

Abstract
Arterial calcification due to CD73 deficiency (ACDC), an autosomal recessive disorder, manifests with extensive mineralization of the lower-extremity arteries as well as of hand and foot joint-capsules. This disease is caused by mutations in the NT5E gene which encodes CD73, a membrane-bound ecto-5′-nucleotidase hydrolyzing 5′-AMP into adenosine and Pi. To gain insight into the pathophysiologic details of ACDC, we have characterized a Nt5e−/− knock out mouse (Nt5etm1Jgsc) deficient in CD73. These mice, when maintained on appropriate strain background, demonstrated stiffening of the joints and micro CT revealed distinct changes in the thoracic skeletal structure with evidence of mineralization at the costochondral junctions. Mineralization was also noted in the juxta-articular spaces of the lower extremities as well as of ligaments and capsules adjacent to the bony structures. No evidence of vascular mineralization was noted either by CT or by microdissection of arteries in the thoracic area or in lower extremities. The Nt5e−/− mutant mice demonstrated significantly increased Pi levels in the serum and significantly reduced PPi concentration in the heparinized plasma, resulting in markedly increased Pi/PPi ratio, thus creating a pro-mineralization environment. In conclusion, the Nt5e−/− targeted mutant mice recapitulate some, but not all, features of ACDC and serve as a model system to study pharmacologic interventions for ectopic mineralization. Collectively, this mouse model deficient in CD73, with other targeted mutant mice with vascular mineralization, attests to the presence of a complex pro-mineralization/anti-mineralization network that under physiologic homeostatic conditions prevents ectopic tissue mineralization.
Keywords: Ectopic mineralization, animal models, mineralization networks
Abbreviations
- ACDC
arterial calcification due to CD73 deficiency
- PXE
pseudoxanthoma elasticum
- GACI
generalized arterial calcification of infancy
- CT
computed tomography
- TNAP
tissue nonspecific alkaline phosphatase
- Pi
inorganic phosphate
- PPi
inorganic pyrophosphate
- ENT1
equilibrative nucleoside transporter
Introduction
CD73 is a membrane-bound ecto-5′-nucleotidase that catalyzes hydrolysis of extracellular nucleoside monophosphates into nucleoside intermediates.1,2 Specifically, CD73 converts 5′-AMP into adenosine and inorganic phosphate (Pi). The newly generated adenosine, acting on 4 G-protein receptors, plays critical physiological roles and can also be involved in pathological processes.3 At the same time, Pi can serve as a pro-mineralization factor, particularly in the context of increased inorganic phosphate/pyrophosphate (Pi/PPi) ratio.4
In 2004, 3 research groups independently generated knock out mice by targeted disruption of the mouse Nt5e, the gene encoding CD73.5-7 These mice were subsequently characterized to show altered thromboregulation and augmented vascular inflammatory response, lack of CD73 was shown to promote arteriogenesis, and the Nt5e−/− mice manifested with fulminant vascular leakage during hypoxia. In 2011, St. Hilare et al. reported on 3 families with 9 affected individuals with mutations in the NT5E gene which encodes human CD73 – this disease is known as arterial calcification due to CD73 deficiency (ACDC).8 These patients, as originally described about 100 y ago,9,10 manifested with extensive calcification of the lower-extremity arteries as well as hand and foot joint-capsules. In particular, contrast-enhanced magnetic-resonance angiography revealed extensive occlusion of the iliac, femoropopliteal and tibial arteries, and X-ray findings showed heavy calcification with areas of arteriomegaly. Interestingly, no vascular calcification was detected by chest X-rays above the diaphragm. Radiography also revealed juxta-articular joint-capsule calcifications in the digits, wrists, ankles and feet. These 3 families were found to have loss-of-function mutations in the NT5E gene, and cultured fibroblasts from affected members from one of the families showed markedly reduced expression of NT5E mRNA, CD73 protein, and enzymatic activity.8,11 These cells were also shown to have increased alkaline phosphatase levels and accumulation of calcium phosphate crystals.
With our long term interest in heritable ectopic mineralization disorders affecting primarily the skin and arterial blood vessels,12 we have now characterized a mutant Nt5e−/− mouse deficient in CD73, and we demonstrate that these mice recapitulate some, but not all, clinical features noted in patients with NT5E mutations.
Results
Generation of Nt5etm1jgsc mice
CD73-deficient mice, generated by targeted inactivation of the Nt5e gene, as described previously,5 were obtained from the National Institutes of Health (courtesy of Dr. Jurgen Schnermann). These mutant mice, Nt5etm1Jgsc, are referred from here on as Nt5e−/− mice. These mice failed to demonstrate Nt5e mRNA sequences beyond the targeted 2nd and 3rd exon, and they lacked CD73 by immunostaining of kidneys, the site depicting the presence of the protein in wild-type mice. The breeding pairs, which were on the 129SvEv strain background, were used to generate homozygous, heterozygous, and wild-type mice with respect to the mutant Nt5e allele. These mice were placed on a specific diet, so-called “acceleration diet” rich in phosphate and low in magnesium, which we have previously shown to accelerate the mineralization in Abcc6tm1Jfk and Enpp1asj mutant mice, animal models of pseudoxanthoma elasticum (PXE) and generalized arterial calcification of infancy (GACI), two ectopic heritable mineralization disorders, respectively.13-15 The animals were sacrificed at different time points up to 11 months of age and examined histologically for systemic mineralization. Initially, all mice, irrespective of the Nt5e gene, demonstrated, extensive mineralization in soft connective tissues, including the dermal sheath of vibrissae and arterial blood vessels, similar to previous demonstrations in a number of mouse strains serving as models of PXE. Specifically, we have previously shown that 4 mouse strains with ectopic mineralization, viz., 129S1/SvImJ, C3H/HeJ, DBA/2J, and KK/HlJ, harbor a single nucleotide polymorphism (SNP; rs32756904) in the Abcc6 gene (Fig. 1).16 This nucleotide variant elicits aberrant splicing of the Abcc6 mRNA, causing deletion of 5 nucleotides at the end of exon 14 and resulting in a frame shift and premature termination codon of translation.17,18 As a consequence ABCC6 protein levels in the liver are markedly reduced, associated with ectopic mineralization of peripheral connective tissues. Examination of the Abcc6 genomic sequence in the Nt5e−/- mice revealed the presence of this SNP (Fig. 1), and consequently, this mutation is likely to explain the mineralization phenotype in these mice independent of the Nt5e mutation status.
Figure 1.
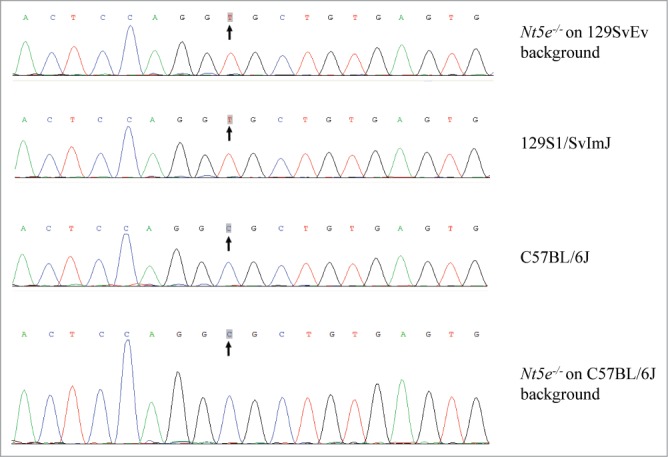
Genomic sequences of a SNP (rs32756904) in the Abcc6 gene of mice on different strain backgrounds. The original Nt5e−/− mouse was developed on 129SvEv background (top line) which harbors a homozygous T nucleotide similar to 129S1/SvImJ (2nd row from top). The T allele is associated with ectopic mineralization. The Nt5e allele was separated from the mutant Abcc6 allele by crossing the mice on to C57BL/6J strain background which is homozygous for the C allele of the SNP (lower rows).
Phenotypic characterization of Nt5e−/− mice
To separate the Nt5e locus in chromosome 9 from the Abcc6 mutant allele in chromosome 7, the Nt5e mutant mice were crossed onto C57BL/6J background by 3 back crosses, which eliminated the mutant allele of the Abcc6 gene (Fig. 1). These mice were subsequently intercrossed to generate homozygous, heterozygous, and wild-type mice with respect to the Nt5e mutant allele. These mice were then placed on the acceleration diet.
To explore the potential of mineralization, complete necropsies of the Nt5e−/− mice were performed at 11–16 months of age.19 These mice, together with heterozygous and wild-type mice, developed mineralization in the kidneys, a finding that we previously demonstrated in mice kept on this particular “acceleration diet.”20 However, no evidence of soft tissue calcification, particularly in arteries, was noted in Nt5e−/− mice. It should be noted, however, that the complete necropsy protocol utilized does not allow detection of mineral deposits on cartilage or on soft tissues adjacent to long bones due to de-calcification of skeletal tissues.
Since ACDC patients develop mineralization of cartilage and juxta-articular joint-capsules as well as in arterial blood vessels of the lower extremities,8 potential development of ectopic mineralization on these sites was monitored in live animals with micro computed tomography (CT) scan at various time points. CT imaging of Nt5e−/− mice revealed consistently distinct changes in the thoracic skeletal structures with evidence of juxta-articular mineralization of the costochondral junctions (Fig. 2). Evidence of mineralization was also noted in the juxta-articular spaces of the lower extremities most notably at the level of the patella and the distal insertion of the fibula and tibia. Mineralization of the Achilles tendon was also observed. In contrast, no juxta-articular mineralization of the upper extremities was found. Careful examination of the extremities by the micro CT scan revealed evidence suggestive of ectopic mineralization of ligaments and capsules adjacent to the bone structures (Fig. 2). No evidence of aberrant mineralization was noted in heterozygous or wild-type littermates.
Figure 2.
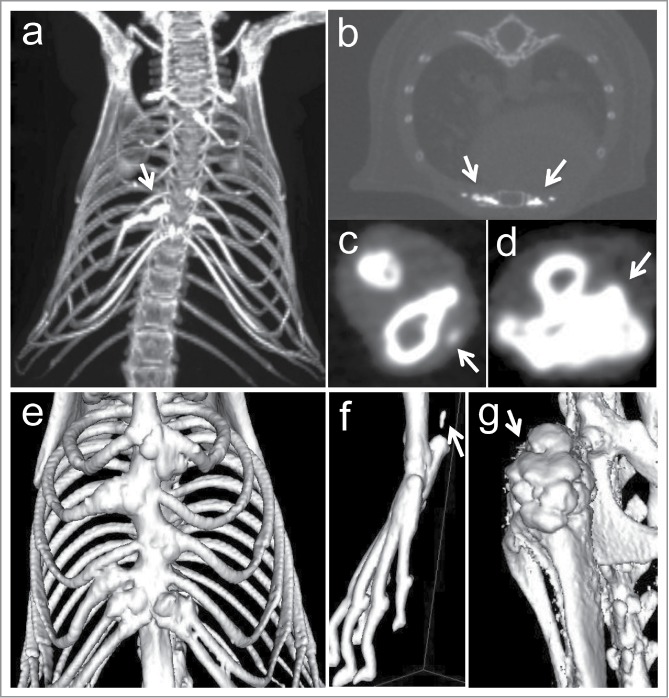
Representative images of micro CT scans of Nt5e−/− mice of 11–16 months of age. Note extensive ectopic mineralization of the sternocostal junctions (A, B), juxta-articular tissues in lower extremities (C), and knee (D) (arrows). Three-D renderings reveal skeletal deformities due to ectopic mineralization of thorax (E), foot (F), and knee (G). Note mineralization of the Achilles tendon in (F) (arrow).
With particular attention to the vascular mineralization in ACDC patients, we performed micro-dissection of saphenous, medial tarsal, medial marginal, and lateral marginal arteries as well as internal mammary and intercostal arteries in Nt5e−/− mice. Histopathologic examination by Hematoxylin-Eosin and Alizarin Red stains failed to provide evidence of vascular mineralization.
Finally, clinical assays were performed in the blood of the Nt5e−/− mutant mice to determine the serum concentration of calcium and inorganic phosphate (Pi) and the plasma inorganic pyrophosphate (PPi) (Table 1). No difference in the serum calcium concentration was noted between the 3 groups of mice. However, the Pi levels in the serum of Nt5e−/− mice were significantly increased, by 43% on the average, as compared to wild-type mice (p < 0.01). At the same time, the plasma PPi concentration was significantly reduced, by ∼50%, in the knock out mice as compared to their wild-type littermates. Consequently the Pi/PPi ratio was increased by 2.27-fold in the knockout mice compared to the corresponding controls. The heterozygous Nt5e+/− mice showed Pi and PPi values intermediate between the corresponding homozygous and wild-type mice, but they were not statistically different from the wild-type mice due to individual variations. No difference was noted in alkaline phosphatase activity in serum.
Table 1.
Blood parameters in Nt5e wild type, as well as heterozygous and homozygous mutant mice
| Nt5e genotype* | Serum/plasma concentration (mean ± S.E.) |
|||||
|---|---|---|---|---|---|---|
| Ca2+ (mg/dl) | Pi (mg/dl) | Ca/Pi ratio | PPi (M) | Pi/PPi ratio | Alp activity (U/L) | |
| WT | 10.20 ± 0.27 | 9.89 ± 0.67 | 1.05 ± 0.04 | 2.47 ± 0.39 | 269.60 ± 48.03 | 90.97 ± 10.00 |
| Het | 9.72 ± 0.11 | 11.83 ± 1.12 | 0.87 ± 0.07 | 2.11 ± 0.52 | 493.21 ± 110.93 | 114.76 ± 17.96 |
| KO+ | 10.05 ± 0.13 | 14.19 ± 0.82# | 0.73 ± 0.04# | 1.30 ± 0.16# | 672.38 ± 111.13* | 95.76 ± 9.67 |
WT, wild type; Het, heterozygous; KO, knock out.
Statistical significance in comparison to wild type mice is indicated: * p < 0.05; # p < 0.01 (n = 5–12). Calcium, Pi and Alp activity were measured in serum samples, and PPi was measured in heparinized plasma samples.
Alp, alkaline phosphatase.
Discussion
A number of heritable disorders have been shown to affect the peripheral connective tissues by ectopic mineralization. The prototype for such conditions is PXE, a multisystem mineralization disorder affecting the skin, the eyes, and the cardiovascular system.21 This disease, caused by a mutations in the ABCC6 gene, characteristically shows late onset and slow progression of the pathological mineralization processes. In contrast, GACI, caused by mutations in the ENNP1 gene, is frequently diagnosed by pre- or perinatal ultrasound and the affected individuals, born with extensive mineralization of arterial blood vessels, die in most cases from cardiovascular complications during the first year of life.22 ACDC, arterial calcification due to CD73 deficiency, has been reported in elderly patients with calcification of arteries in the lower extremities, associated with mineralization of the juxta-articular joint-capsules.8 These patients harbor loss-of-function mutations in the NT5E gene which encodes CD73, an ecto-5′-nucleotidase that catalyzes the hydrolysis of 5′-AMP into adenosine and Pi. It has been proposed that under physiological homeostasis adenosine inhibits the activity of tissue non-specific alkaline phosphatase (TNAP), an enzyme that converts PPi to Pi.8,11 In CD73 deficiency the tissue levels of adenosine are reduced, lessening the inhibition of TNAP, and consequently the generation of Pi from PPi is accelerated, and the ratio of Pi/PPi increases creating a pro-mineralization environment (Fig. 3).
Figure 3.
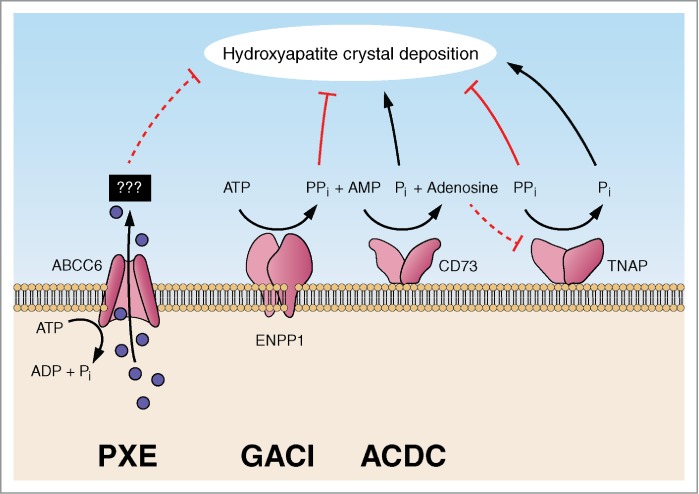
Schematic presentation of factors contributing to thepro-mineralization/anti-mineralization network preventing ectopicmineralization under physiologic conditions. Genetic defects in the genes encoding ABCC6, ENPP1 and CD73 result in ectopic mineralization in pseudoxanthoma elasticum (PXE), generalized arterial calcification of infancy (GACI) and arterial calcification due to CD73 deficiency (ACDC), respectively. (Modified from reference 12).
A number of animals, particularly targeted or spontaneous mutant mice, have served as models of the ectopic mineralization disorders, and they have been extremely helpful in deciphering the pathophysiologic details of many of these conditions.12,23 For example, generation of the Abcc6-/- mice as a model for PXE has greatly facilitated the understanding of the pathomechanistic features of this complex disorder.24,25 Similarly, generation of targeted mutant Enpp1−/− mice or identification of spontaneous Enpp1 mutant mice, such as asj and ttw, have provided novel insight into the mineralization processes in GACI. These mouse models now serve as a platform to test novel treatment modalities for these, currently intractable diseases.15,26,27
Several targeted mutant mice have been developed by disruption of the Nt5e gene resulting in CD73 deficiency, but very little attention has been paid so far on the mineralization aspects of these mice.5-7 In this study, we have characterized a CD73 deficient mouse which was generated by targeted interruption of the Nt5e gene with complete absence of the CD73 protein, a true null mutation.5 Combination of clinical phenotypes and CT imaging studies indicated extensive mineralization of the juxta-articular joint-capsules in the thoracic area with extensive skeletal abnormalities. In addition, extensive mineralization of the joint capsules, particularly in the lower extremities, was evident. Evidence of ectopic calcification was also noted in soft tissues adjacent to the bone, including the Achilles tendon. Thus, this CD73 deficient mouse model, particularly when placed on an “acceleration diet” which facilitates the mineralization processes, recapitulates features noted in patients with ACDC. However, CT scan or extensive histopathologic evaluation of the soft tissues in the lower legs or in the thoracic area failed to reveal mineralization in the arterial blood vessels, a prominent feature in patients with ACDC.8 The reasons for the lack of vascular mineralization in the mice are currently unknown, but could involve the genetic background or epigenetic factors. The former point was clearly demonstrated by changing the strain background to remove the Abcc6 mutation in the original 129SvEv strain to C57BL/6J. Furthermore, the physiological diversity of the cardiovascular system, including the arterial blood vessels, is highly complex and includes positional identities determined by Hox expression in vascular smooth muscle cells and endothelial cells.28 Presumably, such factors could differ between humans and mice. In addition, a number of genetic modifiers could differentially influence the expression of the phenotype in human and mice in diseases which are thought to be single gene disorders, yet demonstrate considerable phenotypic variability.29 Nevertheless, this mouse model can be used to study the pathomechanistic features and metabolic pathways potentially involved in the development of mineralization in ACDC.
As indicated above, the original publication reporting the association of NT5E mutations and arterial calcification in patients with ACDC postulated increased ratio of Pi/PPi as a trigger of ectopic mineralization as a result of increased TNAP activity.8,11 This postulate was based on in vitro analysis of fibroblast cultures established from patients in a family with ACDC in comparison to the controls. However, the patients’ serum phosphate levels and alkaline phosphatase activity were all within normal range.8 In the Nt5e−/− mice, the serum Pi levels were significantly increased and the plasma PPi levels were markedly decreased leading to over 2-fold increase in the Pi/PPi ratio. These observations are consistent with the postulate that in CD73 deficiency, there is an accelerated conversion of PPi to Pi creating a pro-mineralization environment.
Attesting to the role of adenosine in progressive ectopic mineralization of skeletal tissues is a recent study reporting on mice lacking equilibrative nucleoside transporter 1 (ENT1), which transfers nucleosides, such as adenosine, across the plasma membrane.30 This mouse was developed as a model for diffuse idiopathic skeletal hyperostosis (DISH), a non-inflammatory spondyloarthropathy characterized by ectopic mineralization of spinal tissues. This mouse model was generated through targeted deletion of exons 2–4 of the gene encoding ENT1. Micro CT of the homozygous mice revealed ectopic mineralization of perispinal tissues in the cervical-thoracic region and the knock out mice revealed large, irregular accumulation of eosinophilic material in perispinal ligaments and entheses, intervertebral discs, and sternocostal articulations – some of these findings are reminiscent of those in CD73 deficient mice. However, similar to our findings on Nt5e−/− mice, the Ent1-/- mice showed no evidence of mineralization in blood vessels, indicating specificity of the mineralization process in these mice for the axial skeleton.30 Collectively, the mouse models of ectopic mineralization, including those with CD73 and ENT1 deficiency, attest to the presence of a complex pro-mineralization/anti-mineralization network that under physiologic homeostatic conditions prevents ectopic mineralization of soft connective tissues.12 Alterations in the components of this network, such as altered ratio of Pi/PPi and the adenosine concentration, can result in mineralization pathways with phenotypic tissue specificity.
Materials and Methods
Mice and diet
The CD73 deficient mouse was developed by targeted disruption of the Nt5e gene as previously described.5 Wild-type, heterozygous and homozygous mice with respect to the mutant Nt5e allele were generated from heterozygous matings initially on the 129SvEv strain background. Genotyping of Nt5e gene was performed on genomic DNA isolated from tail clips and analyzed using PCR amplification. All the mice were maintained on an ‘acceleration diet’ (Harlan Teklad, Rodent diet TD.00442, Madison, WI, USA), which is enriched in phosphorus and has reduced magnesium content, compared with standard rodent diet (Laboratory Autoclavable Rodent Diet 5010: PMI Nutritional International, Brentwood, MO, USA).
Due to a homozygous Abcc6 mutation (rs32756904) in the 129SvEv strain background, the CD73 deficient mice were backcrossed with C57BL/6J mice for 3 times to replace this mutation in the Abcc6 gene with the wild type allele. Validation of the absence of the Abcc6 mutation in the progeny was performed by sequencing. Wild-type, heterozygous and homozygous mice with respect to the mutant Nt5e allele were generated from heterozygous matings on the C57BL/6J strain background. All the mice were maintained on the “acceleration diet” as described above.
At 11–16 months of age, the mice were euthanized by CO2 asphyxiation and necropsied for further analysis.19 Mice were maintained under standard conditions at the Animal Facility of Thomas Jefferson University. All protocols were approved by the Institutional Animal Care and Use Committee of Thomas Jefferson University. Proper handling and care were practiced according to the animal welfare policies of the Public Health Service.
Small-animal CT (micro CT) scan
Nt5e wild-type, heterozygous and homozygous mice were examined for mineralization at 11–16 months of age by small animal micro CT scan. Briefly, mice were anesthetized with a xylazine-ketamineacetopromazine cocktail (160 μl per 25 g body weight of 10 mg/kg xylazine, 200 mg/kg ketamine, 2 mg/kg acetopromazine) and then scanned with a MicroCAT II (ImTek Inc., Oak Ridge, TN). A 3-dimensional rendering was created for each mouse using Amira software, version 3.1 (Visualization Sciences Group, Burlington, MA, USA).
Complete necropsy
After euthanizing the mice, complete necropsies were performed on a subset of 5 mice at the age of 12 months.19 Briefly, tissues from all organs (Swiss rolls of the duodenum, jejunum, ileum, and colon (with anus and perineal skin), longitudinal section of the stomach with esophagus and cecum (inflated with fixative), cross sections of the left lateral and medial lobes of liver to include the gall bladder, spleen, left and right kidneys with adrenal glands, reproductive organs (testis, epidydimis, accessory sex organs, male; ovary, uterine tube, uterus, mammary glands, female), preputial gland for males/clitoral gland for females, salivary gland cluster with cervical lymph nodes, heart, esophagus and trachea with thyroid and parathyroid glands, tongue, longitudinal sections out of the center of the lobes of both lungs, dorsal skin, ear skin (pinna), ventral skin, muzzle skin, eyelid, longitudinal section of hind leg, include stifle/knee joint, longitudinal section of front leg, include shoulder and elbow joints, longitudinal section of hind foot (soft tissues, bone, and nail unit/footpad), longitudinal section of front foot (soft tissues, bone, and nail unit/footpad), longitudinal section and 1 cross section of the lumbar spine, longitudinal section and 1 cross section of the tail, sections of the lower jaw) were collected and fixed in 10% phosphate-buffered formalin overnight, after which they were transferred and stored in 70% ethanol. Bones were processed in Cal-Ex® (Fisher Thermo Scientific, Waltham, MA, USA). Tissues were then trimmed and embedded in paraffin, cut into 6 μm sections, and stained with Hematoxylin-Eosin (H&E). All tissue slides were reviewed by an experienced, board-certified veterinary pathologist (JPS).
Histopathological analysis of mineralization
Organs from euthanized mice were fixed in 10% phosphate-buffered formalin and embedded in paraffin. The tissues were sectioned (6 μm thick), placed onto glass slides, and stained with Hematoxylin-Eosin (HE) or Alizarin Red (AR) stain using standard procedures. Slides were examined for mineralization under a Nikon (Tokyo, Japan) Te2000 microscope and an AutoQuant imaging system (AutoQuant Imaging, Watervliet, NY, USA).
Chemical quantification of calcium and phosphate in serum
Calcium levels in serum were determined colorimetrically by the ơ-cresolphthalein complexone method (Calcium (CPC) Liquicolor; Stanbio Laboratory, Boerne, TX, USA). Phosphate content in serum was determined with a Malachite Green phosphate assay kit (BioAssay Systems, Hayward, CA, USA).
Plasma collection and inorganic pyrophosphate assay
Whole blood was collected by cardiac puncture into heparin coated blood collection tubes and kept on ice until separation of plasma and erythrocytes by centrifugation. The plasma was collected, depleted of platelets by filtration (2,200 × g at 4°C for 20 min) through a Centrisart I 300,000-kDa mass cutoff filter (Sartorius, New York, NY, USA), and stored at −80°C until further processing. PPi in plasma was measured by an enzymatic assay using uridine-diphosphoglucose (UDPG) pyrophosphorylase, with modifications, as described previously.31-33
Alkaline phosphatase activity in serum
Serum levels of alkaline phosphatase were measured using a quantitative colorimetric assay kit from Abcam (Cambridge, MA, USA).
Statistical analysis
The comparisons between different groups of mice were completed using the 2-sided Kruskal–Wallis nonparametric test, comparable to one-way analysis of variance but without the parametric assumptions. All statistical computations were completed using the SPSS version 5.0 software (SPSS, Chicago, IL, USA).
Funding Statement
This work was supported by NIH/NIAMS grants K01AR064766 (QL), R21AR063781 (JPS), and R01AR28450 (JU).
Acknowledgment
The authors thank Dian Wang, Joshua Kingman, and Kathleen Silva for technical assistance. Dr. Madhukar Thakur and Neil Mehta assisted in micro CT scanning. Dr. Jurgen Schnermann, NIDDK/NIH, provided the original breeding pairs of Nt5e knock out mice. Carol Kelly assisted in manuscript preparation.
References
- 1. Zimmermann H. 5′-Nucleotidase: molecular structure and functional aspects. BiochemJ 1992; 285:345–65; PMID: 1637327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Linden J. Molecular approach to adenosine receptors: receptor-mediated mechanisms of tissue protection. Annu Rev PharmacolToxicol 2001; 41:775–87; PMID: 11264476; http://dx.doi.org/10.1146/annurev.pharmtox.41.1.775 [DOI] [PubMed] [Google Scholar]
- 3. Fredholm BB. Adenosine-a physiological or pathophysiological agent? J MolMed 2014; 92:201–6; PMID: 24362516; http://dx.doi.org/10.1007/s00109-013-1101-6 [DOI] [PubMed] [Google Scholar]
- 4. Nitschke Y, Weissen-Plenz G, Terkeltaub R, Rutsch F. Npp1 promotes atherosclerosis in ApoE knockout mice. J Cell MolMed 2011; 15:2273–83; PMID: 21477221; http://dx.doi.org/10.1111/j.1582-4934.2011.01327.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Castrop H, Huang Y, Hashimoto S, Mizel D, Hansen P, Theilig F, Bachmann S, Deng C, Briggs J, Schnermann J. Impairment of tubuloglomerular feedback regulation of GFR in ecto-5′-nucleotidase/CD73-deficient mice. J ClinInvest 2004; 114:634–42; PMID: 15343381; http://dx.doi.org/10.1172/JCI21851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Koszalka P, Ozuyaman B, Huo Y, Zernecke A, Flogel U, Braun N, Buchheiser A, Decking UK, Smith ML, Sévigny J, et al. Targeted disruption of cd73/ecto-5′-nucleotidase alters thromboregulation and augments vascular inflammatory response. CircRes 2004; 95:814–21; PMID: 15358667; http://dx.doi.org/10.1161/01.RES.0000144796.82787.6f [DOI] [PubMed] [Google Scholar]
- 7. Thompson LF, Eltzschig HK, Ibla JC, Van De Wiele CJ, Resta R, Morote-Garcia JC, Colgan SP. Crucial role for ecto-5′-nucleotidase (CD73) in vascular leakage during hypoxia. J ExpMed 2004; 200:1395–405; PMID: 15583013; http://dx.doi.org/10.1084/jem.20040915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. St Hilaire C, Ziegler SG, Markello TC, Brusco A, Groden C, Gill F, Carlson-Donohoe H, Lederman RJ, Chen MY, Yang D, et al. NT5E mutations and arterial calcifications. N Engl JMed 2011; 364:432–42; PMID: 21288095; http://dx.doi.org/10.1056/NEJMoa0912923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Von Magnus-Levy A. Ueber ungewohnliche Verkalkung der Arterien. Dtsch MeidzinischeWochenschrift 1914; 40:1305–9; http://dx.doi.org/10.1055/s-0029-1190517 [Google Scholar]
- 10. Sharp J. Heredo-familial vascular and articular calcification. Ann RheumDis 1954; 13:15–27; PMID: 13149051; http://dx.doi.org/10.1136/ard.13.1.15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Markello TC, Pak LK, St Hilaire C, Dorward H, Ziegler SG, Chen MY, Chaganti K, Nussbaum RL, Boehm M, Gahl WA. Vascular pathology of medial arterial calcifications in NT5E deficiency: implications for the role of adenosine in pseudoxanthoma elasticum. Mol GenetMetab 2011; 103:44–50; PMID: 21371928; http://dx.doi.org/10.1016/j.ymgme.2011.01.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Li Q, Uitto J. Mineralization/anti-mineralization networks in the skin and vascular connective tissues. Am JPathol 2013; 183:10–18; PMID: 23665350; http://dx.doi.org/10.1016/j.ajpath.2013.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Li Q, Uitto J. The mineralization phenotype in Abcc6 ( -/- ) mice is affected by Ggcx gene deficiency and genetic background – a model for pseudoxanthoma elasticum. J Mol Med (Berl) 2010; 88:173–81; PMID: 19784827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jiang Q, Uitto J. Restricting dietary magnesium accelerates ectopic connective tissue mineralization in a mouse model of pseudoxanthoma elasticum (Abcc6−/−). ExpDermatol 2012; 21:694–9; PMID: 22897576; http://dx.doi.org/10.1111/j.1600-0625.2012.01553.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Li Q, Guo H, Chou DW, Berndt A, Sundberg JP, Uitto J. Mutant Enpp1asj mouse as a model for generalized arterial calcification of infancy. Dis ModelMech 2013; 6:1227–35; PMID: 23798568; http://dx.doi.org/10.1242/dmm.012765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Berndt A, Li Q, Potter CS, Liang Y, Silva KA, Kennedy V, Uitto J, Sundberg JP. A single-nucleotide polymorphism in the Abcc6 gene associates with connective tissue mineralization in mice similar to targeted models for pseudoxanthoma elasticum. J InvestDermatol 2013; 133:833–6; PMID: 23014343; http://dx.doi.org/10.1038/jid.2012.340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Li Q, Berndt A, Guo H, Sundberg J, Uitto J. A novel animal model for pseudoxanthoma elasticum – the KK/HlJ mouse. Am JPathol 2012; 181:1190–6; PMID: 22846719; http://dx.doi.org/10.1016/j.ajpath.2012.06.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Aherrahrou Z, Doehring LC, Ehlers EM, Liptau H, Depping R, Linsel-Nitschke P, Kaczmarek PM, Erdmann J, Schunkert H. An alternative splice variant in Abcc6, the gene causing dystrophic calcification, leads to protein deficiency in C3H/He mice. J BiolChem 2008; 283:7608–15; PMID: 18201967; http://dx.doi.org/10.1074/jbc.M708290200 [DOI] [PubMed] [Google Scholar]
- 19. Silva KA, Sundburg JP. Necropsy Methods. The LaboratoryMouse. London: Academic Press; 2012 [Google Scholar]
- 20. Li Q, Chou DW, Price TP, Sundberg JP, Uitto J. Genetic modulation of nephrocalcinosis in mouse models of ectopic mineralization: the Abcc6tm1Jfk and Enpp1asj mutant mice. LabInvest 2014; 94:623–32; PMID: 24732453; http://dx.doi.org/10.1038/labinvest.2014.52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Uitto J, Li Q, Jiang Q. Pseudoxanthoma elasticum: molecular genetics and putative pathomechanisms. J InvestDermatol 2010; 130:661–70; PMID: 20032990; http://dx.doi.org/10.1038/jid.2009.411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nitschke Y, Rutsch F. Genetics in arterial calcification: lessons learned from rare diseases. Trends CardiovascMed 2012; 22:145–9; PMID: 23122642; http://dx.doi.org/10.1016/j.tcm.2012.07.011 [DOI] [PubMed] [Google Scholar]
- 23. Mackenzie NC, Huesa C, Rutsch F, MacRae VE. New insights into NPP1 function: lessons from clinical and animal studies. Bone 2012; 51:961–8; PMID: 22842219; http://dx.doi.org/10.1016/j.bone.2012.07.014 [DOI] [PubMed] [Google Scholar]
- 24. Klement JF, Matsuzaki Y, Jiang QJ, Terlizzi J, Choi HY, Fujimoto N, Li K, Pulkkinen L, Birk DE, Sundberg JP, et al. Targeted ablation of the Abcc6 gene results in ectopic mineralization of connective tissues. Mol CellBiol 2005; 25:8299–310; PMID: 16135817; http://dx.doi.org/10.1128/MCB.25.18.8299-8310.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gorgels TG, Hu X, Scheffer GL, van der Wal AC, Toonstra J, de Jong PT, van Kuppevelt TH, Levelt CN, de Wolf A, Loves WJP, et al. Disruption of Abcc6 in the mouse: novel insight in the pathogenesis of pseudoxanthoma elasticum. Hum MolGenet 2005; 14:1763–73; PMID: 15888484; http://dx.doi.org/10.1093/hmg/ddi183 [DOI] [PubMed] [Google Scholar]
- 26. Sali A, Favaloro JM, Terkeltaub R, Goding JW. Germline deletion of the nucleoside triphosphate pyrophosphohydrolase (NTPPPH) plasma cell membrane glycoprotein-1 (PC-1) produces abnormal calcification of periarticular tissues. In: Vanduffel LLR, ed. Ecto-ATPases and related ectoenzymes. Maastricht, The Netherlands: Shaker Publishing, 1999:267–82 [Google Scholar]
- 27. Okawa A, Nakamura I, Goto S, Moriya H, Nakamura Y, Ikegawa S. Mutation in Npps in a mouse model of ossification of the posterior longitudinal ligament of the spine. NatureGenet 1998; 19:271–3; PMID: 9662402; http://dx.doi.org/10.1038/956 [DOI] [PubMed] [Google Scholar]
- 28. Pruett ND, Visconti RP, Jacobs DF, Scholz D, McQuinn T, Sundberg JP, Awgulewitsch A. Evidence for Hox-specified positional identities in adult vasculature. BMC DevBiol 2008; 8:93; PMID: 18826643; http://dx.doi.org/10.1186/1471-213X-8-93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sproule TJ, Bubier JA, Grandi FC, Sun VZ, Philip VM, McPhee CG, Adkins EB, Sundberg JP, Roopenian DC. Molecular identification of collagen 17a1 as a major genetic modifier of laminin gamma 2 mutation-induced junctional epidermolysis bullosa in mice. PLoSGenet 2014; 10:e1004068; PMID: 24550734; http://dx.doi.org/10.1371/journal.pgen.1004068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Warraich S, Bone DB, Quinonez D, Ii H, Choi DS, Holdsworth DW, Drangova M, Dixon SJ, Séguin CA, Hammond JR. Loss of equilibrative nucleoside transporter 1 in mice leads to progressive ectopic mineralization of spinal tissues resembling diffuse idiopathic skeletal hyperostosis in humans. J Bone MinerRes 2013; 28:1135–49; PMID: 23184610; http://dx.doi.org/10.1002/jbmr.1826 [DOI] [PubMed] [Google Scholar]
- 31. Lomashvili KA, Khawandi W, O’Neill WC. Reduced plasma pyrophosphate levels in hemodialysis patients. J Am SocNephrol 2005; 16:2495–500; PMID: 15958726; http://dx.doi.org/10.1681/ASN.2004080694 [DOI] [PubMed] [Google Scholar]
- 32. O’Neill WC, Sigrist MK, McIntyre CW. Plasma pyrophosphate and vascular calcification in chronic kidney disease. Nephrol DialTransplant 2010; 25:187–91; PMID: 19633093; http://dx.doi.org/10.1093/ndt/gfp362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tolouian R, Connery SM, O’Neill WC, Gupta A.. Using a filtration technique to isolate platelet free plasma for assaying pyrophosphate. ClinLab 2012; 58:1129–34; PMID: 23289181 [PMC free article] [PubMed] [Google Scholar]