

Abstract
Cyclin-dependent kinase 5 (Cdk5) is a unique member of a family of serine/threonine cyclin-dependent protein kinases. We previously demonstrated disruption of Cdk5 gene expression in mice impairs T-cell function and ameliorates T-cell-mediated neuroinflammation. Here, we show Cdk5 modulates gene expression during T-cell activation by impairing the repression of gene transcription by histone deacetylase 1 (HDAC1) through specific phosphorylation of the mSin3a protein at serine residue 861. Disruption of Cdk5 activity in T-cells enhances HDAC activity and binding of the HDAC1/mSin3a complex to the IL-2 promoter, leading to suppression of IL-2 gene expression. These data point to essential roles for Cdk5 in regulating gene expression in T-cells and transcriptional regulation by the co-repressor mSin3a.
Introduction
Cyclin-dependent kinase 5 (Cdk5) is distinguished from other serine/threonine CDKs as it is known to modify a very broad range of protein substrates in a manner that is dependent on the specific co-activator proteins p35 and/or p39 and independent of classical cyclins.1 These obligate partners of Cdk5 are both constitutively expressed in neuronal stem cells and post-mitotic neurons, suggesting a more lineage-restricted activity for Cdk5. However, there is increasing evidence suggesting a significant function for Cdk5 exists in other lineages including immune cells and this activity has now been linked to disorders of non-neuronal tissues.2 Cellular processes known to be regulated by Cdk5 include neuronal cell migration and survival,3 T cell activation,4 insulin resistance,5 and cancer cell invasion and metastasis.6 Thus, Cdk5 is now recognized as a potential therapeutic target for diseases including neurodegeneration, autoimmunity and cancer.4,7 The relevance of Cdk5 activity to these disorders may be attributed to a growing list of Cdk5 substrates that include transcription factors such as Stat3,8 modulators of cell viability such as Bcl-29, and actin modulators such as coronin-1a and other members of the moeisin family of proteins.4,10
We have recently shown T cells isolated from Cdk5-deficient immune chimeric mice (Cdk5−/-C) or p35 knockout mice (p35−/-) exhibit a diminished response to T cell receptor (TCR) ligation,4 and that induction of Cdk5 activity during T cell activation is necessary for post-translational modification of coronin-1a, an actin co-modulatory protein essential for T cell survival.11 We hypothesized that Cdk5 must act through a series of coordinated mechanisms to control T cell function and differentiation, and that this may include control of the expression of specific genes required for T cell activation. For example, the suppressed proliferative response of T cells deficient in either the expression or activity of Cdk5 may reflect a defect in the expression of autocrine factors,4 such as IL-2, that are known to be essential for an optimal mitogenic response following TCR activation.12 Indeed, the autocrine expression of IL-2 following T cell activation is important for both T-cell differentiation and survival.13,14
Several studies have recently highlighted a role for the classical zinc-dependent histone deacetylases (HDACs) in repressing IL-2 gene expression.15 Previous reports have implicated Cdk5 as a regulator of the HDAC1 complex16 although direct phosphorylation of HDAC1 by Cdk5 has never been demonstrated. Here we explore whether the impaired T cell responses observed in Cdk5 deficient T cells reflect a defect in the autocrine expression of IL-2 and whether this may be linked to Cdk5 regulation of the HDAC1 repressor complex. Our data reveal mSin3a, an essential component of the HDAC1-repressor complex, to be a novel substrate of Cdk5. Disruption of either the expression or the activity of Cdk5 enhances HDAC activity and increases occupancy of the IL-2 promoter by the HDAC1/mSin3a complex, ultimately leading to suppression of IL-2 expression. Our data establish an essential role for Cdk5 in regulating gene expression in T cells through post-translational modification of the co-repressor molecule mSin3a. A precise understanding of these mechanisms will provide a rationale for the therapeutic targeting of either Cdk5 or selected Cdk5 substrates in the setting of T cell mediated disease.
Results
Cdk5 activity is essential for optimal IL-2 expression during T-cell activation
To discern whether induction of Cdk5 activity following T cell receptor (TCR) activation is required for normal T cell production of IL-2, we examined the effects of either Cdk5 gene deletion or pharmacological inhibition of Cdk5 activity on IL-2 production in mouse T-cells (Fig. 1A). Disruption of Cdk5 activity by the selective Cdk inhibitor Roscovitine (ROS) results in a significant decrease in IL-2 production following T cell receptor (TCR) stimulation. Similarly, T cells deficient in Cdk5 gene expression exhibit a significant reduction in autocrine IL-2 production following TCR stimulation with anti-CD3/CD28 antibodies. This reduction in secreted IL-2 correlates with a decreased abundance of IL-2 mRNA transcripts when naïve wild type T cells undergo TCR stimulation in the presence of Roscovitine (Fig. 1B). This relationship between Cdk5 activity and IL-2 mRNA expression is also observed when IL-2 expression is quantified following TCR stimulation of either Cdk5 deficient (Cdk5−/-)T cells or wild type (Cdk5+/+) T cells (Fig. 1C). These results suggest that Cdk5 activity is required for optimal IL-2 expression following TCR stimulation and T cell activation (Fig. 1D).
Figure 1.
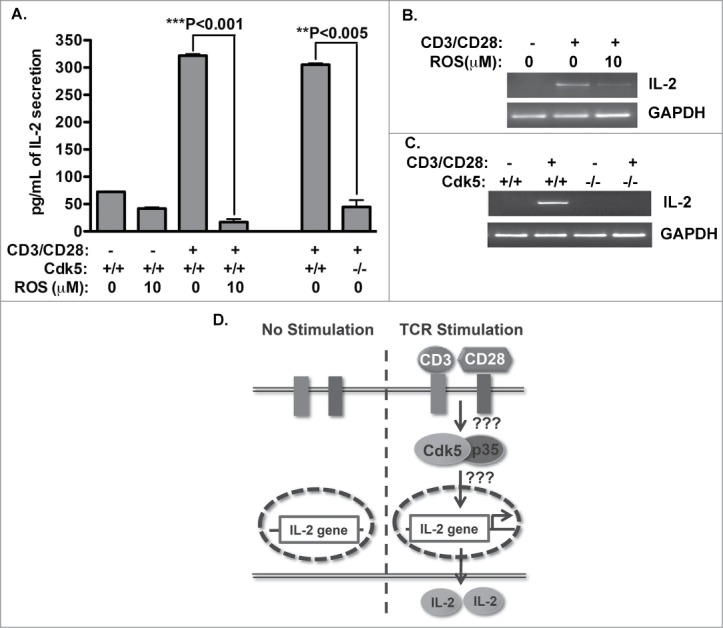
Cdk5 is required for optimal IL-2 expression. (A) Naïve wild type (Cdk5+/+) T cells or Cdk5-deficient (Cdk5−/-) T cells were activated with plate bound anti-CD3 and anti-CD28 antibodies for 48hrs in the presence or absence of Roscovitine. Amounts of IL-2 present in T cell supernatants were detected by ELISA. (B) Semi-quantitative RT-PCR analysis was performed to measure IL-2 mRNA expression in T cells activated by CD3/CD28 antibodies for 12hrs in the presence or absence of Roscovitine, and similarly, (C) IL-2 mRNA expression was determined in either Cdk5+/+ or Cdk5−/-) T cells following anti-CD3/CD28 activation.by semi-quantitative RT-PCR. (D) Summary diagram of IL-2 production after T cell activation with or without the presence of Cdk5 protein or Cdk5 activity.
Cdk5 interacts with HDAC1 and modulates HDAC activity during T cell activation, but does not alter HDAC1 expression
Previous reports indicate an essential role for the HDAC1 complex in repression of the IL-2 gene and recent studies implicate Cdk5 in regulation of the HDAC1 complex in neuronal cells. Thus, we decided to examine whether Cdk5 influences HDAC activity in T cells following T cell receptor stimulation. We first examined the physical association between HDAC1 and Cdk5 during T cell activation. A low level of Cdk5 protein is present in HDAC1 immunoprecipitates prepared from protein lysates of primary wild type naïve T cells and this association increases following TCR activation (Fig. 2A), correlating with the induction of Cdk5 protein expression and activity following TCR stimulation.4 Next, to determine whether Cdk5 activity modulates the expression of HDAC1 in either resting or activated T-cells, we performed RT-PCR and Western blot analysis for HDAC1 with lysates prepared from naïve cells either before or after TCR-activation of either Cdk5−/- or Cdk5+/+ T cells (Fig. 2B). HDAC1 mRNA and protein expression were similar under all conditions, suggesting HDAC1 gene expression is not under the control or influence of Cdk5 signaling. However, the interaction between Cdk5 and HDAC1 observed in activated T cells suggests the potential for a Cdk5-dependent regulation of HDAC activity.
Figure 2.
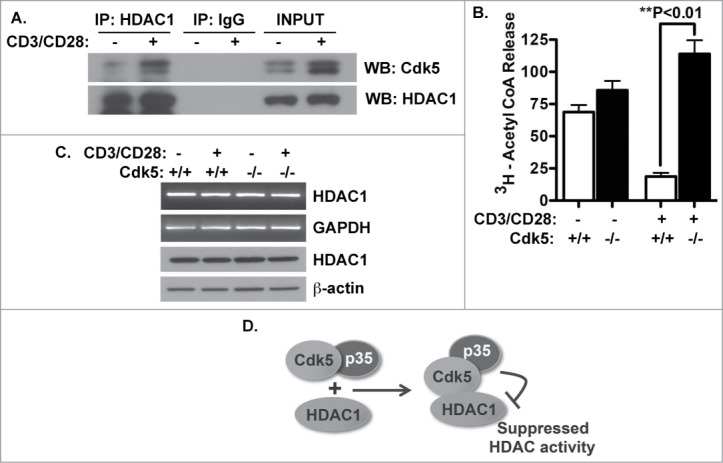
Cdk5 interacts with HDAC1 and alters HDAC activity in activated (T)cells. (A) Immunoprecipitates of HDAC1 protein were prepared from lysates of T cells both before and after CD3/CD28 stimulation for 48 hrs. Precipitates were subjected to Western blot for both Cdk5 and HDAC1 protein. (B) The expression of HDAC1 mRNA and protein was examined in both naïve and activated T cells isolated from both wild type (Cdk5+/+) T cells and Cdk5-deficient (Cdk5−/-) T cells both before and after anti-CD3/CD28 stimulation. (C) HDAC activity was measured in both quiescent and anti-CD3/CD28 stimulated Cdk5+/+ and Cdk5−/- T cells. (D) Model depicting how Cdk5/HDAC1 forms a protein complex and inhibits HDAC activity. Cdk5-deficiency does not alter HDAC1 expression but does relieve the suppression of HDAC activity.
To address this possibility, we performed an HDAC-specific activity assay to determine whether the reduction in HDAC activity that follows TCR ligation is dependent on the function of Cdk5. A significant decrease in HDAC activity follows TCR ligation in Cdk5+/+ T cells, however, this suppression not observed in Cdk5−/- T cells (Fig. 2C). The failure to fully suppress HDAC activity in activated Cdk5−/- T cells supports our hypothesis by directly implicating Cdk5 in the HDAC-dependent regulation of IL-2 gene expression through post-translational modification of the HDAC1 complex (Fig. 2D).
Cdk5 phosphorylates the HDAC co-repressor protein mSin3a at serine residue 861
Although the data above might implicate HDAC1 as a substrate of Cdk5, this possibility has never been directly confirmed. Thus, utilizing an in vitro Cdk5 kinase assay, we combined either HDAC1 protein immunoprecipitated from murine T cells or recombinant HDAC1 protein as substrates with Cdk5 isolated from murine T cells (Fig. 3A). Our data show that HDAC1 is not a direct substrate of Cdk5; neither the recombinant nor the endogenous HDAC1 protein was phosphorylated by the endogenous Cdk5. However, the potential remained that Cdk5 could phosphorylate one of the potent co-repressor proteins that are essential regulators of HDAC activity. Therefore, we examined whether Cdk5 might phosphorylate mSin3a, a crucial component of the HDAC1 repressor complex. Utilizing both recombinant and immunoprecipitated endogenous mSin3a protein as substrates, we found significant phosphorylation of mSin3a by Cdk5 (Fig. 3B).
Figure 3.
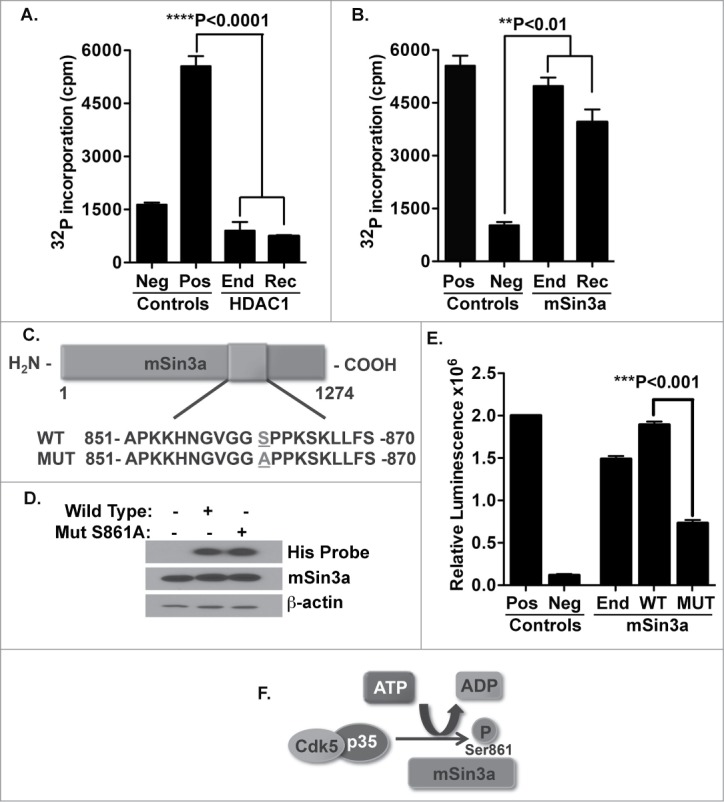
Cdk5 does not phosphorylate HDAC1 but does phosphorylate the mSin3a protein at serine residue 861. (A) Phosphorylation of HDAC1 by the Cdk5 kinase was measured by in vitro kinase assays. Cdk5 protein was immunoprecipitated from murine T cells and combined with either endogenous HDAC1 (End) isolated from murine T cells or with recombinant HDAC1 (Rec). Immunoprecipitates of Cdk5 from Cdk5−/- T cells were used as a negative (Neg) control. Neurofilament H (NF-H) is a known Cdk5 substrate and was used as a positive (Pos) control. (B) Phosphorylation of mSin3a by Cdk5 was determined using the previously described kinase assay, in which immunoprecipitates of Cdk5 isolated from murine T cells were incubated with either the endogenously expressed mSin3a (End) protein or recombinant mSin3a (Rec). (C) Schematic of mSin3a protein showing the amino acid sequence in the wild-type and mutant form of the protein used in these studies. In the mutant mSin3a, serine residue 861 was mutated to an alanine residue. (D) Wild-type or mutant forms of mSin3a were transfected into HEK293T cells and examined for expression using Western blot analysis. (E) Phosphorylation of Wild-type and mutant mSin3a protein was measured using a Cdk5 in vitro kinase assay. Immunoprecipitates of Cdk5 isolated from murine T cells were combined with either endogenous (End), Wild-type (WT) or mutant (MUT) mSin3a protein. Positive (Pos) and negative (Neg) controls were the same as described above. (F) Schematic diagram depicting how Cdk5 phosphorylates the mSin3a protein specifically at the Serin861 residue.
Upon further examination of the mSin3a protein sequence, we found one specific site on the mSin3a protein, serin861, which corresponds to the preferred consensus sequence for established substrates of Cdk5 (proline-directed serine residue followed by a basic residue in the +3 position). To further determine the ability of Cdk5 to phosphorylate mSin3a at this specific site, we created a mutant form of the mSin3a protein (S861A) with its serine861 residue substituted to an alanine (Fig. 3C). Histidine-tagged wild-type and S861A forms of the mSin3a protein were then expressed following transfection into in HEK293T cells (Fig. 3D). In Fig 3E, wild-type or mutant forms of mSin3a were combined as substrates with Cdk5 in an in vitro kinase assay. Our data show a significant decrease in phosphorylation events when mutant mSin3a was used as a substrate, strongly indicating this specific residue as a site for phosphorylation by Cdk5 (Fig. 3F).
Cdk5 activity controls HDAC1/mSin3a complex formation through regulation of mSin3a protein abundance but has no effect on mSin3a mRNA
To further understand the effect of mSin3a phosphorylation by Cdk5, we examined both mSin3a mRNA transcript and protein abundance in either wild type or Cdk5−/- T cells, before and after CD3/CD28 stimulation. As shown in Figure 4A, absence of Cdk5 expression does not alter expression of mSin3a RNA transcripts as examined by semi-quantitative RT-PCR. However, examination of mSin3a protein expression following TCR stimulation in wild type T cells reveal a reduction in mSin3a, whereas this reduction in expression is not observed in Cdk5−/- T cells; implicating an essential role for Cdk5 in mSin3a protein expression. To determine whether phosphorylation of mSin3a by Cdk5 primes mSin3a for proteasomal degradation, cultures of either Cdk5+/+ or Cdk5−/- T cells were activated in either the presence or absence of the proteasome inhibitor MG132. The presence of MG132 allowed for an accumulation of the mSin3a protein (Fig. 4B) in both Cdk5+/+ and Cdk5−/- T cells. To further examine this role of Cdk5 in regulating mSin3a protein expression, we expressed both the wild-type and S861A forms of mSin3a in Jurkat T cells. In the absence of MG132, the level of expression of the S861A mutant protein observed in lysates of Jurkat cells was significantly higher than that of the wild type mSin3a protein. Addition of MG132 to these cultures of Jurkat T cells increased the abundance of both the endogenous mSin3a protein (Fig. 4C, lane 4) and the transfected wild type mSin3A (Fig. 4C, lane 5) to a level comparable to that observed for the S861A mutant in the absence of MG132. This latter observation underlies the augmented expression of mSin3a observed in Jurkat cells transfected with the S861A mutant and exposed to MG132 (Fig. 4C, lane 6). Taken together, these data suggest that mSin3a phosphorylation at serine861 by Cdk5 regulates mSin3a protein expression through proteasomal degradation (Fig. 4D).
Figure 4.
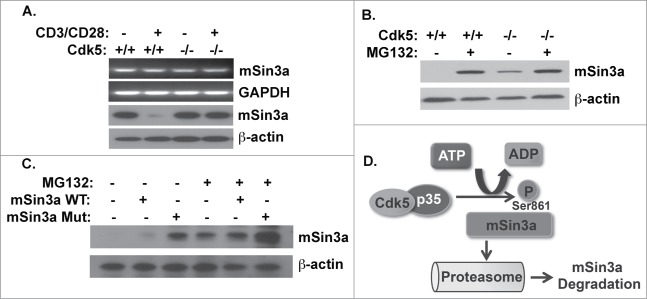
Cdk5 phosphorlyation of mSin3a at Serine861 disrupts the expression of the mSin3a protein. (A) RNA and protein expression for mSin3a were examined with semi-quantitative RT-PCR and Western blot analysis in wild type (Cdk5+/+) T cells or Cdk5−/- T cells with or without anti-CD3/CD28 activation. (B) mSin3a protein expression in either Cdk5+/+ or Cdk5−/- T cells treated with or without the proteasome inhibitor MG132 for 8 hrs. (C) Protein expression for mSin3a were examined in anti-CD3/CD28 activated Jurkat cells transfected with either Wild-type (WT) or mutant (MUT) mSin3a plasmids. Cells were treated in the presence or absence of the proteasome inhibitor MG132. (D) Schematic diagram depicting phosphorylation of the Serine861 residue on mSin3a by Cdk5 and the resultant proteasomal degradation of mSin3a.
Phosphorylation of Serine861 in the mSin3a protein disrupts formation of the HDAC1/mSin3a complex and leads to diminished HDAC1 occupancy of the IL-2 promoter
We hypothesized that phosphorylation of mSin3a by Cdk5 would reduce the capacity to form a complex with HDAC1. This hypothesis is supported by the enhanced abundance of the mSin3a/HDAC1 complex in nuclear extracts of Cdk5−/- T cells following TCR stimulation when compared to extracts of activated Cdk5+/+ T cells. (Fig. 5A). To determine whether mSin3a phosphorylation by Cdk5 influences the capacity of HDAC1 to occupy and thereby repress the IL-2 gene promoter, we performed chromatin immunoprecipitation (ChIP) assays using primary T cells. In unstimulated wild type T cells, HDAC1 clearly occupies the IL-2 gene promoter, however, there is a minimal detectable binding of HDAC1 to the IL-2 promoter in T cells after TCR stimulation. Consistent with our hypothesis, Cdk5−/- T cells exhibit increased binding of HDAC1 to the IL-2 promoter following TCR stimulation (Fig. 5B). Finally, we performed the same ChIP assay using Jurkat T cells transfected with either the wild type or S861A mutant form of the mSin3a protein (Fig. 5C). TCR stimulation in cells expressing wild type mSin3a show no detectable HDAC1 binding to the IL-2 promoter, whereas cells expressing the S861A mutant show significant levels of HDAC1 binding on the IL-2 promoter. In total, the data presented above suggest that Cdk5 phosphorylation of mSin3a negatively influences the occupancy of this region of the IL-2 gene promoter by HDAC1 (Fig. 5D).
Figure 5.
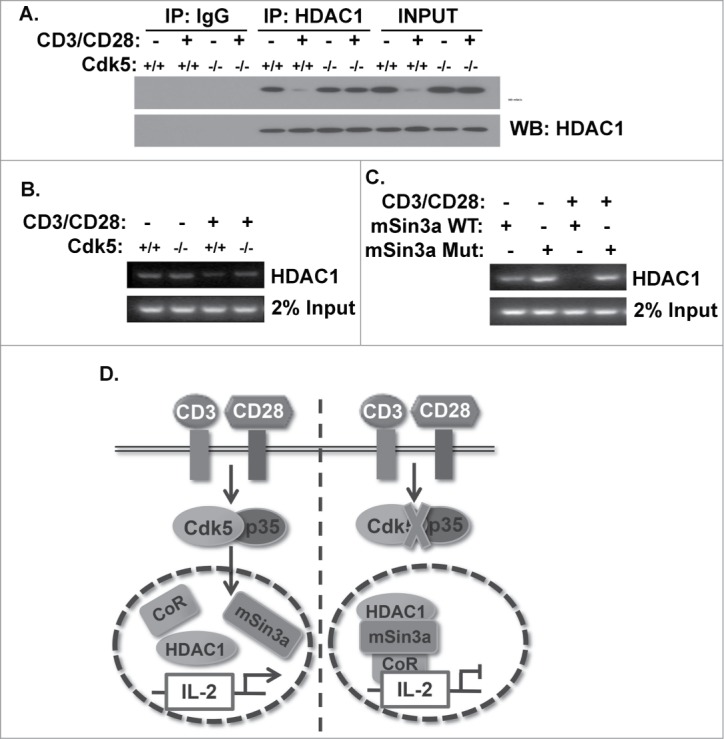
Phosphorylation of mSin3a by Cdk5 disrupts the formation of the HDAC1/mSin3a complex. (A) HDAC1 immunoprecipitates were isolated from nuclear lysates of primary wild type (Cdk5+/+) T cells or Cdk5−/- T cells either before or after stimulation with anti-CD3/CD28 antibodies. Immunoprecipitates were subsequently probed for mSin3a and HDAC1 expression by Western blot. (B) ChIP analysis was performed to assess the binding of HDAC1 to the IL-2 promoter in either Cdk5+/+ or Cdk5−/- T cells, either before or after activation with anti-CD3/CD28 stimulation. (C) Similarly, ChIP analyses were performed on Jurkat cells transfected with either Wild-Type (WT) or mutant (MUT) mSin3a plasmids to determine the binding of HDAC1 to the IL-2 promoter. (D) Diagram depicting the disruption of HDAC1 occupancy of the IL-2 promoter upon TCR stimulation, due to the presence of Cdk5/p35 activity, and the persistence of HDAC1 on the IL-2 promoter when the expression / activity of Cdk5 is disrupted.
Discussion
Here we report a novel mechanism through which Cdk5 controls HDAC activity and reveal that T cells require functional Cdk5 in order to attain optimal IL-2 gene expression following TCR stimulation. Autocrine IL-2 expression is essential for optimal T cell activation following TCR stimulation and is required for the differentiation and survival of T cells. The repression of IL-2 gene expression by classical zinc-dependent histone deacytelases (HDACs)15,17 is relieved during T cell activation. The data shown here demonstrate a requirement for Cdk5-mediated phosphorylation of mSin3a to relieve repression of the IL-2 gene promoter by the HDAC1-mSin3a repressor complex. The relationship between Cdk5 activity in T cells and autocrine IL-2 production is evident as the disruption of either Cdk5 expression or activity results in a significant decrease in IL-2 production and a commensurate reduction in the abundance of IL-2 mRNA transcripts following TCR stimulation. These data suggest that Cdk5 controls the production of IL-2 through a mechanism that includes regulation of IL-2 gene transcription.
Cdk5 is known to regulate gene expression through several mechanisms and our data support the concept that this includes post-translational modification of the HDAC1 repressor complex.18,19 Histone deacetylases regulate gene expression during development and cell differentiation by modulating the accessibility of nucleosomes and by regulating acetylation of proteins within complexes that control gene transcription.20,21 For example, HDAC1 is known to associate with mSin3a and other proteins to form functional repressor complexes capable of silencing selected target genes in a context-dependent manner.22 Aberrant activity of Cdk5 has been linked to neurodegeneration through mechanisms that include inhibition of HDAC1 activity,23 however these studies have not clearly shown HDAC1 to be a direct substrate of Cdk5. Post-translational modification of HDAC proteins can modulate the capacity of HDACs to complex with other co-repressor proteins and potentially influence sub-cellular localization and enzymatic activity.24,25 Our data show that HDAC1 is not a direct substrate of Cdk5 as neither recombinant HDAC1 nor endogenous HDAC1 protein precipitated from T cells were directly phosphorylated by endogenous Cdk5 in vitro. Thus, while Cdk5 physically associates with HDAC1, we show the modification of HDAC activity by Cdk5 instead is mediated through an effect on the HDAC-associated protein mSin3a.
Members of the mammalian class I HDACs such as HDAC1 repress gene transcription as components of large repressor complexes.26,27 HDAC1 associates with mSin3a,22 histone binding proteins and mSin3a-associated polypeptides that influence histone deacetlyase activity.28 In silico analysis of the amino acid sequence of the mouse mSin3a revealed an exact Cdk5 target motif at serine 861 and serine 860 in the human mSin3a. The kinase assays described in this report show Cdk5 directly phosphorylates both the recombinant mSin3a protein and endogenous mSin3a immunoprecipitated from T cells when either is used as a substrate in an in vitro Cdk5 kinase assay. We further examined direct phosphorylation of mSin3a by Cdk5 by utilizing a S861A mutant form of the mSin3a protein. A significant reduction in phosphorylation of the S861A mutant relative to the wild type mSin3a in this in vitro kinase assay with Ckd5 further indicates direct phosphorylation of the mSin3a protein by Cdk5 at the serine 861 residue.
The importance of post-translational modification of mSin3a as a mechanism to regulate mSin3a function has not previously been reported. Our data show that the abundance of mSin3a mRNA in T cells remains constant following TCR stimulation. Specifically, a comparison of mSin3a gene expression in naïve/quiescent T cells versus stimulated T cells shows mSin3a mRNA abundance is not influenced by Cdk5 activity (i.e neither targeted disruption of Cdk5 gene expression in T cells nor pharmacologic inhibition of Cdk5 activity in wild type T cells influences the abundance of mSin3a mRNA). However, the abundance of mSin3a protein decreased during T cell activation, an effect not observed in Cdk5 deficient T cells. These data suggested the potential for Cdk5 activity to regulate the stability of the mSin3a protein, an effect we further confirmed by the addition of the proteasomal inhibitor, MG132. These data were corroborated in studies in activated Jurkat T cells, where the expression of the wild-type mSin3a protein was reduced relative to that of the S861A mutant. While our data with the proteasome inhibitor confirms that phosphorylation of mSin3a at Serine 861 causes the degradation of mSin3a, there exists the possibility that Cdk5-directed phosphorylation may also cause mSin3a localization to change. Studying the potential for this phenomenon as an additional function of Cdk5-directed phosphorylation would be an important aspect to address in the future.
In addition to an effect on mSin3a abundance, we found formation of the mSin3a/HDAC1 complex in activated T-cells was enhanced in nuclear lysates of T cells lacking Cdk5 when compared to wild type T cells. This effect of Cdk5 on formation of the mSin3a/HDAC1 complex directly impact on the ability for HDAC1 to occupy the IL-2 promoter. A ChIP analysis revealed increased occupancy of HDAC1 on the IL-2 promoter both in Cdk5−/- T cells cells in T cells expressing the S861A mutant version of the mSin3a protein. In sum, these observations clearly demonstrate that phosphorylation of mSin3a at Serine861 by Cdk5 leads to degradation of mSin3a, thereby repressing formation of a functional mSin3a/HDAC1 complex during T-cell activation.
Disruption of mSin3a protein expression and function has been shown to impair the normal transcriptional repression of specific target genes by the HDAC1 complex.29 The significance of this post-translational modification of mSin3a in T cells is supported by previous studies that have shown the repression of IL-2 gene transcription by the lineage determining factor Ikaros is dependent on the association of Ikaros with mSin3 proteins,30, an event which is thought to be important in T cell anergy.31 The results presented here reveal a pivotal role of Cdk5 in controlling IL-2 gene expression during T cell activation, through post-translational modification of mSin3a, an event that impairs stability of the mSin3a-HDAC1 complex and thereby relieves repression of the IL-2 gene promoter by HDAC1.
Our data add to accumulating evidence pointing to the importance of the interaction between Cdk5 and the nuclear machinery regulating gene expression. Cdk5 has a prominent nuclear localization32 and is now known to modulate the activity of several transcription factors, including p53,33 pRB1 and Stat3.8 A number of reports have also indicated the expression of its activator p35 in the nuclear compartment of cells, albeit at a level lower than in its cytoplasmic fraction.34-36 For instance, the N-terminal region of p35 has been found to interact and co-localize with the nuclear protein SET in the nucleus of the neurons. Additionally, Cdk5 phosphorylation of its nuclear substrate MEF2 has been shown; where Cdk5 specific phosphorylation of MEF2 inhibits this pro-survival transcription factor. It has been found that p35 nuclear import requires direct interaction with soluble import factors; and Imp-β, Imp-5, and Imp-7 have all been shown to transport p35 into the nucleus through an energy dependent manner. Our data here further corroborates these reports for Cdk5-p35 expression and activity in the nucleus of the cell. A role for Cdk5 in histone modification has been suggested by observations that include the demonstration that the chromatin modulating SET protein affects Cdk5/p35 activity,34 the observation that NCoR serves as an adaptor protein that enhances association of Cdk5 with PPARgamma,37 and by the demonstration that the co-activator p35 binds to mouse Sds3, a core component of the HDAC-mSin3a complex.19 It is interesting that the latter interaction leads to Cdk5-mediated phosphorylation of Sds3 at serine 228, thereby enhancing transcriptional repression by this complex in neuronal and muscle lineages, suggesting that this control of HDAC function by Cdk5 is very context-dependent and potentially bidirectional.
Previous reports have shown inhibition of HDAC activity in T-cells suppresses IL-2 gene transcription and impairs proliferation and survival of T cells.15 These effects of HDAC inhibitors may also be attributed to increased acetylation of non-histone proteins, including transcription factors such as Stat3,38 which is a known substrate of Cdk5. Furthermore, the potential clinical utility of HDAC inhibitors in the treatment of autoimmune and inflammatory disorders has also been partially attributed to the ability of these molecules to affect the acetylation status of Foxp3 and the induction of regulatory T-cells.39 Acetylation of Foxp3 is also known to enhance the ability of Foxp3 to bind to the IL-2 gene promoter and repress IL-2 expression.
The onset of a number of pathological conditions including chronic inflammation and autoimmunity is often the result of aberrant gene transcription and thus the regulatory role of HDACs in immunity is of significant interest. Recently, roles for HDACs have been studied as important for innate immunity as well as in lymphocyte development and function.40 Existing literature has documented a role for class I HDACs in negatively regulating inflammatory cytokine production. By contrast, in the context of immunity, the multitude of HDAC isoforms are known to exert differential effects and often resulting in both positive and negative regulation of the immune response. Compounds that have been developed to inhibit HDACs act through chelating Zn2+ ions at the active site, and they have been examined for therapeutic use in a number of diseases. Three HDAC inhibitors are now in clinical use for T cell lymphoma, epilepsy and bipolar disorders whereas a number of additional inhibitors are in clinical trials. Additionally, ongoing studies of HDAC inhibitor use are being performed on animal models in inflammatory diseases such as graft vs. host disease, septic shock and inflammatory bowel diseases. Although these broad-spectrum inhibitors display some degree of selectivity for different HDAC enzymes, ultimately they target multiple isoforms and classes of HDACs. Indeed the pleiotropic and often contrasting effects of the different HDACs on the immune response, it is expected that use of these inhibitors have shows evidence of contraindications. Thus our identification of Cdk5 as a novel target molecule able to modulate the HDAC1-mSin3a repressor complex offers an exciting and novel approach to the treatment of immune mediated disease without the above caveat associated with current HDAC directed therapies. In sum, our data demonstrate the importance of Cdk5 as a modulator of HDAC activity in T-cells and suggest a potential mechanism through which small molecule inhibitors of Cdk5 activity may serve to impair the progression of T-cell mediated autoimmune and inflammatory diseases.
Material and Methods
Animals
C57BL/6 mice, ages 6–8 weeks, were purchased from The Jackson Laboratory and were used for T cell isolations. Cdk5-null immune chimeric mice used in these studies were generated in our research facility.4 All animals were housed in micro-isolator cages and maintained in climate/light controlled rooms with free access to food and water. Studies performed were in compliance with the procedures approved by the Case Western Reserve University School of Medicine's Institutional Animal Care and Use Committee.
Antibodies
The Cdk5 antibody (C8), mSin3a antibody, His-probe antibody and rabbit IgG antibody were purchased from Santa Cruz Biotechnology and used for both immunoprecipitation as well as Western blotting. HDAC1 antibody used for both Western blotting, immunoprecipitation and ChIP assays was purchased from Abcam. The MPM-2 antibody used for both immunoprecipitation and western blot experiments was purchased from Millipore Inc… Monoclonal mouse β-actin antibody was purchased from Sigma.
Cell line
Jurkat T cell lines were cultured in RPMI medium containing 10% (vol/vol) fetal bovine serum and grown according to recommended protocol (ATCC).
Cdk5 kinase assay
Radioactive Cdk5 kinase activity assays were performed as previously described.4 In brief, total protein cell lysates were prepared and then incubated overnight at 4°C with Cdk5 antibody. Afterwards, protein A-Agarose beads were added to samples for 3–5 hours at 4°C. Following immuno-precipitation, samples were combined with desired substrates and incubated at 30°C after the addition of ATP. Similarly, non-radioactive Cdk5 kinase assays were performed using ADP-Glo Kinase Assay detection kit purchased from Promega to detect the occurrences of phosphorylation events.
Chromatin immunoprecipitation (ChIP) assay
Chromatin immunoprecipitation (ChIP) analysis was performed according to the manufacturer's instructions (Upstate Biotechnology) with T cells under the indicated stimulation and treatment conditions. Immunoprecipitated samples (DNA) were then subjected to semi-quantitative RT-PCR analysis of the IL-2 proximal promoter region.
3H-acetyl CoA release assay
Histone deacetylase activity was analyzed using a histone deacetlyase assay kit (Upstate, Millipore) according to procedures described in the manufacturer's protocol. [3H]-radiolabelled Histone H4 peptides were incubated with cell extracts obtained from T cells under various stimulation conditions. Released [3H]-acetate was assessed on a MicroBeta TriLux Counter to determine the HDAC activity in each sample.
ELISA
Cells were first cultured on culture dishes with or without CD3/CD28 stimulation for 48 hours; supernatants of cultured cells were then collected and subjected to Enzyme-Linked Immunosorbent Assays (ELISAs). ELISAs were performed as described in the manufacturer's protocol for IL-2 ELISA set kits purchased from BD Biosciences.
Immunoprecipitation assays
Protein lysates were first diluted to a concentration of 1μg/uL and subsequently pre-cleared with protein A-Agarose beads (Santa Cruz) and then incubated overnight with rotation at 4°C with the immunoprecipitating antibody. Lysates were then incubated with protein A-Agarose beads for 3–5 hours with rotation at 4°C. After incubation, protein A-Agarose beads were collected and washed with lysis buffer. Samples were then resolved by SDS-PAGE and followed by Western blot analysis.
Semi-quantitative RT-PCR
Total RNA was extracted using Trizol reagent (Invitrogen) and cDNA was then prepared using cDNA first-strand synthesis kit (Invitrogen). PCR was performed with a Px2 Thermal Cycler (Thermo).
Statistical analyses
Statistical evaluations were performed using the Prism computer program (GraphPad Software). Significant differences between experiments were assessed by comparing the means of data sets using the Student t test, with a p value of <0 .05 considered significant.
T cell receptor stimulation
Cell culture plates were coated by incubating with anti-CD3(3ug/mL) and anti-CD28 (1ug/ML) antibodies (BD Biosciences) diluted in PBS overnight at 4°C. Prior to use, plates were washed with fresh PBS twice.
Roscovitine
Roscovitine was purchased from Enzo Life Sciences. Stock solutions of 10 mM were first prepared in DMSO and working concentrations of 10 μM were used in experiments.
T lymphocyte isolation
A mixed population of cells was collected by first isolating cells from spleen and regional lymph nodes of mice and creating single cell suspensions by passing these tissues through a 40 μm cell strainer (BD Biosciences). Cells were then incubated with ACK lysing buffer (Lonza) on ice for 5 minutes to deplete the mixed cell population of erythrocytes. T lymphocytes were purified through a process of negative isolation by passing mixed cells through MACS separation columns using a pan T isolation kit (MiltenyiBiotec) in accordance to the manufacturer's protocol.
Western blot analysis
Protein samples were prepared from cellular lysates made in RIPA buffer (Thermo Fisher Scientific) containing a protease inhibitor cocktail tablet (Roche) in addition to a phosphatase inhibitor phosSTOP (Roche). Proteins were denatured by heating for 10 min at 94°C in sample loading buffer (2% SDS, 10% glycerol 80 mM Tris, pH 6.8, and 1 mM DTT). 50 μg of total protein was separated by electrophoresis in 4–20% Tris-Glycine gels (Invitrogen). Proteins were transferred to a 0.2 μm nitrocellulose membrane (Invitrogen) and subsequently blocked for 1 hour in blocking buffer (TBS containing 10% non-fat dry milk and 0.05% Tween 20). Membranes were incubated overnight at 4°C with a primary antibody and probed with horse radish peroxidase-conjugated secondary antibody for 1 hour in room temperature.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
We would like to acknowledge support from the Jane and Lee Seidman Chair in Pediatric Cancer Innovation and grants from the NIH (5R01HL111682–02 and R01 EY022937-01), Hyundai Hope on Wheels Foundation, and the St. Baldrick's Foundation for Pediatric Cancer Research.
References
- 1. Dhavan R, Tsai LH. A decade of CDK5. Nat Rev Mol Cell Biol 2001; 2:749-59; PMID:11584302; http://dx.doi.org/ 10.1038/35096019 [DOI] [PubMed] [Google Scholar]
- 2. Arif A. Extraneuronal activities and regulatory mechanisms of the atypical cyclin-dependent kinase Cdk5. Biochem Pharmacol 2012; 84:985-93; PMID:22795893; http://dx.doi.org/ 10.1016/j.bcp.2012.06.027 [DOI] [PubMed] [Google Scholar]
- 3. Jessberger S, Gage FH, Eisch AJ, Lagace DC. Making a neuron: Cdk5 in embryonic and adult neurogenesis. Trends Neurosci 2009; 32:575-82; PMID:19782409; http://dx.doi.org/ 10.1016/j.tins.2009.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pareek TK, Lam E, Zheng X, Askew D, Kulkarni AB, Chance MR, Huang AY, Cooke KR, Letterio JJ. Cyclin-dependent kinase 5 activity is required for T cell activation and induction of experimental autoimmune encephalomyelitis. J Exp Med 2010; 207:2507-19; PMID:20937706; http://dx.doi.org/ 10.1084/jem.20100876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nohara A, Okada S, Ohshima K, Pessin JE, Mori M. Cyclin-dependent kinase-5 is a key molecule in tumor necrosis factor-alpha-induced insulin resistance. J Biol Chem 2011; 286:33457-65; PMID:21813649; http://dx.doi.org/ 10.1074/jbc.M111.231431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Strock CJ, Park JI, Nakakura EK, Bova GS, Isaacs JT, Ball DW, Nelkin BD. Cyclin-dependent kinase 5 activity controls cell motility and metastatic potential of prostate cancer cells. Cancer Res 2006; 66:7509-15; PMID:16885348; http://dx.doi.org/ 10.1158/0008-5472.CAN-05-3048 [DOI] [PubMed] [Google Scholar]
- 7. Cheung ZH, Ip NY. Cdk5: a multifaceted kinase in neurodegenerative diseases. Trends Cell Biol 2012; 22:169-75; PMID:22189166; http://dx.doi.org/ 10.1016/j.tcb.2011.11.003 [DOI] [PubMed] [Google Scholar]
- 8. Lin H, Chen MC, Chiu CY, Song YM, Lin SY. Cdk5 regulates STAT3 activation and cell proliferation in medullary thyroid carcinoma cells. J Biol Chem 2007; 282:2776-84; PMID:17145757; http://dx.doi.org/ 10.1074/jbc.M607234200 [DOI] [PubMed] [Google Scholar]
- 9. Brinkkoetter PT, Wu JS, Ohse T, Krofft RD, Schermer B, Benzing T, Pippin JW, Shankland SJ. p35, the non-cyclin activator of Cdk5, protects podocytes against apoptosis in vitro and in vivo. Kidney Int 2010; 77:690-9; PMID:20130526; http://dx.doi.org/ 10.1038/ki.2009.548 [DOI] [PubMed] [Google Scholar]
- 10. Yang HS, Alexander K, Santiago P, Hinds PW. ERM proteins and Cdk5 in cellular senescence. Cell Cycle 2003; 2:517-20; PMID:14504464; http://dx.doi.org/ 10.4161/cc.2.6.582 [DOI] [PubMed] [Google Scholar]
- 11. Kaminski S, Hermann-Kleiter N, Meisel M, Thuille N, Cronin S, Hara H, Fresser F, Penninger JM, Baier G. Coronin 1A is an essential regulator of the TGFbeta receptor/SMAD3 signaling pathway in Th17 CD4(+) T cells. J Autoimmun 2011; 37:198-208; PMID:21700422; http://dx.doi.org/ 10.1016/j.jaut.2011.05.018 [DOI] [PubMed] [Google Scholar]
- 12. Popmihajlov Z, Xu D, Morgan H, Milligan Z, Smith KA. Conditional IL-2 gene deletion: consequences for T cell proliferation. Front Immunol 2012; 3:102; PMID:22590468; http://dx.doi.org/ 10.3389/fimmu.2012.00102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Castro I, Yu A, Dee MJ, Malek TR. The basis of distinctive IL-2- and IL-15-dependent signaling: weak CD122-dependent signaling favors CD8+ T central-memory cell survival but not T effector-memory cell development. J Immunol 2011; 187:5170-82; PMID:21984699; http://dx.doi.org/ 10.4049/jimmunol.1003961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Liao W, Lin JX, Leonard WJ. IL-2 family cytokines: new insights into the complex roles of IL-2 as a broad regulator of T helper cell differentiation. Curr Opin Immunol 2011; 23:598-604; PMID:21889323; http://dx.doi.org/ 10.1016/j.coi.2011.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Koyama Y, Adachi M, Sekiya M, Takekawa M, Imai K. Histone deacetylase inhibitors suppress IL-2-mediated gene expression prior to induction of apoptosis. Blood 2000; 96:1490-5; PMID:10942396 [PubMed] [Google Scholar]
- 16. Ikiz B, Przedborski S. A sequel to the tale of p25/Cdk5 in neurodegeneration. Neuron 2008; 60:731-2; PMID:19081365; http://dx.doi.org/ 10.1016/j.neuron.2008.11.020 [DOI] [PubMed] [Google Scholar]
- 17. Kametani Y, Wang L, Koduka K, Sato T, Katano I, Habu S. Rapid histone deacetylation and transient HDAC association in the IL-2 promoter region of TSST-1-stimulated T cells. Immunol Lett 2008; 119:97-102; PMID:18606456; http://dx.doi.org/ 10.1016/j.imlet.2008.05.006 [DOI] [PubMed] [Google Scholar]
- 18. Fu AK, Hung KW, Wong HH, Fu WY, Ip NY. Cdk5 phosphorylates a component of the HDAC complex and regulates histone acetylation during neuronal cell death. Neurosignals 2013; 21(1-2):55-60; PMID:22398430; http://dx.doi.org/ 10.1159/000335158 [DOI] [PubMed] [Google Scholar]
- 19. Li Z, David G, Hung KW, DePinho RA, Fu AK, Ip NY. Cdk5/p35 phosphorylates mSds3 and regulates mSds3-mediated repression of transcription. J Biol Chem 2004; 279:54438-44; PMID:15489224; http://dx.doi.org/ 10.1074/jbc.M411002200 [DOI] [PubMed] [Google Scholar]
- 20. Nicolas E, Yamada T, Cam HP, Fitzgerald PC, Kobayashi R, Grewal SI. Distinct roles of HDAC complexes in promoter silencing, antisense suppression and DNA damage protection. Nat Struct Mol Biol 2007; 14:372-80; PMID:17450151; http://dx.doi.org/ 10.1038/nsmb1239 [DOI] [PubMed] [Google Scholar]
- 21. Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet 2009; 10:32-42; PMID:19065135; http://dx.doi.org/ 10.1038/nrg2485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Grzenda A, Lomberk G, Zhang JS, Urrutia R. Sin3: master scaffold and transcriptional corepressor. Biochim Biophys Acta 2009; 1789:443-50; PMID:19505602; http://dx.doi.org/ 10.1016/j.bbagrm.2009.05.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kim D, Frank CL, Dobbin MM, Tsunemoto RK, Tu W, Peng PL, Guan JS, Lee BH, Moy LY, Giusti P, et al. Deregulation of HDAC1 by p25/Cdk5 in neurotoxicity. Neuron 2008; 60:803-17; PMID:19081376; http://dx.doi.org/ 10.1016/j.neuron.2008.10.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Segre CV, Chiocca S. Regulating the regulators: the post-translational code of class I HDAC1 and HDAC2. J Biomed Biotechnol 2011; 2011:690848; PMID:21197454; http://dx.doi.org/ 10.1155/2011/690848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pflum MK, Tong JK, Lane WS, Schreiber SL. Histone deacetylase 1 phosphorylation promotes enzymatic activity and complex formation. J Biol Chem 2001; 276:47733-41; PMID:11602581; http://dx.doi.org/ 10.1074/jbc.M105590200 [DOI] [PubMed] [Google Scholar]
- 26. Wang J, Lee S, Teh CE, Bunting K, Ma L, Shannon MF. The transcription repressor, ZEB1, cooperates with CtBP2 and HDAC1 to suppress IL-2 gene activation in T cells. Int Immunol 2009; 21:227-35; PMID:19181930; http://dx.doi.org/ 10.1093/intimm/dxn143 [DOI] [PubMed] [Google Scholar]
- 27. Nagy L, Kao HY, Chakravarti D, Lin RJ, Hassig CA, Ayer DE, Schreiber SL, Evans RM. Nuclear receptor repression mediated by a complex containing SMRT, mSin3A, and histone deacetylase. Cell 1997; 89:373-80; PMID:9150137; http://dx.doi.org/ 10.1016/S0092-8674(00)80218-4 [DOI] [PubMed] [Google Scholar]
- 28. Spain MM, Caruso JA, Swaminathan A, Pile LA. Drosophila SIN3 isoforms interact with distinct proteins and have unique biological functions. J Biol Chem 2010; 285:27457-67; PMID:20566628; http://dx.doi.org/ 10.1074/jbc.M110.130245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cowley SM, Iritani BM, Mendrysa SM, Xu T, Cheng PF, Yada J, Liggitt HD, Eisenman RN. The mSin3A chromatin-modifying complex is essential for embryogenesis and T-cell development. Mol Cell Biol 2005; 25:6990-7004; PMID:16055712; http://dx.doi.org/ 10.1128/MCB.25.16.6990-7004.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Koipally J, Renold A, Kim J, Georgopoulos K. Repression by Ikaros and Aiolos is mediated through histone deacetylase complexes. EMBO J 1999; 18:3090-100; PMID:10357820; http://dx.doi.org/ 10.1093/emboj/18.11.3090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bandyopadhyay S, Dure M, Paroder M, Soto-Nieves N, Puga I, Macian F. Interleukin 2 gene transcription is regulated by Ikaros-induced changes in histone acetylation in anergic T cells. Blood 2007; 109:2878-86; PMID:17148585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhang J, Li H, Herrup K. Cdk5 nuclear localization is p27-dependent in nerve cells: implications for cell cycle suppression and caspase-3 activation. J Biol Chem 2010; 285:14052-61; PMID:20189989; http://dx.doi.org/ 10.1074/jbc.M109.068262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ajay AK, Upadhyay AK, Singh S, Vijayakumar MV, Kumari R, Pandey V, Boppana R, Bhat MK. Cdk5 phosphorylates non-genotoxically overexpressed p53 following inhibition of PP2A to induce cell cycle arrest/apoptosis and inhibits tumor progression. Mol Cancer 2010; 9:204; PMID:20673369; http://dx.doi.org/ 10.1186/1476-4598-9-204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Qu D, Li Q, Lim HY, Cheung NS, Li R, Wang JH, Qi RZ. The protein SET binds the neuronal Cdk5 activator p35nck5a and modulates Cdk5/p35nck5a activity. J Biol Chem 2002; 277:7324-32; PMID:11741927; http://dx.doi.org/ 10.1074/jbc.M107270200 [DOI] [PubMed] [Google Scholar]
- 35. Fu X, Choi YK, Qu D, Yu Y, Cheung NS, Qi RZ. Identification of nuclear import mechanisms for the neuronal Cdk5 activator. J Biol Chem 2006; 281:39014-21; PMID:17060323; http://dx.doi.org/ 10.1074/jbc.M512663200 [DOI] [PubMed] [Google Scholar]
- 36. Gong X, Tang X, Wiedmann M, Wang X, Peng J, Zheng D, Blair LA, Marshall J, Mao Z. Cdk5-mediated inhibition of the protective effects of transcription factor MEF2 in neurotoxicity-induced apoptosis. Neuron 2003; 38:33-46; PMID:12691662; http://dx.doi.org/ 10.1016/S0896-6273(03)00191-0 [DOI] [PubMed] [Google Scholar]
- 37. Li P, Fan W, Xu J, Lu M, Yamamoto H, Auwerx J, Sears DD, Talukdar S, Oh D, Chen A, et al. Adipocyte NCoR knockout decreases PPARgamma phosphorylation and enhances PPARgamma activity and insulin sensitivity. Cell 2011; 147:815-26; PMID:22078880; http://dx.doi.org/ 10.1016/j.cell.2011.09.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Shen L, Ciesielski M, Ramakrishnan S, Miles KM, Ellis L, Sotomayor P, Shrikant P, Fenstermaker R, Pili R. Class I histone deacetylase inhibitor entinostat suppresses regulatory T cells and enhances immunotherapies in renal and prostate cancer models. PLoS One 2012; 7:e30815; PMID:22303460; http://dx.doi.org/ 10.1371/journal.pone.0030815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tao R, de Zoeten EF, Ozkaynak E, Chen C, Wang L, Porrett PM, Li B, Turka LA, Olson EN, Greene MI, et al. Deacetylase inhibition promotes the generation and function of regulatory T cells. Nat Med 2007; 13:1299-307; PMID:17922010; http://dx.doi.org/ 10.1038/nm1652 [DOI] [PubMed] [Google Scholar]
- 40. Shakespear MR, Halili MA, Irvine KM, Fairlie DP, Sweet MJ. Histone deacetylases as regulators of inflammation and immunity. Trends Immunol 2011; 32:335-43; PMID:21570914; http://dx.doi.org/ 10.1016/j.it.2011.04.001 [DOI] [PubMed] [Google Scholar]