

Abstract
E2F transcription factors regulate a wide range of biological processes, including the cellular response to DNA damage. In the present study, we examined whether E2F family members are transcriptionally induced following treatment with several genotoxic agents, and have a role on the cell DNA damage response. We show a novel mechanism, conserved among diverse species, in which E2F1 and E2F2, the latter specifically in neuronal cells, are transcriptionally induced after DNA damage. This upregulation leads to increased E2F1 and E2F2 protein levels as a consequence of de novo protein synthesis. Ectopic expression of these E2Fs in neuronal cells reduces the level of DNA damage following genotoxic treatment, while ablation of E2F1 and E2F2 leads to the accumulation of DNA lesions and increased apoptotic response. Cell viability and DNA repair capability in response to DNA damage induction are also reduced by the E2F1 and E2F2 deficiencies. Finally, E2F1 and E2F2 accumulate at sites of oxidative and UV-induced DNA damage, and interact with γH2AX DNA repair factor. As previously reported for E2F1, E2F2 promotes Rad51 foci formation, interacts with GCN5 acetyltransferase and induces histone acetylation following genotoxic insult. The results presented here unveil a new mechanism involving E2F1 and E2F2 in the maintenance of genomic stability in response to DNA damage in neuronal cells.
Keywords: DNA damage response, DNA repair, E2F transcription factor, genomic stability, neuronal cells
Abbreviations
- ODN
oligodeoxynucleotide
- NCS
neocarzinostatin
- ASCAT
CAT antisense
- ASE2F1
E2F1 antisense
- ASE2F2
E2F2 antisense
- wt E2F DO
wild-type E2F decoy oligodeoxynucleotide
- mut E2F DO
mutant E2F decoy oligodeoxynucleotide
Introduction
The E2F family of transcription factors is encoded by 8 genes, E2F1-E2F8, which give rise to 9 different proteins. The 2 E2F3 proteins –E2F3a and E2F3b– are the product of alternative use of promoters of the E2F3 locus.1-2 Traditionally, E2F family members have been subdivided into 2 groups based on their transcriptional activities, structures and interactions with the pocket proteins Retinoblastoma protein (pRB), Retinoblastoma-like protein 1 (p107) and Retinoblastoma-like protein 2 (p130). E2F1, E2F2 and E2F3a, that only bind pRB, constitute the ‘activator’ E2Fs due to their ability to activate transcription of E2F target genes. E2F3b and E2F4-E2F8 are considered the ‘repressor’ E2Fs since they are capable of repressing the expression of mostly overlapping sets of target genes. E2F3b, E2F4 and E2F5 exert their repressive function in association with a pRB family member, while E2F6-E2F8 repress transcription in a pocket protein-independent manner as they lack the pocket protein binding domain.3-6
Despite the fact that E2Fs were originally described to play a pivotal role in cell cycle control,7-8 it has become clear that they participate in the regulation of a plethora of biological processes, including the cellular response to DNA damage. It was first reported that E2F1 protein levels increase upon treatment with several DNA damaging agents.9-12 Further studies revealed that E2F4 levels decrease whereas E2F3a, E2F7 and E2F8 are upregulated following DNA damage.13-15
E2F1 is the best studied family member with respect to its regulation and function following genotoxic stress. Its participation in the DNA damage response can be described from 3 different angles.16 First, E2F1 undergoes posttranslational modifications in response to DNA damage. E2F1 is phosphorylated on serine 31 by ataxia-telangiectasia mutated (ATM) and ataxia-telangiectasia and Rad3-related (ATR) kinases,12 and on serine 364 by Checkpoint kinase 2 (Chk2).17 It is also acetylated on lysines 117, 120 and 125 by either p300/CREB-binding protein (p300/CBP) or p300/CREB-binding protein-associated factor (P/CAF) acetyltransferases.18-19 These modifications contribute to E2F1 stabilization and explain the elevated E2F1 protein levels observed after genotoxic insult.12,17,18 Second, E2F1 transactivation ability following DNA damage is regulated through its interaction with specific protein partners. E2F1-pRB complexes repress transcription when they are associated with nucleosome remodeling proteins, methyltransferases, or histone deacetylases,20-22 but activate it upon interaction with histone acetyltransferases.23-24 Finally, E2F1 is recruited to sites of DNA lesions and promotes the recruitment of repair factors, suggesting a role for E2F1 in DNA repair.25-27
It was also shown that UV irradiation induces the transcription of E2F1, resulting in increased E2F1 protein levels.28 Therefore, 2 parallel mechanisms contribute to E2F1 induction in response to DNA damage: the posttranslational modifications and consequent protein stabilization, and the enhanced transcription that leads to de novo protein synthesis. Recent observations have also demonstrated that other E2F family members are transcriptionally induced upon treatment with doxorubicin.14 These findings suggest that DNA damage itself, and not a signal generated by a particular genotoxic agent, is at the origin of the upregulation of E2F1 gene transcription. The aim of the present work is to examine whether various E2F family members are transcriptionally induced following treatment with several genotoxics in different species –with special interest on a neuronal cell based system– and have a role on the cell DNA damage response.
Here, we show that E2F1 and E2F2, the latter specifically in neuronal cells, are transcriptionally induced upon DNA damage. This upregulation contributes to the augmentation of E2F1 and E2F2 protein levels, which are active in their transcription regulation functions. Importantly, ectopic expression of these E2Fs in neuronal cells reduces the accumulation of DNA damage following treatment with genotoxic agents. Conversely, ablation of E2F1 and E2F2 leads to increased levels of DNA damage and apoptotic response, and also reduces cell viability and DNA repair capability upon genotoxic stress. Moreover, we show that E2F1 and E2F2 accumulate at sites of oxidative and UV-induced DNA damage, and associate with γH2AX DNA repair factor. Finally, as it was formerly established for E2F1,25,27 we demonstrate that E2F2 promotes Rad51 foci formation, interacts with GCN5 acetyltransferase and induces histone acetylation following genotoxic insult. In summary, the evidence presented here establishes a new mechanism involving E2F1 and E2F2 in the response to DNA damage and the maintenance of genomic integrity in neuronal cells.
Results
E2F1 and E2F2 are induced upon DNA damage in neuronal cells
In order to evaluate the response of E2F genes to DNA damage, we first performed a time course study of the mRNA levels of E2F1–5 after exposure of cells to several genotoxic agents differing in their mechanism of action and the resulting lesions. The genotoxics used were neocarzinostatin (NCS) –a radiomimetic drug that generates double strand breaks–, hydrogen peroxide (H2O2) –known to produce oxidative stress and consequently single-strand breaks (SSBs) and base damage– and UV-C (UV) irradiation –which causes essentially pyrimidine dimers–. Northern blot assays on HepG2 cells revealed an increase of E2F1 mRNA levels upon treatment with each of the genotoxic agents used (Fig. S1A). No such changes were detected for the other family members studied. E2F1 mRNA raise was also observed in HEK293 cells (Fig. S1B). Next, we used the same approach on neuronal cell lines and found that both E2F1 and E2F2 transcripts were augmented after treatment with each of the DNA damaging agents in SH-SY5Y (Fig. 1A), Neuro-2a, HN9 and PC12 cells (Fig. S2A-C). Finally, E2F1 and E2F2 transcript levels were increased in rat primary hippocampal neuron cultures irradiated with UV (Fig. S2D), strengthening the notion that the E2F2 mRNA augmentation in response to DNA damaging agents is characteristic of neuronal cells. Together, these results indicate that in response to DNA damage there is an E2F1 mRNA increase in all cell types, while E2F2 mRNA raise would be restricted to neuronal cells. As proposed, this response appears to be independent of the type of DNA damage induced and shared by several species. The specificity of the probes used in Northern blot assays is shown in Fig. S3. Next, to rule out the possibility that E2F1 and E2F2 mRNA increase observed after the genotoxic stress was a consequence of transcript stabilization, we studied the effect of blocking transcription through actinomycin D treatment. To examine this, cells were incubated with actinomycin D for 3 h, exposed to genotoxic agents and harvested for a Northern blot assay at the time the maximum mRNA levels had been observed. Transcriptional inhibition prevented E2F1 and E2F2 mRNA accumulation induced by the 3 types of DNA damage (Fig. 1B). These data suggest that the upregulation of E2F1 and E2F2 mRNA by genotoxics is due to enhanced transcription.
Figure 1.

E2F1 and E2F2 mRNA and protein levels increase following DNA damage in neuronal cells. (A and B) Northern blot analysis of SH-SY5Y cells treated with NCS, H2O2 or UV and harvested at the specified times. Total RNA was extracted from cells and subjected to Northern blot with the [32P]-labeled probes shown in the left margin. In (B), cells were pre-incubated 3 h with 1 μM actinomycin D (Act D). The numbers under the bands indicate E2F1–5 quantitation normalized to β-tubulin and control in (A), and E2F1 and E2F2 quantitation normalized to β-tubulin and None (-) condition in (B). (C) Western blot of E2F1 and E2F2 in SH-SY5Y cells treated with NCS, H2O2 or UV and harvested at the indicated times. The numbers under the bands indicate E2F1 and E2F2 quantitation normalized to β-actin and control. (D–F) Immunoblot of GFP in SH-SY5Y cells expressing E2F1-GFP (D), E2F2-GFP (E) or pEGFP-C1 empty vector (F) and harvested at the indicated times post-UV. (G and H) Western blot of E2F1 and E2F2 in SH-SY5Y cells pre-incubated 3 h with 10 μM cycloheximide (CHX) and harvested at the specified times after genotoxic treatment, as shown in (G). In (D–F, H), data represent the mean±S.E.M. of at least 4 independent experiments for (D and E) and n = 3 for (F and H). In (D–F), P-values were calculated by one-way ANOVA, Dunnett's: *P < 0.05, **P < 0.01, n.s. not significant. C, control mock-treated cells.
Figure 2.
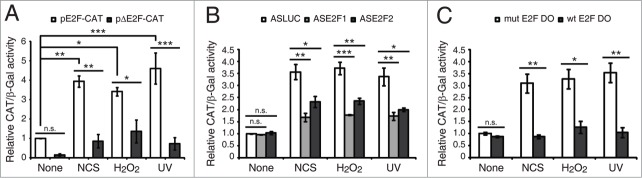
DNA damage induced E2F1 and E2F2 are transcriptionally active. (A) CAT activity of Neuro-2a cells transfected with pE2F-CAT or pΔE2F-CAT along with pCEFL-β-galactosidase, and harvested 24 h post-genotoxic treatment. (B and C) CAT activity of Neuro-2a cells transfected with pE2F-CAT, pCEFL-β-galactosidase and 1 μM of the indicated ODN, and harvested 24 h after DNA damage. In all cases, CAT activity was normalized to β-galactosidase activity. In (B and C), results are expressed relative to None-ASLUC or None-mut E2F DO conditions. Data represent the mean±S.E.M. of 3 independent experiments performed in triplicate. P-values were obtained using one-way ANOVA with Tukey's posttest in (A), one-way ANOVA with Dunnett's posttest in (B) and Student's t-test in (C): *P < 0.05, **P < 0.01, ***P < 0.001, n.s. not significant. DO, decoy oligodeoxynucleotide.
Figure 3.
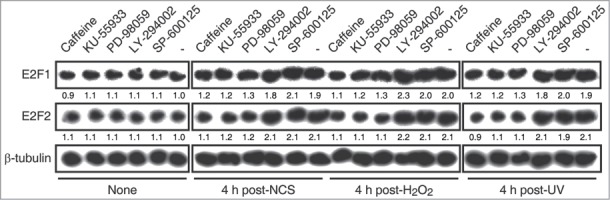
E2F1 and E2F2 transcriptional upregulation requires ATM/ATR and MEK kinases activity. SH-SY5Y cells incubated 1 h with 5 mM caffeine, 10 μM KU-55933, 10 μM PD-98059, 50 μM LY-294002 or 25 μM SP-600125 and harvested after a 4 h treatment with NCS, H2O2 or UV. Total RNA was extracted and subjected to Northern blot analysis with the [32P]-labeled probes shown in the left margin. The numbers under the bands indicate E2F1 and E2F2 quantitation normalized to β-tubulin and None (-) condition.
In light of these findings, we examined whether the increases in E2F1 and E2F2 mRNA levels resulted in an elevation of their protein levels. Western blot assays on SH-SY5Y cells revealed an increase in E2F1 and E2F2 proteins upon exposure to each of the genotoxics tested (Fig. 1C). Surprisingly, while the induction of E2F1 was expected due to its increased transcription and the reported stabilization of the protein,12,17,18 the induction of E2F2 was detected not only in neuronal cells, but also in HEK293 cells (Fig. S4) where we did not observe an increase in E2F2 mRNA level in response to the DNA damaging treatments (Fig. S1B), suggesting a stabilization of the protein after the genotoxic insult. To test this notion, we evaluated whether ectopically expressed E2F2 could be stabilized by UV light. Western blot assays on SH-SY5Y cells transfected either with E2F1-GFP or E2F2-GFP vectors and exposed to UV showed a significant increase in the exogenously expressed E2F1 or E2F2 detected with anti-GFP tag antibody (Fig. 1D, E). Since the expression of these E2Fs is driven by a promoter that does not respond to UV treatment (Fig. 1F), these results suggest that E2F2 levels are increased by a posttranslational mechanism in response to DNA damage, and confirm E2F1 stabilization by genotoxic stress as previously reported.12,17,18
Figure 4.

E2F1 and E2F2 upregulation reduces γH2AX intensity following UV irradiation. (A) SH-SY5Y cells expressing E2F1-GFP, E2F2-GFP or pEGFP-C1 empty vector, fixed 30 minutes post-UV and immunostained with anti-γH2AX antibody. Nuclei were visualized with DAPI staining. Scale bar, 10 μm. (B and C) Percentage of damaged cells obtained by measurement of γH2AX intensity levels. Quantifications were carried out classifying cells according to the E2F expression level: no E2F, low E2F or high E2F in (B), or to the GFP expression level: no GFP, low GFP or high GFP in (C). Results are expressed relative to mock-treated no E2F condition in (B) or mock-treated no GFP condition in (C), which represent the 10% of the maximum γH2AX intensity detected, and UV treatment was normalized to mock-treatment for each of the E2F (B) or GFP (C) intensity levels. Data represent the mean±S.E.M. of at least 4 independent experiments, in which 250 to 400 cells were analyzed for each condition. P-values were calculated by one-way ANOVA, Tukey's: **P < 0.01, ***P < 0.001, n.s. not significant. (D) Immunoblot of E2F1 or E2F2 in SH-SY5Y cells transfected with E2F1-GFP or E2F2-GFP respectively, and in control not transfected (NT) cells. Cells were harvested at the indicated times post-UV. E2F bands correspond to the endogenous protein in NT and to the exogenously expressed protein in E2F-GFP. The numbers under the bands indicate E2F1 and E2F2 quantitation normalized to β-actin and NT-None condition.
Finally, to assess whether E2F1 and E2F2 increases upon genotoxic stress were a consequence –at least partially– of de novo protein synthesis, we examined the effect of inhibiting this process by treating cells with cycloheximide. Cells pre-incubated for 3 h with cycloheximide were harvested and analyzed by Western blot assays 0, 60 and 90 minutes after the times maximum proteins levels had been detected post-genotoxic treatment: 8 h for E2F1 and 2 h for E2F2 (Fig. 1G). Results revealed that de novo protein synthesis inhibition blocked E2F1 and E2F2 induction in response to UV irradiation (Fig. 1H). E2F1 increase in mock-treated cells might be due to the unspecific stabilization by cycloheximide of some mRNAs.29 Therefore, these results indicate that E2F1 and E2F2 are de novo synthesized following DNA damage.
E2F1 and E2F2 induced by DNA damage are transcriptionally active
To address whether the induced E2F1 and E2F2 are active in their transcription regulation functions, we initially transfected neuronal cells with pE2F-CAT or pΔE2F-CAT reporter plasmids and treated them with the DNA damaging agents. pE2F-CAT encodes the chloramphenicol acetyltransferase (CAT) reporter gene driven by adenovirus E2 core promoter and 4 copies of the E2F DNA binding sequence, while pΔE2F-CAT lacks them.30 When compared to mock-treated control cells, we observed an increase in pE2F-CAT activity in cells exposed to DNA damage (Fig. 2A). Consequently, we sought to distinguish between the contributions to this increased transactivation capability of the de novo synthesized E2F1 and E2F2 from that of the stabilized E2F after genotoxic treatment. To this purpose cells were transfected with pE2F-CAT along with E2F1 or E2F2 antisense oligodeoxynucleotides (ODNs) –ASE2F1 and ASE2F2 respectively– (Fig. S5), which were designed to block protein synthesis, or with wild-type E2F decoy ODN (wt E2F DO) containing E2F consensus sequences that would sequester cellular E2F away from its target gene promoters,31 hence abolishing both contributions. After subjecting these cells to DNA damaging agents, we observed that transfection with ASE2F1 or ASE2F2 led to an impaired CAT induction in response to the 3 genotoxic agents tested (Fig. 2B). Interestingly, a more pronounced diminution on the reporter activity was detected with wt E2F DO (Fig. 2C). Taken together, these data indicate that both transcription followed by de novo protein synthesis and protein stabilization contribute to the pools of transcriptionally active E2F1 and E2F2.
Figure 5.

Blockade of E2F1 and E2F2 induction increases γH2AX intensity and reduces DNA repair capability in response to DNA damage. (A–E) Neuro-2A cells transfected with 1 μM of the indicated ODN, exposed to NCS, H2O2 or UV (A) for 4 h (B and D) or 10 h (C and E), fixed and immunostained using anti-γH2AX antibody. Nuclei were visualized with DAPI staining. Scale bar, 10 μm. In (B–E), data represent the mean±S.E.M. of at least 3 independent experiments, in which 300 to 1000 cells were analyzed for each condition. The percentage of damaged cells was obtained by measurement of γH2AX intensity levels. Quantifications were carried out so that data is expressed relative to the control ODNs ASCAT or mut E2F DO, which represent the 10% of the maximum γH2AX intensity detected. P-values were obtained by one-way ANOVA followed by Dunnett's posttest in (B and C) and Student's t-test in (D and E): *P < 0.05, **P < 0.01, ***P < 0.001, n.s. not significant. (F) SH-SY5Y cells transfected with 1 μM of the indicated ODN, UV-irradiated and harvested immediately (0 h) or at 6, 24 or 48 h post-irradiation. Genomic DNA was slot-blotted and analyzed by immunoblot for CPD photoproducts. Methylene Blue staining for total DNA was used as a loading control. The table indicates the average of 2 independent experiments of the percentage of remaining CPD photoproducts, obtained by CPD quantitation and normalization to total DNA. DO, decoy oligodeoxynucleotide.
E2F1 and E2F2 transcriptional induction in response to genotoxic stress requires ATM/ATR and MEK kinases
The cellular response to genomic instability implies the activation of a network of transduction pathways. To investigate which pathways are involved in E2F1 and E2F2 mRNA induction after genotoxic treatment, SH-SY5Y cells were incubated with specific inhibitors before exposure to the DNA damaging agents and harvested 4 h later, which was the time the maximum induction had been detected (Fig. 1A). Inhibition of both ATM and ATR kinases, ATM kinase alone or MEK kinase –with caffeine, KU-55933 or PD-98059 respectively– abrogated E2F1 and E2F2 mRNA increase after genotoxic stress (Fig. 3). Inhibition of PI3K or JNK kinases with LY-294002 or SP-600125 didn't have an effect on E2F1 and E2F2 transcriptional induction in response to DNA damage. Similar results were obtained with Neuro-2A and HN9 cells (Fig. S6). Therefore, these results imply that ATM/ATR and MEK kinases activities are required for the E2F1 and E2F2 transcriptional upregulation following DNA damage.
Figure 6.
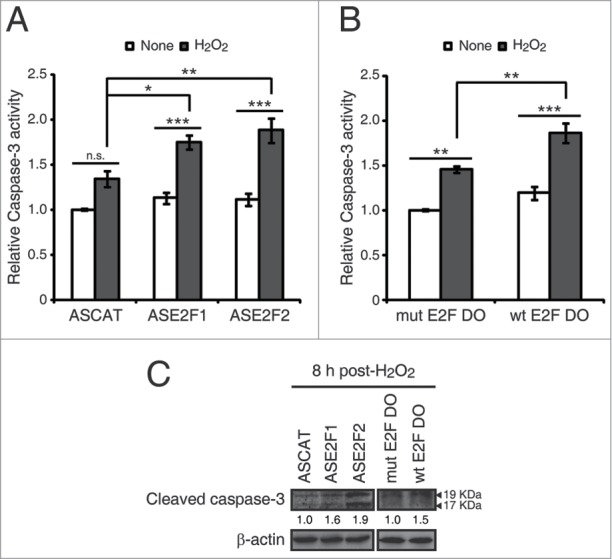
E2F1 and E2F2 reduce apoptotic response after genotoxic stress. SH-SY5Y cells transfected with 1 μM of the specified ODN and treated with H2O2. (A and B) Cell lysates examined for caspase-3 activity 24 h post-H2O2. (C) Western blot of anti-cleaved caspase-3 8 h post-H2O2. The numbers under the bands indicate cleaved caspase-3 quantitation normalized to β-actin and control ODN. In (A and B), data represent the mean±S.E.M. of 4 independent experiments performed in duplicate. One-way ANOVA, Tukey's: *P < 0.05, **P < 0.01, *** P < 0.001, n.s. not significant. DO, decoy oligodeoxynucleotide.
E2F1 and E2F2 induction prevents increased cellular DNA damage upon genotoxic stress
In view of these findings, we addressed the possibility that E2F1 and E2F2 may play a functional role in the neuronal cell response to genotoxic stress. Phosphorylation of histone H2AX on serine 139 (γH2AX) is a well-known indicator of genomic injury.32 We first analyzed the levels of DNA damage by measuring the intensity of γH2AX in cells that overexpressed E2F1 or E2F2. To do this, cells were transfected either with E2F1-GFP, E2F2-GFP or GFP empty vector, fixed 30 minutes after UV irradiation and immunostained with anti-γH2AX antibody (Fig. 4A). For the data analysis, cells were classified according to the E2F fluorescence intensity observed –no E2F, low E2F or high E2F–, which reflects the protein's expression level. Significantly lower γH2AX intensity levels were observed in irradiated cells when E2F1 or E2F2 were upregulated (Fig. 4B). Besides, it is interesting to note the inverse correlation between E2F1 expression and γH2AX staining. No reduction on γH2AX intensity levels was detected by different expression levels of the GFP empty vector –no GFP, low GFP or high GFP– (Fig. 4C), indicating that the observed decrease is indeed a consequence of E2F1 and E2F2 upregulation. Finally, overexpression was confirmed by E2F1 and E2F2 immunoblot of the endogenous and exogenously expressed proteins. There was a 8.4-fold and 6.7-fold increase in E2F1 and E2F2 exogenous proteins respectively compared to the endogenous proteins in basal conditions. These increases were higher than the levels observed for the endogenous proteins at the time post-UV cells were fixed (30 minutes) and also at the time maximum protein levels had been previously observed post-UV irradiation: 8 h for E2F1 and 2 h for E2F2 (Fig. 4D). Thus, these results suggest a role for E2F1 and E2F2 in protecting neuronal cells against the accumulation of DNA damage in cells exposed to UV.
Next, we decided to discriminate between the de novo protein synthesis from the stabilization contributions to this E2F protection capability against DNA damage. To assess this, cells were transfected with ASE2F1, ASE2F2 or wt E2F DO, exposed to genotoxics and analyzed for γH2AX immunostaining (Fig. 5A; Fig. S7). Either at 4 h or 10 h post-genotoxic stress we observed an increased percentage of damaged cells in the presence of ASE2F1 or ASE2F2 (Fig. 5B and C), as well as with wt E2F DO (Fig. 5D and E). It is worth noting that the temporal analysis using the antisense ODNs revealed that E2F1 and E2F2 transcriptional induction plays a role in regulating the level of DNA damage in neuronal cells after genotoxic insult at different time points: E2F2 at an early and E2F1 at a later phase after DNA damage. Moreover, our data suggests that de novo protein synthesis is the major contribution in this regulation. To summarize, these results demonstrate that the E2F1 and E2F2 increase upon genotoxic treatment, either from de novo protein synthesis or protein stabilization, reduces the accumulation of DNA damage.
Figure 7.

E2F1 and E2F2 confer cellular resistance to genotoxic stimuli. (A and B) Cell survival assessed by MTT reduction assay in SH-SY5Y cells transfected with 1 μM of the specified ODN and exposed to UV irradiation. Data is representative of 4 independent experiments carried out in octuplicate. (C–E) Clonogenic assay in SH-SY5Y cells transfected with 1 μM of the indicated ODN and treated with the DNA damaging agent. In (D and E), results are expressed relative to the control mock-treated cells for each ODN, and data represent the mean±S.E.M. of 4 independent experiments performed in cuadruplicate. P-values were calculated using one-way ANOVA with Dunnett's posttest in (D) and Student's t-test in (E): *P < 0.05, **P < 0.01, ***P < 0.001, n.s. not significant. DO, decoy oligodeoxynucleotide.
Finally, to assess whether E2F1 and E2F2 affect DNA repair capability in global genomic repair, we measured the levels of UV induced-cyclobutane pyrimidine dimers (CPDs) in total DNA extracted from neuronal cells that have been transfected with ASE2F1 or ASE2F2 and exposed to UV light. Downregulation of E2F1 and E2F2 led to a delayed removal rate of CPDs (Fig. 5F). While removal of CPD lesions started at 24 h post-UV irradiation and were almost completely repaired at 48 h post-UV in control cells (ASCAT), significant amounts of CPD lesions were still detected at this time point in E2F1 and E2F2 downregulated cells (ASE2F1 and ASE2F2). Therefore, these results show that E2F1 and E2F2 enhance the efficient removal of UV-induced CPD lesions.
E2F1 and E2F2 upregulation reduces apoptotic response after genotoxic injury
Cell death triggered by apoptosis is a common consequence of the excessive accumulation of DNA damage. We therefore investigated whether E2F1 and E2F2 downregulation affected the apoptotic response after genotoxic insult. To this purpose, we first measured caspase-3 activity in neuronal cells exposed to each of the 3 genotoxics. The results revealed that all treatments led to an increase in caspase-3 activity (Fig. S8). Next, we transfected cells with ASE2F1, ASE2F2 or wt E2F DO and treated them with H2O2, the genotoxic agent that triggered the maximum caspase-3 activity. Higher caspase-3 activity levels were observed in the presence of ASE2F1 and ASE2F2 or wt E2F DO after H2O2 exposure (Fig. 6A and B). To confirm these findings, we also analyzed caspase-3 cleavage through a Western blot assay in cells transfected with the ODNs and exposed to H2O2. Cells treated with ASE2F1, ASE2F2 or wt E2F DO had increased levels of cleaved caspase-3 (Fig. 6C), suggesting that impairment of E2F1 and E2F2 induction after genotoxic treatment triggers caspase-3 activation. Taken together, these findings point out a role for E2F1 and E2F2 in protecting neuronal cells from apoptosis induced by DNA damage.
Figure 8.

Accumulation of E2F1 and E2F2 at sites of DNA damage. (A) E2F1 and E2F2 Western blot of cytoplasmic and chromatin fractions of SH-SY5Y cells treated with genotoxic agents for the indicated times. GAPDH and H3 were used as cytoplasmic and chromatin specific markers respectively. Data represents the cytoplasm and chromatin-associated E2F relative percentages for each condition, obtained by normalization to GAPDH and H3 correspondingly. (B–E) Live-cell imaging of SH-SY5Y cells expressing E2F1-GFP or E2F2-GFP microirradiated with a 405 nm laser and pre-incubated or not with the photosensitizer Ro 19–8022 (Ro). In (C and E), data represent the mean±S.D. of 2 independent experiments, in which 10 cells were analyzed for each condition. Arrows indicate the site of microirradiation. Scale bar, 2 μm. (F) SH-SY5Y cells expressing E2F1-HA or E2F2-HA (upper panel) or JNK-HA (lower panel) were UV-irradiated or mock-treated, fixed, lysed and ChIP was carried out with anti-HA antibody. Pulled-down DNA was slot-blotted and analyzed by immunoblot for CPD photoproducts. Methylene Blue staining for total DNA was used as a loading control. (G) Co-immunoprecipitation assays of whole-cell lysates from SH-SY5Y cells harvested 1 h post-NCS treatment. Immunoprecipation (IP) was performed with anti-E2F1, anti-E2F2 and anti-E2F4 antibodies and associated proteins were detected by immunoblot (IB). Non specific IgG isotype antibody served as IP control.
E2F1 and E2F2 confer increased cellular resistance to DNA damaging agents
We next investigated a potential physiological function of E2F1 and E2F2 in neuronal cell response to genotoxic stress. Our findings implicating E2F1 and E2F2 in the reduction of the apoptotic response and in the protection of neuronal cells from DNA damage after genotoxic stress, raise the hypothesis that impairment of E2F1 and E2F2 induction might also affect cell viability in response to DNA damage. To test this, we measured cell survival by MTT reduction assay at different times during the 7 d following UV irradiation on cells that have been previously transfected with ASE2F1, ASE2F2 or wt E2F DO. A significant sensitization of cells transfected with ASE2F2 was observed, but not of those transfected with ASE2F1 (Fig. 7A). Cells transfected with wt E2F DO also showed a reduced percentage of viable cells (Fig. 7B).
To further analyze E2F1 and E2F2s long-term effects and biological relevance on cell viability, we performed a clonogenic assay. Neuronal cells were transfected with ASE2F1, ASE2F2 or wt E2F DO and treated with each of the 3 genotoxics (Fig. 7C). Ten days later, we observed a reduced colony formation capability in cells transfected with ASE2F1 or ASE2F2, and also in cells treated with wt E2F DO (Fig. 7D and E). In summary, the experiments described above unveil a role for E2F2, and to a minor extent for E2F1, in cellular resistance to different genotoxic stresses.
E2F1 and E2F2 accumulate at sites of DNA lesion
To investigate the mechanism of action of E2F1 and E2F2 following DNA damage, we examined whether these proteins localize to the sites of DNA injury in neuronal cells. As a first approach, we analyzed whether E2F1 and E2F2 were recruited to chromatin upon genotoxic stress. To do this, we subjected cells to subcellular fractionation following genotoxic insult, and collected the cytoplasmic and chromatin fractions which were analyzed by electrophoresis. Immunoblots against E2F1 and E2F2 revealed an increase in E2F1 and E2F2 proteins in the chromatin insoluble fraction after treatment with each of the genotoxic agents, which peaks at 30 minutes post-DNA damage (Fig. 8A). Besides, quantification of the cytoplasm and chromatin-associated E2F relative percentages shows an enrichment of E2F1 and E2F2 in the chromatin fraction versus the cytoplasmic fraction following genotoxic injury.
To address the possibility that E2F2 –as well as E2F1– accumulates at sites of DNA lesion, we performed microirradiation experiments with a 405 nm laser coupled with the photosensitizer Ro 19–8022 that promotes local formation of oxidative DNA damage,33 in neuronal cells previously transfected with E2F1-GFP or E2F2-GFP. We observed recruitment of E2F1 and E2F2 to sites of DNA damage, but only in the presence of the photosensitizer (Fig. 8B-E; Movies S1, S2). These results suggest that both proteins localize to the sites of induced oxidized bases but not in the SSBs generated by the 405 nm laser.34 Hence, in order to determine if E2F1 and E2F2 are recruited to sites of UV-induced lesions, we carried out a modified chromatin immunoprecipitation assay on cells transfected with E2F1-HA or E2F2-HA, after exposure to UV light. E2F1 and E2F2 were capable of pulling down DNA fragments that contained the CPD DNA photoproduct, characteristic of UV-induced DNA damage (Fig. 8F). JNK was used as a negative control of precipitation. Taken together, these results indicate that E2F1 and E2F2 accumulate at sites of oxidative and UV-induced DNA damage.
Finally, to study whether E2F1 and E2F2 interact with factors of the DNA repair machinery such as γH2AX, we performed co-immunoprecipitation assays. Whole-cell extracts from neuronal cells harvested 1 h post-NCS treatment were immunoprecipitated with anti-E2F1 or anti-E2F2 antibodies and analyzed by immunoblot against γH2AX. Results showed that γH2AX co-immunoprecipitates with endogenous E2F1 and E2F2 (Fig. 8G). E2F4 family member was used as a negative control of co-immunoprecipitation since it didn't associate with γH2AX in response to NCS-induced DNA damage. These results indicate an E2F1-γH2AX and E2F2-γH2AX interaction following genotoxic injury.
E2F2 promotes Rad51 foci formation and induces histone acetylation following DNA damage
E2F1's role at sites of DNA lesion has been described in previous work, but E2F2's nontranscriptional function is still unknown. It has been established that E2F1 promotes the recruitment of DNA repair factors, such as Rad51, to sites of DNA double-strand breaks.25 Rad51 is the central recombinase in homologous recombination pathways.35 To determine if E2F2 is also involved in Rad51 recruitment to sites of DNA lesion, we performed immunofluorescence assays in neuronal cells transfected with ASE2F2, fixed 1 h post-NCS treatment and analyzed for Rad51 immunostaining (Fig. 9A). Downregulation of E2F2 impaired Rad51 foci formation in response to NCS-induced DNA damage (Fig. 9B). To further confirm this result, Rad51 redistribution to sites of DNA damage was detected by biochemical fractionation with Triton X-100.36 Neuronal cells with ASE2F2 harvested 1 h post-NCS treatment were fractionated into Triton soluble fraction, containing soluble cytoplasmic proteins, or Triton insoluble fraction that contained chromatin-bound proteins. Equal amounts of proteins from each fraction were analyzed by immunoblot against Rad51. Results revealed a decrease in Rad51 recruitment to chromatin in the Triton insoluble fraction in DNA-damaged cells transfected with ASE2F2 (Fig. 9C). Taken together, these results indicate that E2F2, as it was reported for E2F1, is also implicated in genomic stability maintenance through the recruitment of DNA repair factors to DNA double-strand breaks.
Figure 9.
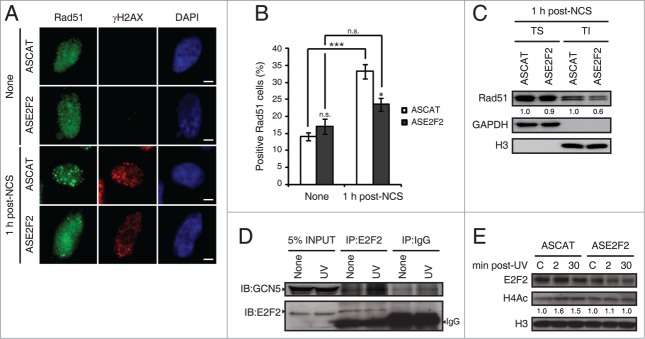
E2F2 promotes Rad51 foci formation and induces histone acetylation in response to DNA damage. (A and B) SH-SY5Y cells transfected with 1 μM of ASE2F2, fixed 1 h post-NCS treatment and immunostained using anti-Rad51 and anti-γH2AX antibodies. Nuclei were visualized with DAPI staining. Scale bar, 10 μm. In (B) data represent the mean±S.E.M. of 4 independent experiments, in which 100 to 250 cells were analyzed for each condition. P-values were calculated using one-way ANOVA with Tukey's posttest: *P < 0.05, *** P < 0 .001, n.s. not significant. Cells with 5 or more Rad51 foci were considered as positive Rad51 cells. (C) Rad51 immunoblot of Triton soluble (TS) and insoluble (TI) fractions of SH-SY5Y cells transfected with 1 μM of ASE2F2 and harvested 1 h post-NCS treatment. GAPDH and H3 were used to detect soluble cytoplasmic and chromatin-bound proteins respectively. The numbers under the bands indicate Rad51 quantitation normalized to GAPDH or H3 in TS or TI fractions correspondingly, and ASCAT condition for each fraction. (D) Co-immunoprecipitation assay of whole-cell lysates from SH-SY5Y cell harvested 30 minutes post-UV irradiation. Immunoprecipation (IP) was performed with anti-E2F2 antibody and associated proteins were detected by immunoblot (IB). Non specific IgG isotype antibody served as IP control. (E) Western blot of acetylated H4 (H4Ac) in SH-SY5Y cells transfected with 1 μM of ASE2F2 and harvested 2 or 30 minutes following UV light exposure. The numbers under the bands indicate H4Ac quantitation normalized to H3 and ASCAT-control condition. C, control mock-treated cells.
Previous work has demonstrated that E2F1 interacts with GCN5 acetyltransferase, promoting its recruitment to sites of DNA damage, and that both E2F1 and GCN5 are necessary to induce H3K9 acetylation in response to UV irradiation.27 To evaluate if E2F2 is involved in a similar epigenetic mechanism, we first performed co-immunoprecipitation assays to determine if E2F2 also interacts with GCN5 following UV-induced DNA damage. Results showed an association between E2F2 and GCN5 upon UV exposure (Fig. 9D). GCN5 has been implicated in the acetylation of both H3 and H4 histones after UV irradiation.37 To assess whether loss of E2F2 affected global H4 acetylation in response to UV-induced DNA damage, we analyzed acetylated H4 protein levels by Western blot in neuronal cells downregulated for E2F2 and harvested 2 or 30 minutes following UV light exposure. We observed that H4 acetylation induction in response to UV irradiation is impaired in E2F2 downregulated cells (Fig. 9E). Therefore, these results suggest that E2F2, like E2F1, is involved in an epigenetic mechanism that promotes histone acetylation upon DNA damage, which in turn would facilitate repair by increasing DNA repair machinery accessibility to sites of damage.
Discussion
In this work we report that E2F1 and E2F2, the latter specifically in neuronal cells, are transcriptionally induced in response to DNA damage. This novel mechanism, which is common to the response to various genotoxic stresses and is conserved in several species, contributes to increase E2F1 and E2F2 protein levels. Therefore, there are 2 parallel mechanisms that lead to the upregulation of E2F1 and E2F2 following DNA damage: the posttranslational modifications of the already synthesized E2F and consequent protein stabilization and, on the other hand, the transcriptional E2F gene induction and de novo protein synthesis. The resulting E2F1 and E2F2 act to promote DNA repair, leading to a reduced apoptotic response and an increased cell survival capability, thereby conferring resistance to genotoxic insult and cooperating in the maintenance of the genome integrity. It should be emphasized that we show for the first time that E2F2 is upregulated following genotoxic stress and plays a critical role in the DNA damage response.
E2F1 response to DNA damage has been subject of interest for many years. It has been well established that E2F1 undergoes posttranslational modifications –such as phosphorylation and acetylation– resulting in protein stabilization.12,17,18 Here, we show evidence that suggests that E2F2 is also stabilized by a posttranslational mechanism in response to genotoxic stress. Further studies to determine the types of modifications and enzymes responsible for these modifications are required.
Our findings highlight a transcriptional mechanism for E2F1 and E2F2 induction upon DNA damage, which depends on ATM/ATR and MEK kinases activities. A recent study has reported an increase of the mRNAs of the 3 activating E2Fs –E2F1, E2F2 and E2F3a– in Saos2 cells upon overnight doxorubicin treatment.14 We were unable to detect any E2F3a induction with the cell lines and genotoxic insults used. Besides, we only observed E2F2 mRNA upregulation in neuronal cells. We don't know the reason for the discrepancy between the results, although we can speculate differences are due to the cell type analyzed and the dose of the DNA damaging agent used. Anyway, since we performed a broad study on the time course changes of the mRNA levels of E2F1–5 in a variety of cells (diverse tissues and species) after exposure to several genotoxic agents, we propose that in response to DNA damage there is a general mechanism resulting in E2F1 and E2F2 transcriptional induction, the latter being restricted to neuronal cells. We presume that E2F2 neuronal specifity upregulation might rely on EGR-1 activity. EGR-1 transcription factor, which is expressed in the nervous system, suits as a potential candidate since it has been shown to be upregulated –mRNA and protein– in response to DNA damage, and to behave as a prosurvival factor following ionizing and UV radiation.38-42 Considering these evidence, we suggest EGR-1 as an upstream regulator of E2F1 and E2F2 induction upon DNA damage. Although EGR-1 has many potential binding sites in both E2F1 and E2F2 promoters, we speculate that in E2F2 promoter in neuronal cells it might associate with coactivators necessary to promote its transcription upon genotoxic stress. Additional studies are required to evaluate this hypothesis and elucidate the mechanism underlying the basis of neuronal specificity of E2F2 transcriptional induction in response to DNA damage.
E2Fs transcriptional functions depend on the signals elicited by a particular type of DNA damaging agent.43-44 Many lines of evidence have indicated that the E2F1 protein increase in response to genotoxic insult is associated to DNA damage-induced apoptosis.12,17,43,45 Although it was always believed that the induction of apoptosis is a unique function of E2F1, it was demonstrated that E2F2 and E2F3a can also activate pro-apoptotic genes.46-47 In contrast, here we show that the induced E2F1 and E2F2 reduce the apoptotic response following DNA damage. This is consistent with the dual role these E2Fs may play upon genotoxic stress, either promoting apoptosis or cell survival, and reinforces the notion that this depends on the cell type and the source and dose of DNA damage.48 In line with our study, previous work has indicated a prosurvival role for E2F1. Transgenic E2F1 -/- mice show enhanced levels of keratinocyte apoptosis following UV-B radiation, whereas basal layer keratinocyte specific overexpression of E2F1 suppresses UV-B-induced apoptosis. Besides, inhibition of apoptosis induced by UV-B is correlated with increased efficiency on removal of DNA photoproducts.49,50 In addition, experiments have shown that in response to DNA damage and ATM phosphorylation, E2F1 apoptotic activities are inhibited by binding to TopBP1, and that this interaction results in E2F1 relocalization to BRCA1-containing repair complexes.51 E2F1 also participates in the recruitment of the Mre11 recombination/repair complex to replication forks.52 Furthermore, E2F1 regulates the transcription of the base excision repair gene XRCC1 and thus contributes to DNA repair.53 Collectively, these findings point out a role for E2F1 in DNA damage checkpoints and/or repair. Consistently, our results provide evidence that E2F1 and E2F2 upregulation upon genotoxic insult protects neuronal cells from the accumulation of DNA damage. We can speculate that the observed reduction in the apoptotic response following genotoxic stress is a consequence of an increased DNA repair efficiency.
Interestingly, we show that E2F1 and E2F2 upregulation contributes to the maintenance of the genome stability. Activator E2Fs can behave both as oncogenes and tumor suppressors depending on the cellular context.54,56 The oncogene activity is probably due to their ability to promote cell proliferation, while the tumor suppression is believed to be a consequence of their pro-apoptotic functions. The fact that we observed that E2F1 and E2F2 reduce apoptosis, localize to the sites of DNA lesions and stimulate DNA repair in response to genotoxic stress, is in agreement with earlier evidence that supports that E2F1 tumor suppressor activity is in some cases unrelated to its apoptotic regulation but rather an outcome of its nontranscriptional functions that facilitate DNA repair. E2F1 is localized to sites of DNA double-strand breaks and UV-induced DNA damage, and promotes the recruitment of DNA repair factors and chromatin modifying enzymes.25,27,51 Our results add a new component to this puzzle, E2F2, which as E2F1, is recruited to sites of oxidative and UV-induced DNA damage, interacts with γH2AX DNA repair factor and GCN5 acetyltransferase, induces histone acetylation and promotes Rad51 foci formation. We also demonstrate that upregulation of E2F1 and E2F2 protects against the accumulation of DNA damage in cells exposed to UV light, whereas downregulation of either E2F1 or E2F2 leads to increased levels of γH2AX following NCS, H2O2 or UV exposure, and impairs the removal of CPD lesions after UV treatment. Therefore, these results suggest 2 possible roles for E2F1 and E2F2 in DNA damage repair. First, a nontranscriptional function in which these E2Fs localize to sites of DNA lesion upon genotoxic stress, and promote the recruitment of DNA repair factors and chromatin modifying enzymes. Second, a transcriptional role involving the expression of prosurvival genes in response to DNA damage. Given that we show that the induced E2F1 and E2F2 are transcriptionally active, further experiments designed to determine which are the E2F1 and E2F2 target genes that promote cell survival following genotoxic insult are required.
DNA damage is a causal factor in neurodegenerative syndromes such as Alzheimer and Parkinson diseases, with patients having an impaired DNA repair ability in their neural tissues.57 E2F1 and E2F2 represent potential targets for therapies to ameliorate the neurological symptoms of these diseases. Thereby, a deeper understanding of the fine molecular mechanisms that regulate these transcription factors’ participation in the DNA damage cellular response is needed.
Materials and Methods
Cell culture, genotoxic agents and transfections
Neuro-2a murine neuroblastoma, HN9 murine hippocampal, HepG2 human hepatoma, and HEK293 human embryonic kidney cells were grown in DMEM (Life Technologies) supplemented with 10% fetal bovine serum (FBS), 100 U/ml penicillin, 100 μg/ml streptomycin, 2 mM glutamine and 1 mM sodium pyruvate at 37°C in a 5% CO2 humidified atmosphere. The human neuroblastoma SH-SY5Y and rat pheochromocytoma PC12 cells were maintained in DMEM (45%) and HAM F-12 (45%) (Life Technologies) at 37°C in 5% CO2 supplemented as indicated above. Rat primary hippocampal neuron cultures were obtained from Wistar embryos of 18–19 gestation days as previously described,58 and were grown in Neurobasal medium (GIBCO) containing N2 supplement, B27 supplement, 100 U/ml penicillin, 100 μg/ml streptomycin, 2 mM glutamine and 1 mM sodium pyruvate at 37°C in 5% CO2.
When indicated, cells were treated with 1 μM actinomycin D (Sigma-Aldrich), 10 μM cycloheximide (Sigma-Aldrich), 50 ng/ml neocarzinostatin (Sigma-Aldrich), 100 μM hydrogen peroxide, or were irradiated in open dishes with UV-C (UV) 40 J/m2, 254 nm (range 240–280 nm) at room temperature from a Philips ultraviolet lamp (TUV15WG15T8) calibrated to deliver 2.5 J/m2 sec. After UV exposure the medium was replaced and cells were maintained at 37°C in 5% CO2.
Cells were transfected either with polyethylenimine (PEI, Polysciences) or Lipofectamine 2000 reagent (Life Technologies), according to the manufacturer's instructions.
Oligodeoxynucleotides and plasmids
Single-strand oligodeoxynucleotides (ODNs) were synthesized with phosphodiester linkage by Bio-Synthesis (Lewisville, TX). Antisense ODN sequences are: ASE2F1: 5´-CCCGAGCAGGGCCTCCAGCGC-3´ and ASE2F2: 5´-TCTGTGGG GCTCATCGCG-3´. ASCAT: 5´-TGAAACTCACCCAGGGATTG-3´ and ASLUC: 5´-GCATACGACGATTCTGTGA-TTTG-3´ were used as internal controls. Circular dumbbell double-stranded decoy ODN 31 wild-type E2F decoy: 5´-ATGCGCGAAACGCGTTTTCGCGTTTCGCGCATAGTTTTCT-3´ and mutant E2F decoy: 5´-ATCTAAACGCGTTTTCGCGTTTAGATTATAGTTTTCT-3´ were annealed and ligated for 24 h at 16°C with 1 unit of T4 DNA ligase (Life Technologies). The underlined sequences correspond to the E2F consensus binding sites. In all cases, cells were transfected at a final concentration of 1 μM ODN.
pE2F-CAT and pΔE2F-CAT plasmids were kindly provided by M. Imperiale.59 The human E2F1 expression plasmid E2F1-GFP was generously supplied by D. Johnson.26 To obtain E2F2-GFP fusion protein, the open reading frame of human E2F2 was inserted into a pEGFP-C1 vector (Clontech) at the BglII/XbaI sites. E2F1-HA and E2F2-HA plasmids were a gift from M. Campanero.60
RNA extraction and Northern blotting analysis
Total cellular RNA was isolated from cultures as described previously.61 Briefly, for Northern blotting analysis, 10 μg of total RNA were denatured, electrophoresed in 1% glyoxal-agarose gels, and transferred to nylon membranes (Hybond N+, GE Healthcare). Membranes were sequentially hybridized with the indicated [32P]-labeled probes and radioactivity was detected using a PhosphorImager (FujiFilm BAS-1800II). Densitometric analysis was performed using the NIH ImageJ software.
Western blotting analysis
Cells were lysed with RIPA buffer (50 mM Tris-HCl pH 7, 150 mM NaCl, 1% Triton X-100, 0.25% sodium deoxycholate, 1 mM EDTA pH 8 and 1X protease inhibitor cocktail (Calbiochem)) and equal amounts of protein were resolved on SDS-PAGE. After transfer to a nitrocellulose membrane (Hybond-ECL, GE Healthcare), analysis by immunoblotting was performed using 1:100 E2F1 (sc-251), 1:100 E2F2 (sc-9967), 1:1000 cleaved caspase-3 (Cell Signaling 9664), 1:3000 β-actin (sc-47778), 1:1000 GAPDH (sc-32233), 1:1000 Histone H3 (sc-8654-R), 1:1000 phospho-Histone H2A.X Ser139 (Upstate 05–636), 1:200 E2F4 (sc-866), 1:1000 Rad51 (sc-8349), 1:1000 GCN5 (sc-20698) and 1:2000 affinity purified polyclonal anti-GFP antibodies. Secondary antibodies were from Sigma-Aldrich. The signal was visualized with enhanced chemiluminiscence reagent (GE Healthcare) and LAS-1000 Image Analyzer (Fujifilm). Densitometric analysis was performed using the NIH ImageJ software.
Chloramphenicol acetyltransferase assay
Neuro-2a cells were transfected with pE2F-CAT or pΔE2F-CAT along with pCEFL-β-galactosidase for chloramphenicol acetyltransferase (CAT) assay. After 18 h, cells were exposed to the genotoxic agents and harvested 24 h later. CAT activity was determined as previously described62 and normalized to β-galactosidase activity.
Immunofluorescence
SH-SY5Y or Neuro-2a cells were seeded onto glass coverslips and allowed to attach 24 h before transfection with the specified plasmid or ODN. Genotoxic treatment was performed 18 h later and cells were fixed and immunostained as described,63 using 1:500 anti-phospho-Histone H2A.X Ser139 (Upstate 05–636) and 1:2000 Alexa Fluor 555 anti-mouse (Life Technologies) antibodies, or 1:1000 anti-Rad51 (Calbiochem PC130) and 1:2000 Alexa Fluor 488 anti-rabbit (Life Technologies) antibodies, and 1 μg/μl DAPI to visualize nuclei. SH-SY5Y cells image acquisition was performed with a Leica confocal microscope SPE, and Neuro-2a slides were analyzed using an Eclipse E600W Nikon microscope and images were acquired with a Coolpix 5000 Nikon digital camera. γH2AX intensity measurements and Rad51 foci number determinations were performed with CellProfiler cell image analysis software.
Slot-blot DNA repair assay
SH-SY5Y cells were UV-irradiated and harvested at different time points post-irradiation. Genomic DNA was isolated and equal amounts (200 ng) of DNA were spotted onto a nylon membrane (Hybond N+, GE Healthcare) with a slot-blot device (Life Technologies). DNA was denatured by incubation of the membrane in 0.4 M NaOH for 20 minutes at room temperature. The filter was further baked at 80°C for 2 h. UV-induced DNA lesions were detected by immunoblot with 1:500 CPD antibody (Kamiya Biomedical, clone KTM53). The membrane was also stained with Methylene Blue (Merck Millipore) for loading control, according to the manufacturer's protocol.
Caspase-3 activity assay
SH-SY5Y cells were transfected with the specified ODN when indicated and exposed to genotoxic agents. Cells were harvested 24 h later and incubated with lysis buffer (50 mM Tris-HCl pH 7.4, 1 mM EDTA pH 8, 10 mM EGTA and 0.5 mM PMSF) at 37°C for 1 h with vigorous vortexing every 15 min, and centrifuged at 10000 x g for 15 min. The activity of caspase-3 in 150 μl cell lysate was determined using 150 μM of the synthetic caspase-3 substrate Ac-DEVD-pNA (Sigma-Aldrich) in reaction buffer (100 mM HEPES pH 7.5, 0.5 mM EDTA pH 8, 20% glycerol and 5 mM dithiothreitol) in a final volume of 300 μl and incubated at 37°C for 10 h. Color development was measured at 405 nm and caspase-3 activity was estimated as A405/μg protein h.
MTT assay
SH-SY5Y cells were transfected with the specified ODN as described above. After 18 h, UV irradiation was carried out and cells were further incubated for the indicated times. Cell activity was assessed by 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT, Sigma-Aldrich) assay as described previously.63
Clonogenic assay
SH-SY5Y cells plated as single cell in 24-well plates (aprox. 100 cells/well), were transfected with the indicated ODN and 18 h later were treated with the specified genotoxics. After 10 days, colonies were stained as previously described.64
Chromatin isolation
Chromatin isolation was carried out as described,65 with minor modifications. Briefly, 1×107 SH-SY5Y cells were resuspended in 300 μl of buffer A (10 mM HEPES pH 7.9, 10 mM KCl, 1.5 mM MgCl2, 0.34 M sucrose, 10% glycerol, 1 mM DTT and 1X protease inhibitor cocktail (Calbiochem)), 0.1% Triton X-100 was added and the cells were incubated on ice for 5 min. Nuclei were obtained in the pellet after low-speed centrifugation (4 min, 1300 x g, 4°C). The supernatant was clarified following high-speed centrifugation (15 min, 16,000 x g, 4°C) to collect the cytoplasmic soluble fraction. Nuclei were washed in buffer A, and lysed in 200 μl of buffer B (3 mM EDTA, 0.2 mM EGTA, 1 mM DTT and 1X protease inhibitor cocktail (Calbiochem)) 30 min on ice. Insoluble chromatin was obtained by centrifugation (5 min, 1700 x g, 4°C), washed in buffer B, centrifuged again and the final chromatin pellet was resuspended in 200 μl of Laemmli buffer and sonicated twice for 15 sec in a Fisher Sonic Dismembrator Model 300 sonicator at 50% power. Cytoplasmic and chromatin fractions were analyzed by immunoblotting (see Western blotting analysis).
Live-cell imaging and microirradiation
SH-SY5Y cells were transfected with E2F1-GFP or E2F2-GFP plasmids. Live-cell imaging was carried out as previously described,34 with a Nikon A1 inverted confocal microscope with an environmental chamber that allows the control of temperature, humidity and gas conditions. Microirradiation was performed for ∼6 sec in preselected regions of 1 μm2, with a 405 nm diode laser at 10% power. Confocal image series of a mid z-section were acquired every 1 sec with the 488 nm laser at 5% power, during the 6 sec before irradiation and for 1 min following irradiation. Fluorescence intensities of the microirradiated region were expressed relative to the immediate post-irradiation intensity for the recruitment kinetics analysis. Five minutes before irradiation, 5 μM Ro 19–8022 photosensitizer was added to the medium when indicated.
Chromatin Immunoprecipitation and Slot-blot
SH-SY5Y cells were transfected with E2F1-HA, E2F2-HA or JNK-HA plasmids, UV-irradiated or mock-treated and 30 min later in vivo cross-linking and chromatin immunoprecipitation (ChIP) was performed as previously reported,66 with some modifications. In brief, cells were fixed with 1% formaldehyde for 10 min at room temperature and further incubated for 5 min with 0.125 M glycine solution at room temperature. Cells were lysed in RIPA buffer and sonicated 12 times for 15 sec in a Fisher Sonic Dismembrator Model 300 sonicator at 50% power. ChIP was carried out at 4°C overnight with 4 μl of HA antibody (Covance, clone 16B12) along with 20 μl of protein A/G PLUS-agarose beads (sc-2003). The precipitated DNA was reverse cross-linked, purified and quantified. UV-induced DNA lesions were detected as described for the Slot-blot DNA repair assay.
Co-immunoprecipitation
Whole-cell lysates (1 mg of protein diluted to 1 ml RIPA buffer) from SH-SY5Y cells treated with the DNA damaging agent and harvested at the indicated times were subjected to immunoprecipitation at 4°C overnight with 1 μg of E2F1 (sc-251), E2F2 (sc-9967) or E2F4 (sc-866) antibodies, along with 20 μl of protein A/G PLUS-agarose beads (sc-2003). Isotype IgG control antibody (sc-2025) served as control. The beads were washed 3 times in PBS, 20 μl of Laemmli buffer was added, and the samples were analyzed by immunoblotting (see Western blotting analysis).
Triton X-100 cell fractionation
SH-SY5Y cells were transfected with the indicated ODN and 18 h later were exposed to UV irradiation and harvested at the indicated times. Biochemical fractionation into Triton X-100 soluble and insoluble fractions was performed as previously described.67 Equal amounts of proteins from each fraction were analyzed by immunoblotting (see Western blotting analysis).
Data analysis
Data analysis was performed with GraphPad Prizm 5.0 (GraphPad Software, La Jolla, CA, USA). Statistical differences were assessed by analysis of variance (ANOVA) with Tukey post hoc analysis for multiple comparisons or with Dunnett post hoc analysis for multiple comparisons to one control group. Student's t-test was applied when only 2 independent groups were compared. P-values of <0 .05 were considered significant.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgements
We thank Thierry Kortulewski at the IRCM Microscopy Facility for his help with the use of the video confocal microscope.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
Funding
This study was supported by research grants from Agencia Nacional de Promoción Científica y Tecnológica [PICT622]; Universidad de Buenos Aires [UBACYT20020100100284]; and Fondation ARC [PJA20131200165] to JPR. Exchanges between the Argentinian and French laboratories were supported by the Ministerio de Ciencia, Tecnología e Innovación Productiva and the French Ministry of Foreign Affairs through an Ecos-Sud Project [A09B01].
References
- 1. Leone G, Nuckolls F, Ishida S, Adams M, Sears R, Jakoi L, Miron A, Nevins JR. Identification of a novel E2F3 product suggests a mechanism for determining specificity of repression by Rb proteins. Mol Cell Biol 2000; 20:3626-32; PMID:10779352; http://dx.doi.org/ 10.1128/MCB.20.10.3626-3632.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. He Y, Armanious MK, Thomas MJ, Cress WD. Identification of E2F-3B, an alternative form of E2F-3 lacking a conserved N-terminal region. Oncogene 2000; 19:3422-33; PMID:10918599 [DOI] [PubMed] [Google Scholar]
- 3. Cartwright P, Muller H, Wagener C, Holm K, Helin K. E2F-6: a novel member of the E2F family is an inhibitor of E2F-dependent transcription. Oncogene 1998; 17:611-23; PMID:9704927 [DOI] [PubMed] [Google Scholar]
- 4. Gaubatz S, Wood JG, Livingston DM. Unusual proliferation arrest and transcriptional control properties of a newly discovered E2F family member, E2F-6. Proc Natl Acad Sci U S A 1998; 95:9190-5; PMID:9689056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Di Stefano L, Jensen MR, Helin K. E2F7, a novel E2F featuring DP-independent repression of a subset of E2F-regulated genes. EMBO J 2003; 22:6289-98; PMID:14633988; http://dx.doi.org/ 10.1093/emboj/cdg613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Christensen J, Cloos P, Toftegaard U, Klinkenberg D, Bracken AP, Trinh E, Heeran M, Di Stefano L, Helin K. Characterization of E2F8, a novel E2F-like cell-cycle regulated repressor of E2F-activated transcription. Nucleic Acids Res 2005; 33:5458-70; PMID:16179649; http://dx.doi.org/ 10.1093/nar/gki855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bagchi S, Raychaudhuri P, Nevins JR. Adenovirus E1A proteins can dissociate heteromeric complexes involving the E2F transcription factor: a novel mechanism for E1A trans-activation. Cell 1990; 62:659-69; PMID:2143697; http://dx.doi.org/ 10.1016/0092-8674(90)90112-R [DOI] [PubMed] [Google Scholar]
- 8. Lavia P, Jansen-Durr P. E2F target genes and cell-cycle checkpoint control. Bioessays 1999; 21:221-30; PMID:10333731; http://dx.doi.org/ 10.1002/(SICI)1521-1878(199903)21:3%3c221::AID-BIES6%3e3.0.CO;2-J [DOI] [PubMed] [Google Scholar]
- 9. Huang Y, Ishiko T, Nakada S, Utsugisawa T, Kato T, Yuan ZM. Role for E2F in DNA damage-induced entry of cells into S phase. Cancer Res 1997; 57:3640-3; PMID:9288762 [PubMed] [Google Scholar]
- 10. Blattner C, Sparks A, Lane D. Transcription factor E2F-1 is upregulated in response to DNA damage in a manner analogous to that of p53. Mol Cell Biol 1999; 19:3704-13; PMID:10207094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. O'Connor DJ, Lu X. Stress signals induce transcriptionally inactive E2F-1 independently of p53 and Rb. Oncogene 2000; 19:2369-76; PMID:10828878 [DOI] [PubMed] [Google Scholar]
- 12. Lin WC, Lin FT, Nevins JR. Selective induction of E2F1 in response to DNA damage, mediated by ATM-dependent phosphorylation. Genes Dev 2001; 15:1833-44; PMID:11459832 [PMC free article] [PubMed] [Google Scholar]
- 13. Ma Y, Freeman SN, Cress WD. E2F4 deficiency promotes drug-induced apoptosis. Cancer Biol Ther 2004; 3:1262-9; PMID:15611646; http://dx.doi.org/ 10.4161/cbt.3.12.1239 [DOI] [PubMed] [Google Scholar]
- 14. Martinez LA, Goluszko E, Chen HZ, Leone G, Post S, Lozano G, Chen Z, Chauchereau A. E2F3 is a mediator of DNA damage-induced apoptosis. Mol Cell Biol 2010; 30:524-36; PMID:19917728; http://dx.doi.org/ 10.1128/MCB.00938-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zalmas LP, Zhao X, Graham AL, Fisher R, Reilly C, Coutts AS, La Thangue NB. DNA-damage response control of E2F7 and E2F8. EMBO Rep 2008; 9:252-9; PMID:18202719; http://dx.doi.org/ 10.1038/sj.embor.7401158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Biswas AK, Johnson DG. Transcriptional and nontranscriptional functions of E2F1 in response to DNA damage. Cancer Res 2012; 72:13-7; PMID:22180494; http://dx.doi.org/ 10.1158/0008-5472.CAN-11-2196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Stevens C, Smith L, La Thangue NB. Chk2 activates E2F-1 in response to DNA damage. Nat Cell Biol 2003; 5:401-9; PMID:12717439 [DOI] [PubMed] [Google Scholar]
- 18. Ianari A, Gallo R, Palma M, Alesse E, Gulino A. Specific role for p300/CREB-binding protein-associated factor activity in E2F1 stabilization in response to DNA damage. J Biol Chem 2004; 279:30830-5; PMID:15123636; http://dx.doi.org/ 10.1074/jbc.M402403200 [DOI] [PubMed] [Google Scholar]
- 19. Galbiati L, Mendoza-Maldonado R, Gutierrez MI, Giacca M. Regulation of E2F-1 after DNA damage by p300-mediated acetylation and ubiquitination. Cell Cycle 2005; 4:930-9; PMID:15917652 [DOI] [PubMed] [Google Scholar]
- 20. Ferreira R, Magnaghi-Jaulin L, Robin P, Harel-Bellan A, Trouche D. The three members of the pocket proteins family share the ability to repress E2F activity through recruitment of a histone deacetylase. Proc Natl Acad Sci U S A 1998; 95:10493-8; PMID:9724731; http://dx.doi.org/ 10.1073/pnas.95.18.10493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Vandel L, Nicolas E, Vaute O, Ferreira R, Ait-Si-Ali S, Trouche D. Transcriptional repression by the retinoblastoma protein through the recruitment of a histone methyltransferase. Mol Cell Biol 2001; 21:6484-94; PMID:11533237; http://dx.doi.org/ 10.1128/MCB.21.19.6484-6494.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Strober BE, Dunaief JL, Guha , Goff SP. Functional interactions between the hBRM/hBRG1 transcriptional activators and the pRB family of proteins. Mol Cell Biol 1996; 16:1576-83; PMID:8657132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Martinez-Balbas MA, Bauer UM, Nielsen SJ, Brehm A, Kouzarides T. Regulation of E2F1 activity by acetylation. EMBO J 2000; 19:662-71; PMID:10675335; http://dx.doi.org/ 10.1093/emboj/19.4.662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Morris L, Allen KE, La Thangue NB. Regulation of E2F transcription by cyclin E-Cdk2 kinase mediated through p300/CBP co-activators. Nat Cell Biol 2000; 2:232-9; PMID:10783242; http://dx.doi.org/ 10.1038/35041123 [DOI] [PubMed] [Google Scholar]
- 25. Chen J, Zhu F, Weaks RL, Biswas AK, Guo R, Li Y, Johnson DG. E2F1 promotes the recruitment of DNA repair factors to sites of DNA double-strand breaks. Cell Cycle 2011; 10:1287-94; PMID:21512314; http://dx.doi.org/ 10.4161/cc.10.8.15341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Guo R, Chen J, Zhu F, Biswas AK, Berton TR, Mitchell DL, Johnson DG. E2F1 localizes to sites of UV-induced DNA damage to enhance nucleotide excision repair. J Biol Chem 2010; 285:19308-15; PMID:20413589; http://dx.doi.org/ 10.1074/jbc.M110.121939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Guo R, Chen J, Mitchell DL, Johnson DG. GCN5 and E2F1 stimulate nucleotide excision repair by promoting H3K9 acetylation at sites of damage. Nucleic Acids Res 2011; 39:1390-7; PMID:20972224; http://dx.doi.org/ 10.1093/nar/gkq983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Carcagno AL, Ogara MF, Sonzogni SV, Marazita MC, Sirkin PF, Ceruti JM, Canepa ET. E2F1 transcription is induced by genotoxic stress through ATM/ATR activation. IUBMB Life 2009; 61:537-43; PMID:19391166; http://dx.doi.org/ 10.1002/iub.197 [DOI] [PubMed] [Google Scholar]
- 29. Dogra SC, Hahn CN, May BK. Superinduction by cycloheximide of cytochrome P4502H1 and 5-aminolevulinate synthase gene transcription in chick embryo liver. Arch Biochem Biophys 1993; 300:531-4; PMID:7678728; http://dx.doi.org/ 10.1006/abbi.1993.1073 [DOI] [PubMed] [Google Scholar]
- 30. Hiebert SW, Chellappan SP, Horowitz JM, Nevins JR. The interaction of RB with E2F coincides with an inhibition of the transcriptional activity of E2F. Genes Dev 1992; 6:177-85; PMID:1531329; http://dx.doi.org/ 10.1101/gad.6.2.177 [DOI] [PubMed] [Google Scholar]
- 31. Park KK, Deok Ahn J, Lee IK, Magae J, Heintz NH, Kwak JY, Lee YC, Cho YS, Kim HC, Chae YM, et al. . Inhibitory effects of novel E2F decoy oligodeoxynucleotides on mesangial cell proliferation by coexpression of E2F/DP. Biochem Biophys Res Commun 2003; 308:689-97; PMID:12927774; http://dx.doi.org/ 10.1016/S0006-291X(03)01455-4 [DOI] [PubMed] [Google Scholar]
- 32. Sharma A, Singh K, Almasan A. Histone H2AX phosphorylation: a marker for DNA damage. Methods Mol Biol 2012; 920:613-26; PMID:22941631; http://dx.doi.org/ 10.1007/978-1-61779-998-3_40 [DOI] [PubMed] [Google Scholar]
- 33. Will O, Gocke E, Eckert I, Schulz I, Pflaum M, Mahler HC, Epe B. Oxidative DNA damage and mutations induced by a polar photosensitizer, Ro19-8022. Mutat Res 1999; 435:89-101; PMID:10526220 [DOI] [PubMed] [Google Scholar]
- 34. Campalans A, Kortulewski T, Amouroux R, Menoni H, Vermeulen W, Radicella JP. Distinct spatiotemporal patterns and PARP dependence of XRCC1 recruitment to single-strand break and base excision repair. Nucleic Acids Res 2013; 41:3115-29; PMID:23355608; http://dx.doi.org/ 10.1093/nar/gkt025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Krejci L, Altmannova V, Spirek M, Zhao X. Homologous recombination and its regulation. Nucleic Acids Res 2012; 40:5795-818; PMID:22467216; http://dx.doi.org/ 10.1093/nar/gks270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mladenov E, Anachkova B, Tsaneva I. Sub-nuclear localization of Rad51 in response to DNA damage. Genes Cells 2006; 11:513-24; PMID:16629903; http://dx.doi.org/ 10.1111/j.1365-2443.2006.00958.x [DOI] [PubMed] [Google Scholar]
- 37. Brand M, Moggs JG, Oulad-Abdelghani M, Lejeune F, Dilworth FJ, Stevenin J, Almouzni G, Tora L. UV-damaged DNA-binding protein in the TFTC complex links DNA damage recognition to nucleosome acetylation. EMBO J 2001; 20:3187-96; PMID:11406595; http://dx.doi.org/ 10.1093/emboj/20.12.3187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Beckmann AM, Wilce PA. Egr transcription factors in the nervous system. Neurochem Int 1997; 31:477-510; discussion 7-6; http://dx.doi.org/ 10.1016/S0197-0186(96)00136-2 [DOI] [PubMed] [Google Scholar]
- 39. Quinones A, Dobberstein KU, Rainov NG. The egr-1 gene is induced by DNA-damaging agents and non-genotoxic drugs in both normal and neoplastic human cells. Life Sci 2003; 72:2975-92; PMID:12706485; http://dx.doi.org/ 10.1016/S0024-3205(03)00230-3 [DOI] [PubMed] [Google Scholar]
- 40. Shin SY, Kim CG, Lee YH. Egr-1 regulates the transcription of the BRCA1 gene by etoposide. BMB Rep 2013; 46:92-6; PMID:23433111; http://dx.doi.org/ 10.5483/BMBRep.2013.46.2.202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hallahan DE, Dunphy E, Virudachalam S, Sukhatme VP, Kufe DW, Weichselbaum RR. C-jun and Egr-1 participate in DNA synthesis and cell survival in response to ionizing radiation exposure. J Biol Chem 1995; 270:30303-9; PMID:8530452; http://dx.doi.org/ 10.1074/jbc.270.51.30303 [DOI] [PubMed] [Google Scholar]
- 42. Huang RP, Fan Y, deBelle I, Ni Z, Matheny W, Adamson ED. Egr-1 inhibits apoptosis during the UV response: correlation of cell survival with Egr-1 phosphorylation. Cell Death Differ 1998; 5:96-106; PMID:10200450; http://dx.doi.org/ 10.1038/sj.cdd.4400322 [DOI] [PubMed] [Google Scholar]
- 43. Pediconi N, Ianari A, Costanzo A, Belloni L, Gallo R, Cimino L, Porcellini A, Screpanti I, Balsano C, Alesse E, et al. . Differential regulation of E2F1 apoptotic target genes in response to DNA damage. Nat Cell Biol 2003; 5:552-8; PMID:12766778; http://dx.doi.org/ 10.1038/ncb998 [DOI] [PubMed] [Google Scholar]
- 44. Yoshida K, Inoue I. Expression of MCM10 and TopBP1 is regulated by cell proliferation and UV irradiation via the E2F transcription factor. Oncogene 2004; 23:6250-60; PMID:15195143; http://dx.doi.org/ 10.1038/sj.onc.1207829 [DOI] [PubMed] [Google Scholar]
- 45. Kowalik TF, DeGregori J, Leone G, Jakoi L, Nevins JR. E2F1-specific induction of apoptosis and p53 accumulation, which is blocked by Mdm2. Cell Growth Differ 1998; 9:113-8; PMID:9486847 [PubMed] [Google Scholar]
- 46. Vigo E, Muller H, Prosperini E, Hateboer G, Cartwright P, Moroni MC, Helin K. CDC25A phosphatase is a target of E2F and is required for efficient E2F-induced S phase. Mol Cell Biol 1999; 19:6379-95; PMID:10454584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ziebold U, Reza T, Caron A, Lees JA. E2F3 contributes both to the inappropriate proliferation and to the apoptosis arising in Rb mutant embryos. Genes Dev 2001; 15:386-91; PMID:11230146; http://dx.doi.org/ 10.1101/gad.858801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. DeGregori J. E2F and cell survival: context really is key. Dev Cell 2005; 9:442-4; PMID:16198285; http://dx.doi.org/ 10.1016/j.devcel.2005.09.006 [DOI] [PubMed] [Google Scholar]
- 49. Wikonkal NM, Remenyik E, Knezevic D, Zhang W, Liu M, Zhao H, Berton TR, Johnson DG, Brash DE. Inactivating E2f1 reverts apoptosis resistance and cancer sensitivity in Trp53-deficient mice. Nat Cell Biol 2003; 5:655-60; PMID:12833065; http://dx.doi.org/ 10.1038/ncb1001 [DOI] [PubMed] [Google Scholar]
- 50. Berton TR, Mitchell DL, Guo R, Johnson DG. Regulation of epidermal apoptosis and DNA repair by E2F1 in response to UV B radiation. Oncogene 2005; 24:2449-60; PMID:15735727; http://dx.doi.org/ 10.1038/sj.onc.1208462 [DOI] [PubMed] [Google Scholar]
- 51. Liu K, Lin FT, Ruppert JM, Lin WC. Regulation of E2F1 by BRCT domain-containing protein TopBP1. Mol Cell Biol 2003; 23:3287-304; PMID:12697828; http://dx.doi.org/ 10.1128/MCB.23.9.3287-3304.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Maser RS, Mirzoeva OK, Wells J, Olivares H, Williams BR, Zinkel RA, Farnham PJ, Petrini JH. Mre11 complex and DNA replication: linkage to E2F and sites of DNA synthesis. Mol Cell Biol 2001; 21:6006-16; ; PMID:11486038; http://dx.doi.org/ 10.1128/MCB.21.17.6006-6016.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Chen D, Yu Z, Zhu Z, Lopez CD. E2F1 regulates the base excision repair gene XRCC1 and promotes DNA repair. J Biol Chem 2008; 283:15381-9; PMID:18348985; http://dx.doi.org/ 10.1074/jbc.M710296200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Field SJ, Tsai FY, Kuo F, Zubiaga AM, Kaelin WG, Jr., Livingston DM, Orkin SH, Greenberg ME. E2F-1 functions in mice to promote apoptosis and suppress proliferation. Cell 1996; 85:549-61; PMID:8653790 [DOI] [PubMed] [Google Scholar]
- 55. Yamasaki L, Jacks T, Bronson R, Goillot E, Harlow E, Dyson NJ. Tumor induction and tissue atrophy in mice lacking E2F-1. Cell 1996; 85:537-48; PMID:8653789 [DOI] [PubMed] [Google Scholar]
- 56. Zhu JW, Field SJ, Gore L, Thompson M, Yang H, Fujiwara Y, Cardiff RD, Greenberg M, Orkin SH, DeGregori J. E2F1 and E2F2 determine thresholds for antigen-induced T-cell proliferation and suppress tumorigenesis. Mol Cell Biol 2001; 21:8547-64; PMID:11713289; http://dx.doi.org/ 10.1128/MCB.21.24.8547-8564.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. McKinnon PJ. DNA repair deficiency and neurological disease. Nat Rev Neurosci 2009; 10:100-12; PMID:19145234; http://dx.doi.org/ 10.1038/nrn2559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Caceres A, Banker GA, Binder L. Immunocytochemical localization of tubulin and microtubule-associated protein 2 during the development of hippocampal neurons in culture. J Neurosci 1986; 6:714-22; PMID:3514816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Harris KF, Christensen JB, Radany EH, Imperiale MJ. Novel mechanisms of E2F induction by BK virus large-T antigen: requirement of both the pRb-binding and the J domains. Mol Cell Biol 1998; 18:1746-56; PMID:9488491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Campanero MR, Flemington EK. Regulation of E2F through ubiquitin-proteasome-dependent degradation: stabilization by the pRB tumor suppressor protein. Proc Natl Acad Sci U S A 1997; 94:2221-6; PMID:9122175; http://dx.doi.org/ 10.1073/pnas.94.6.2221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem 1987; 162:156-9; PMID:2440339; http://dx.doi.org/ 10.1016/0003-2697(87)90021-2 [DOI] [PubMed] [Google Scholar]
- 62. Varone CL, Giono LE, Ochoa A, Zakin MM, Canepa ET. Transcriptional regulation of 5-aminolevulinate synthase by phenobarbital and cAMP-dependent protein kinase. Arch Biochem Biophys 1999; 372:261-70; PMID:10600163; http://dx.doi.org/ 10.1006/abbi.1999.1470 [DOI] [PubMed] [Google Scholar]
- 63. Ceruti JM, Scassa ME, Flo JM, Varone CL, Canepa ET. Induction of p19INK4d in response to ultraviolet light improves DNA repair and confers resistance to apoptosis in neuroblastoma cells. Oncogene 2005; 24:4065-80; PMID:15750620; http://dx.doi.org/ 10.1038/sj.onc.1208570 [DOI] [PubMed] [Google Scholar]
- 64. Ceruti JM, Scassa ME, Marazita MC, Carcagno AC, Sirkin PF, Canepa ET. Transcriptional upregulation of p19INK4d upon diverse genotoxic stress is critical for optimal DNA damage response. Int J Biochem Cell Biol 2009; 41:1344-53; PMID:19130897; http://dx.doi.org/ 10.1016/j.biocel.2008.12.005 [DOI] [PubMed] [Google Scholar]
- 65. Mendez J, Stillman B. Chromatin association of human origin recognition complex, cdc6, and minichromosome maintenance proteins during the cell cycle: assembly of prereplication complexes in late mitosis. Mol Cell Biol 2000; 20:8602-12; PMID:11046155; http://dx.doi.org/ 10.1128/MCB.20.22.8602-8612.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Fousteri M, Vermeulen W, van Zeeland AA, Mullenders LH. Cockayne syndrome A and B proteins differentially regulate recruitment of chromatin remodeling and repair factors to stalled RNA polymerase II in vivo. Mol Cell 2006; 23:471-82; PMID:16916636; http://dx.doi.org/ 10.1016/j.molcel.2006.06.029 [DOI] [PubMed] [Google Scholar]
- 67. Gospodinov A, Tsaneva I, Anachkova B. RAD51 foci formation in response to DNA damage is modulated by TIP49. Int J Biochem Cell Biol 2009; 41:925-33; PMID:18834951; http://dx.doi.org/ 10.1016/j.biocel.2008.09.004 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.