

Mature naïve B cells are quiescent until activated by antigen receptor engagement. Once activated, follicular B cells rapidly proliferate and undergo immunoglobulin (IG) locus gene modifications to generate high-affinity antibodies in a transient anatomical structure in peripheral lymphoid organs called a germinal center (GC). During a GC reaction, B cells interact with neighboring T cells and dendritic cells to ultimately differentiate into antibody-secreting plasma cells or memory B cells.1,2 The GC reaction is tightly controlled as errors contribute to lymphomagenesis—the majority of immune system malignancies are thought to originate by transformation of GC B cells.2
Previously, we discovered a role for liver kinase B1 (LKB1) in B cells exiting the GC reaction. The LKB1 serine/threonine kinase regulates signal transduction pathways that control cell metabolism, polarity, and additional features.3 Terminal B cell differentiation into plasma cells requires signaling from activation induced cytidine deaminase (AID) during IG class switch recombination via activated LKB1 to inactivate CREB regulated transcriptional coactivator 2 (CRTC2). LKB1 dependent CRTC2 inactivation represses a GC B cell gene expression program that precedes or coincides with the initiation of the plasma cell transcriptional program. Defects in this pathway prolong the GC reaction and inhibit GC B cell to plasma cell differentiation in an in vitro model system.4
To investigate this pathway further, we generated B cell specific Lkb1 knockout (BKO) mice. BKO mice deleted Lkb1 from ∼40% of mature B cells on average, but surprisingly exhibited robust GC formation in the absence of exogenous antigenic stimulation. Mature, naïve Lkb1 deficient B cells expressed activation biomarkers, displayed enhanced proliferation, and secreted a panel of pro-inflammatory cytokines and chemokines that included IL-6. These factors, specifically IL-6, polarized the differentiation and expansion of neighboring T cells into T follicular helper (TFH) cells to support GC formation and expansion. This pro-inflammatory state also led to the activation of co-existing wild-type B cells and their recruitment into GCs.5 These data reveal that LKB1 maintains mature pre-GC B cell quiescence and that loss of LKB1 in B cells promotes B cell activation, which can trigger the start of the GC reaction without exogenous antigen.
We also showed that in wild-type B cells, LKB1 was phosphorylated at Ser431 in a signaling pathway downstream of surface IgM engagement, a modification that inhibits LKB1 activity in a physiologically relevant context.5 Lkb1 is frequently inactivated by mutation in its kinase domain, particularly in epithelial cancers, non-small cell lung cancers, and cervical cancers.3 Importantly, we have also shown that Lkb1 expression is decreased in many human lymphoma samples,4 suggesting that LKB1 opposes lymphomagenesis.
Our studies suggest that LKB1 inactivation propels mature, naïve follicular B cells to exit a quiescent state and progress into highly proliferative GC B cells. As we have shown previously, LKB1 activation in B cells is also required at the end of the GC reaction to generate plasma cells. We therefore propose that LKB1 acts as a switch in B cells, with inactivation of LKB1 helping to start the GC reaction, followed by activation of LKB1 to help return GC B cells to a non-proliferative state in antibody-secreting plasma cells (Fig. 1).
Figure 1.
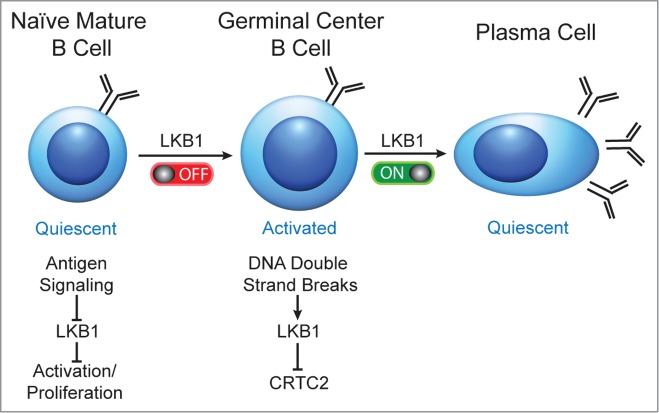
Proposed model in which LKB1 acts as an on/off switch to sequentially activate and inactivate B cells during a T cell dependent humoral immune response. In mature naïve B cells, LKB1 is active. Antigenic stimulation is proposed to inactivate LKB1, enabling cellular activation and the differentiation of highly proliferative GC B cells. To exit a GC, DNA double strand breaks during IG class switch recombination activate LKB1 to inactivate CRTC2 and promote the terminal differentiation of GC B cells into quiescent plasma cells.
Recently, the regulation of cellular quiescence by LKB1 has been demonstrated in other immune cell types including T cells6 and hematopoietic stem cells (HSCs).7 Deletion of Lkb1 in HSCs causes an increase in proliferation and HSC expansion, and in T cells Lkb1 deletion causes hyper-activation and increased cytokine production, similar to our findings in LKB1 deficient B cells.
The mechanism(s) for how constitutively active LKB1 maintains quiescence of these hematopoietic cell types remains unclear. Loss of Lkb1 in B cells led to the activation of NF-κB, which contributed to IL-6 production that promoted TFH cell differentiation and GC formation.5 Additional LKB1 downstream target proteins may also be involved in maintaining quiescence. A leading candidate is 5′-AMP activated protein kinase (AMPK), which regulates metabolism and the cell cycle. In the absence of nutrients or reduced intracellular ATP, AMPK becomes activated to inhibit anabolic processes, such as lipid and protein biosynthesis, through regulation of ACC, mTORC, and other effector proteins. Additionally, AMPK can phosphorylate p53 and halt cell cycle progression.3 Both of these activities may help LKB1 maintain quiescence in haematopoietic cell types. In B cells, inactivation of LKB1 during antigen encounters may allow protein, nucleotide and lipid biosynthesis and also prevent p53 phosphorylation to permit the rapid proliferation of GC B cells.
In other Lkb1 cell-specific knockout models, complementation or pharmacological activation of AMPK has not been sufficient to reverse the activation and proliferative phenotype.6-7 LKB1 targets 12 additional AMPK-related family member proteins that may also regulate quiescence. The functions of these other kinases include controlling anoikis and cell polarity,3 but other functions remain unknown and are thus a rich area for future studies of AMPK-independent activities of LKB1 deficient B cells.
References
- 1.Tooze RM. Front Immunol 2013. PMID: 24385976; http://dx.doi.org/ 10.3389/fimmu.2013.00460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Basso K, Dalla-Favera R. Nat Rev Immunol 2015. PMID: 25712152; http://dx.doi.org/ 10.1038/nri3814 [DOI] [PubMed] [Google Scholar]
- 3.Alessi DR. et al.. Ann Rev Biochem 2006; 75:137–163. PMID: 16756488; http://dx.doi.org/ 10.1146/annurev.biochem.75.103004.142702 [DOI] [PubMed] [Google Scholar]
- 4.Sherman MH. et al.. Mol Cell 2010; 39:873–885. PMID: 20864035; http://dx.doi.org/ 10.1016/j.molcel.2010.08.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Walsh NC, et al.. EMBO Rep 2015; 16(6):753-68; PMID: 25916856; http://dx.doi.org/ 10.15252/embr.201439505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blagih J, et al.. Immunol Rev 2012; 249:59–71. PMID: 22889215; http://dx.doi.org/ 10.1111/j.1600-065X.2012.01157 [DOI] [PubMed] [Google Scholar]
- 7.Krock B, et al.. Cell Metab 2011; 13:8–10. PMID: 21195344; http://dx.doi.org/ 10.1016/j.cmet.2010.12.015 [DOI] [PubMed] [Google Scholar]