer progression and invasion. These mice develop digestive tumors with dissemination of EMT-like epithelial malignant cells to the distant organs. The model allows following a tumor cell through its way from the initial steps of EMT up to the metastatic sites, providing clear evidence that metastasis is indeed the consequence of EMT.7
Finally, we have shown that in invasive human colon cancer samples and their related metastases, EMT markers were enhanced together with a negative regulation of epithelial markers. This phenotype in human samples is associated with modulation of Notch, Wnt and p53 downstream targets expression similarly to the mice model, supporting a synergy between these genes to permit EMT induction also in human disease.7
There are several significant achievements in the study: first, the prediction of synergy between Notch activation and p53 downregulation, which we confirmed experimentally, demonstrated that there are ways to reach permissive conditions that induce EMT in addition to those already described in the literature. Second, the comprehensive signaling map is a bioinformatics resource on the relationship between EMT and other cellular processes. The map can be used in further studies on the role of mutations not only in colon cancer but also in other cancer types. Third, the new EMT-prone mouse model, thanks to the lineage tracing technology, provides a compelling evidence for causal relationship between EMT and the consequent appearance of metastases. Fourth, the developed mouse model can be used to decipher the role of the microenvironment in metastases development. Indeed, it is possible to follow the evolution of cancer cells using the lineage tracer, and in parallel, to dissect the composition of stroma and extracellular matrix surrounding the invading cancer cell. Fifth, the Notch+/+ p53−/− mouse is a relevant model mimicking invasive human colon cancer with a potential for therapeutic anti-metastatic drug discovery.7
References
- 1.Valastyan S, Weinberg RA. Cell 2011; 147:275-92; PMID:22000009; http://dx.doi.org/ 10.1016/j.cell.2011.09.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Davies EJ, et al.. J Pathol 2013; 233:27-38; PMID: 24293351; http://dx.doi.org/25688112 10.1002/path.4312 [DOI] [PubMed] [Google Scholar]
- 3.Thiery JP, Sleeman JP. Mol Cell Biol 2006; 7:131-142; PMID: 16493418.25688112 [DOI] [PubMed] [Google Scholar]
- 4.Kuperstein I, et al.. Mutagenesis 2015; 30(2):191-204; PMID:25688112; http://dx.doi.org/ 10.1093/mutage/geu078 [DOI] [PubMed] [Google Scholar]
- 5.Kuperstein I, et al.. BMC Syst Biol 2013; 7:100; PMID:24099179; http://dx.doi.org/ 10.1186/1752-0509-7-100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bonnet E, et al. BMC Syst Biol 2013; 7:18; PMID:23453054; http://dx.doi.org/ 10.1186/1752-0509-7-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chanrion M, et al.. Nat Commun 2014; 5:5005; PMID:25295490; http://dx.doi.org/ 10.1038/ncomms6005 [DOI] [PMC free article] [PubMed] [Google Scholar]