

Abstract
The California mussel, Mytilus californianus, adheres in the highly oxidizing intertidal zone with a fibrous holdfast called the byssus using 3, 4-dihydroxyphenyl-l-alanine (DOPA)-containing adhesive proteins. DOPA is susceptible to oxidation in seawater and, upon oxidation, loses adhesion. Successful mussel adhesion thus depends critically on controlling oxidation and reduction. To explore how mussels regulate redox during their functional adhesive lifetime, we tracked extractable protein concentration, DOPA content and antioxidant activity in byssal plaques over time. In seawater, DOPA content and antioxidant activity in the byssus persisted much longer than expected—50% of extractable DOPA and 30% of extractable antioxidant activity remained after 20 days. Antioxidant activity was located at the plaque–substrate interface, demonstrating that antioxidant activity keeps DOPA reduced for durable and dynamic adhesion. We also correlated antioxidant activity to cysteine and DOPA side chains of mussel foot proteins (mfps), suggesting that mussels use both cysteine and DOPA redox reservoirs for controlling interfacial chemistry. These data are discussed in the context of the biomaterial structure and properties of the marine mussel byssus.
Keywords: redox, antioxidant activity, mussels
1. Introduction
The California mussel (Mytilus californianus) adheres to surfaces in highly oxidizing seawater via plaques within a versatile, opportunistic holdfast known as the byssus (figure 1a), which represents an excellent model system for understanding adaptive mechanisms of adhesion under wet, oxidizing conditions. The mussel secretes byssal precursors into the distal depression of its foot, creating attachment plaques fused to threads (thread–plaque pairs) during the deposition process. These byssal plaques mediate sessile attachment through proteins with ‘sticky’ catecholic side chains of 3,4-dihydroxyphenyl-l-alanine (DOPA) [1,2]. Yet, DOPA is prone to oxidation in alkaline (pH approx. 8) seawater, whereupon it loses adhesion and becomes primarily cohesive through cross-linking [3–5]. Given the vast applications of adhesion in wet, oxidizing environments, understanding and implementing relevant design concepts from mussels can benefit new materials in medical, dental and maritime industries.
Figure 1.
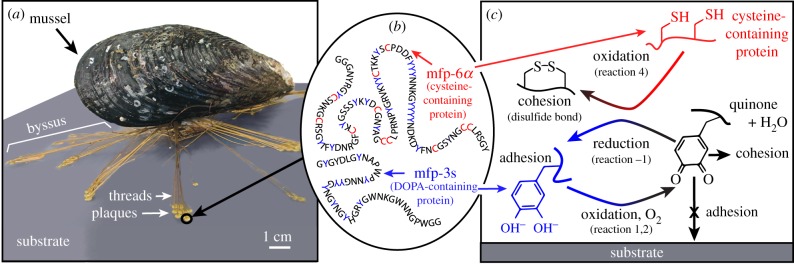
Adaptive aspects of M. californianus adhesion in oxidizing seawater conditions. (a) Thread–plaque pairs forming the adhesive byssus that attaches M. californianus to the substrate. (b) Schematic of representative DOPA-containing adhesive proteins (mfp-3s) and cysteine-containing antioxidant proteins (mfp-6α) in the plaques. (c) DOPA (blue) oxidizing to dopaquinone (black) in a DOPA-containing protein (e.g. mfp-3s), reducing O2 to H2O in the process. Reduced DOPA adhering readily to a substrate where its oxidized form, dopaquinone, is primarily cohesive. Cysteine thiols (red) in an antioxidant protein (e.g. mfp-6α) reducing dopaquinone back to DOPA.
DOPA, a post-translationally modified amino acid in adhesive proteins, can adhere to a variety of surfaces underwater [1,2,6]. DOPA adheres through multiple bidentate H-bonds or coordination complexes on polar surfaces, such as minerals or metals with near-covalent bond energy [2,6,7]. DOPA also achieves dynamic, reversible cohesion through hydrogen bonding, metal ion chelation and π–cation interactions [8–12]. In seawater (pH 8), DOPA (QH2) in solution oxidizes readily to dopaquinone (Q), reducing molecular O2 to H2O in the process (reactions 1–3 at standard conditions of pH 7, 1 atm, 1 M, 25°C) [13,14]. Thermodynamically, DOPA oxidation is favourable over a wide range of pHs, but the high activation energy at low pH (pH 3) makes it kinetically stable. At high pH, dopaquinone formation triggers mussel foot proteins (mfps) to solidify thereby increasing cohesion [3,4,15]. Uncurtailed oxidation of adhesive mfps (mfp-3 and mfp-5) decreases adhesion by more than 80% on titania and more than 90% on polysiloxane (mica) surfaces [1,2,5]. In this way, DOPA redox modulates the balance between adhesion and cohesion; yet, maintaining DOPA is challenging given seawater's highly oxidizing conditions.
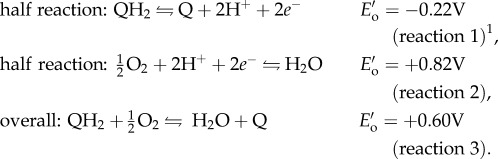 |
To preserve DOPA over dopaquinone in adhesive proteins during plaque deposition, mussels co-secrete H3O+ and antioxidants into the distal depression with mfps [5,15,16]. The H3O+ lowers the pH of the distal depression, shielding mfps from oxidation by dissolved oxygen and preserving adhesive DOPA. The antioxidant protein (e.g. mfp-6α) contains cysteine thiols (R-SH) that reduce dopaquinone (Q) back to DOPA (QH2) in adhesive proteins, such as mfp-3s, linking disulfides (R-S-S-R) and increasing both adhesion and cohesion in the process (reactions −1, 4–5 at standard conditions, figure 1b, c). Yu et al. [5] hypothesized that both these protective mechanisms, H3O+ and antioxidants, maintain DOPA just long enough to allow a critical number of surface-binding engagements, after which the remaining DOPA oxidizes and cross-links to increase cohesion. Although the pH appears to equilibrate quickly with seawater [17], it is not known whether antioxidant activity remains after pH equilibration; if it does, the duration is not well understood
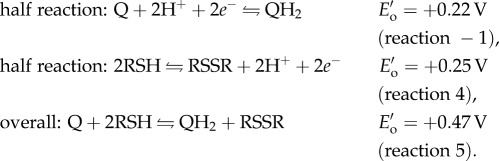 |
To test whether reducing conditions introduced during plaque deposition equilibrate quickly with seawater, we tracked the levels of soluble protein, DOPA content and antioxidant activity in mussel byssal plaques over time. We identified a persistent reservoir of antioxidant (reducing) activity in the plaques that correlates to both cysteine and DOPA side chains of mfps (e.g. mfp-6α and mfp-3s). This correlation suggests that mussels use both cysteine and DOPA redox potentials for favourable long-term interfacial chemistry. The slow oxidation rate of DOPA in the byssus compared with the rate in solution suggests that plaque redox is non-equilibrium and under kinetic control. This effective insulation from oxidation may be an adaptive, self-healing strategy that contributes to the mussel's dynamic and durable adhesive interface.
2. Material and methods
2.1. Experimental animals and plaque collection
Mytilus californianus were collected offshore around Goleta Pier (34°24′54″ N by 199°49′30″ W, Goleta, CA, USA) and we transferred them to mariculture tanks with a flow rate of approximately 4 gallons min−1 of seawater (50% filtered and 50% circulating). To track mussel byssus formation, we selected 10 of the largest mussels (approx. 9 cm) and secured them (using elastic bands) to poly(methyl methacrylate) (PMMA, a.k.a. Plexiglas™) sheets (20 × 20 cm). We tracked the length of time each plaque was in use (age) for 20 days. Plaques deposited on the first day of tracking resulted in 20-day-old plaques; those deposited on the last day of tracking resulted in 1-day-old plaques. At harvest, we severed the mussel byssal thread 1 cm from its plaque–adhesive interface and shaved the plaque attachment off the PMMA surface using a single-edged razor. After harvest, we washed (500 volumes of Milli-Q water) the thread–plaque segments (further referred to as plaques) of any excess salt before lyophilizing them in a Labconco FreeZone Plus freeze dryer (Kansas City, MO, USA). We sorted plaques by mussel of origin and length of time in use, pooling plaques deposited on the same day. Plaques were stored at −20°C for no more than two weeks.
2.2. Soluble protein extraction and quantification
To extract the soluble protein from mussel plaques, we homogenized the lyophilized plaques with 5% (v/v) acetic acid (Thermo Fisher Scientific, Waltham, MA, USA) in a Kontes tissue grinder (Kimble Chase, Rockwood, TN, USA). To ensure particulate removal, we centrifuged (10 min at 13 000 r.p.m.) the mixture twice using an Eppendorf MiniSpin microcentrifuge (Hauppauge, NY, USA) and decanted between spins. To determine the extracted soluble protein concentration of mussel plaques, we used the Bradford assay to generate a standard curve. We generated a standard curve using bovine serum albumin (Thermo Fisher Scientific) as a standard and combining it at varying concentrations with a Coomassie stock solution (0.1 g Coomassie Brilliant Blue G-250 (Bio-Rad, Hercules, CA, USA) in 50 ml ethanol (Thermo Fisher Scientific) and 100 ml phosphoric acid (Thermo Fisher Scientific)). We then quantified the soluble protein by combining it with the Coomassie stock solution and extrapolated the soluble protein concentration using the established standard curve. Finally, we normalized the soluble protein concentration to 80 µg ml−1 in 5% (v/v) acetic acid. Herein, extractable protein (soluble protein) refers to protein solubilized by mechanical means under acidic conditions, prepared the same day we assayed for DOPA content and antioxidant activity.
2.3. DOPA quantification
To quantify DOPA content in plaque extracts, we used a borate-shift assay that specifically and non-destructively measures DOPA content in a complex protein mixture [18]. The borate-shift assay relies on measuring the absorbance difference at 296 nm (Δɛ of 2900 to 3200 M−1 cm−1) of a sample in acidic versus basic conditions and extrapolating DOPA concentration from the borate-shift of a standard. Using l-DOPA (23 µM–1.5 mM (Sigma-Aldrich, St Louis, MO, USA)) as a standard, we determined DOPA concentration of normalized plaque extracts by monitoring the absorbance difference in acidic (pH approx. 2, 0.1 M HCl) versus borate-buffered (pH 8, 0.1 M sodium borate) solutions (Thermo Fisher Scientific) using a Nanodrop 2000c spectrophotometer (Thermo Fisher Scientific).
2.4. Antioxidant activity assay
We tested for antioxidant activity using the free radical redox sensor 2, 2-diphenyl-1-picrylhydrazyl (DPPH (Sigma-Aldrich)). When DPPH is exposed to antioxidants (reagents with reducing power), DPPH's intense purple colour is bleached to a pale yellow [16,19,20]. Based on this colour shift, we tested for antioxidant activity by first mixing a stock solution of DPPH (2 mM, Sigma-Aldrich) and methanol (Fisher Scientific) for 1 h at room temperature. We combined DPPH stock solution (100 µM) with normalized plaque extracts (8 µg ml−1) in a pH 3.0 citric acid–phosphate buffer (68 mM citric acid monohydrate (Sigma-Aldrich), 34 mM sodium phosphate dihydrate (EMD Millipore, Merck KGaA, Darmstadt, Germany), 0.3% (v/v) Triton X-100 (Sigma-Aldrich)). We then measured absorbance differences of plaque extracts versus a control (5% (v/v) acetic acid) at 515 nm using a Nanodrop 2000c spectrophotometer at 15-min time intervals (1, 15, 30 min). DPPH reduction occurred primarily in the first 15 min. Reported absorbance measurements are at t = 30 min, at the point which the readings stabilized. For comparison, we measured the antioxidant activity of vitamin C, a known antioxidant, under the same conditions.
2.5. Antioxidant activity localization
To locate antioxidant activity in whole, intact thread–plaques, we exposed them to surfaces doped with DPPH. First, we cut thin layer chromatography (TLC) plates (Analtech Uniplate GS, Newark, DE, USA) to size (6 × 6 cm) and activated them in methanol before saturating the plates in DPPH stock solution and drying the plates on aluminium foil in an oven (37°C, 2 min) thereby creating DPPH-doped surfaces. To locate in situ antioxidant activity, we removed 4-day-old plaques from the PMMA plates and immediately (less than 1 min) placed them on the DPPH-doped plates, covered the plate with a glass slide, secured the plate to the slide with a rubber band and wrapped it in aluminium foil. After 24 h of equilibration at room temperature, we separated the DPPH-doped plate from the slide and imaged the DPPH-doped TLC plate both before and after plaque removal using an Olympus BX60 microscope (Waltham, MA, USA). To determine if soaking the whole, intact thread–plaque pairs removed the antioxidant activity, both soaked (500 equivalents of milli-Q H2O) and unsoaked plaques were tested for in situ antioxidant activity.
2.6. Cysteine contribution to antioxidant activity
We investigated to what extent protein cysteine residues (thiols) contribute to antioxidant activity by specifically blocking the thiols in plaque extracts [21]. The soluble portion of freshly homogenized 4-day-old plaques was mixed with either pH 3 citric acid–phosphate buffer or 0.22 µM iodoacetamide (Sigma-Aldrich) in pH 7.0 citric acid–phosphate buffer (0.7 mM citric acid monohydrate, 14 mM sodium phosphate dihydrate, 0.3% (v/v) Triton X-100), and equilibrated for 15 min. Using HCl as needed, we adjusted the pH to 3.0 in plaque extracts both with and without blocking agent (iodoacetamide), and added DPPH (100 µM) before monitoring the absorbance change (30 min post-mixing) at 515 nm. Because buffered iodoacetamide did not significantly affect the DPPH assay, we used it as a control.
3. Results
3.1. Soluble protein extraction and quantification
We collected M. californianus plaques and demonstrated that all plaque extracts contained soluble protein that is directly correlated to plaque age. Using the Bradford assay, we determined that the protein concentration decreased significantly (p < 0.0001) from 11.5 ± 2.3% (w/w) at days 1–4 to 2.8 ± 0.5% at days 17–20 (n = 13–20, figure 2a).
Figure 2.

Plaque ageing as indicated by (a) soluble protein concentration, (b,d) DOPA content and (c,d) antioxidant activity decreasing with plaque age. (a) Soluble protein (plaque extract) concentration decreasing as determined by the Bradford assay using Coomassie Brilliant Blue in a 1 : 2 ethanol : phosphoric acid solution. Ordinates denote weight/plaque dry weight % over time. (b,d) DOPA content of plaque extracts decreasing as determined by the borate-shift assay (296 nm) in acidic (pH approximately 2, 0.1 M HCl) versus borate-buffered (pH 8, 0.1 M sodium borate) solutions over time. Ordinates denote mmol DOPA per milligram of protein. (c,d) Antioxidant activity of plaque extracts decreasing as determined by the DPPH assay (515 nm) using a citric acid–phosphate buffer (68 mM citric acid monohydrate, 34 mM sodium phosphate dihydrate, 0.3% (v/v) Triton X-100, pH 3.0), 100 µM DPPH and 5% methanol (v/v). Ordinates denote normalized % of antioxidant activity ((sample absorbance change—negative control absorbance change)/(largest absorbance change—negative control absorbance change) × 100) over time. (d) Antioxidant activity and DOPA content in plaque extracts correlate (R2 = 0.66) linearly (1 : 1) over time. The arrow indicates the direction of increasing plaque age. (a–d) Data are represented as means and standard errors (n = 13–20).
3.2. DOPA content in plaque extracts
Using the borate-shift assay, we detected DOPA in all plaque extracts and found that the DOPA content is indirectly correlated to plaque age. Whereas DOPA content in extracts of younger plaques (days 1–4) averaged 67 ± 10 µmol mg−1 of protein, DOPA content in older plaques (days 17–20) decreased significantly (p < 0.001) to 27 ± 5 µmol mg−1 of protein (n = 13–20, figure 2b).
3.3. Antioxidant activity in plaque extracts and in situ
Using the DPPH assay, we demonstrated that antioxidant activity solubilized from plaques and the degree of antioxidant activity indirectly correlated to plaque age (significant (p < 0.001) decrease in antioxidant activity between days 1–4 and days 17–20). Antioxidant activity (normalized % of antioxidant activity [(sample DPPH activity − negative control)/full DPPH reduction] × 100) was maintained at 60–70% over days 1–12, and then stabilized at approximately 30% over days 13–20 (no significant (p > 0.87) change between the days 13–16 and days 17–20, n = 13–20, figure 2c). We then compared the antioxidant activity of plaque extracts with the antioxidant activity of vitamin C under the same conditions (pH 3) and found that antioxidant activity of plaque extracts was 250-fold higher (w/w).
To locate antioxidant activity, we presented whole, intact thread–plaque pairs to DPPH-doped TLC plates and found that only some areas of the plaque reduced DPPH. All areas of direct contact between the plaque underside and TLC plate reduced DPPH, whereas the plaque-top and thread contact did not reduce DPPH (figure 3). To determine whether soaking intact thread–plaque pairs removed the antioxidant activity from the plaque undersides, we tested for in situ antioxidant activity in both soaked and unsoaked plaques. Antioxidant activity on the plaque undersides was not released by soaking in water.
Figure 3.
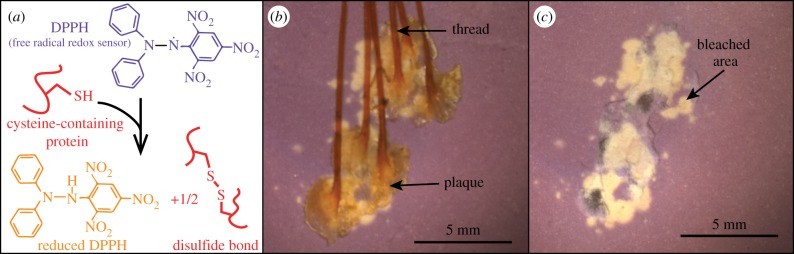
(a) Schematic of the DPPH assay showing a DPPH radical changing colour from purple to yellow upon reduction via a cysteine residue, and forming a disulfide bond in the process. (b,c) In situ DPPH assay of whole, intact thread–plaque pairs on a DPPH-doped TLC plate (b) before and (c) after thread–plaque removal localizing antioxidant activity to the plaque underside. (b,c) Small patches of antioxidant activity to the side of plaques denote areas of transient contact with the plate before equilibration. (Online version in colour.)
3.4. Cysteine contribution to antioxidant activity
To determine the dependence of antioxidant activity on cysteine residues (thiols) in extracted plaque proteins, we specifically blocked thiols using iodoacetamide. We observed a significant decrease in antioxidant activity with cysteine residues blocked. We blocked the cysteine residues in extracts from 4-day-old plaques and antioxidant activity decreased by 50% (n = 3, p < 0.002), compared with untreated extracts as measured by the DPPH assay (figure 4).
Figure 4.
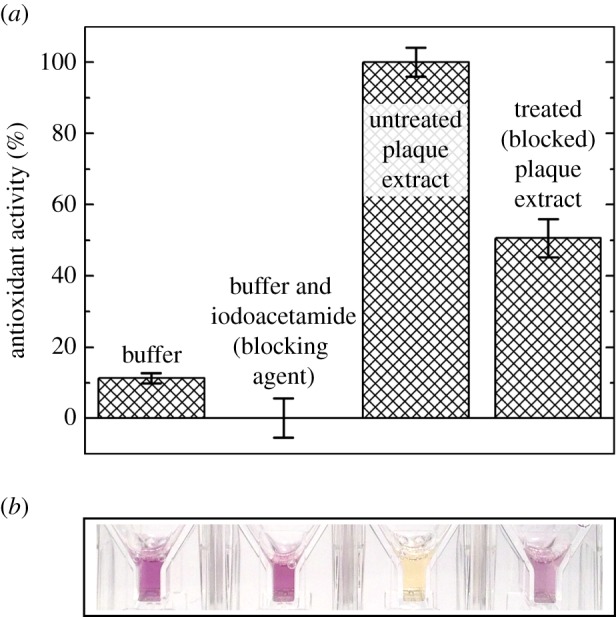
Cysteine contribution to antioxidant activity in mussel plaque extracts. (a,b) Comparing antioxidant activity in untreated plaque extracts, iodoacetamide treated (cysteine blocked) plaque extracts, buffer and buffer with iodoacetamide. Final solution conditions were pH 3.0 buffer (0.7 mM citric acid monohydrate, 14 mM sodium phosphate dihydrate, 0.3% (v/v) Triton X-100, pH 3.0) either with or without 0.22 µM iodoacetamide. Data are represented as both (a) normalized % ((sample absorbance change—negative control absorbance change)/(largest absorbance change—negative control absorbance change) × 100) with standard errors (n = 3) and (b) representative sample absorbance. (Online version in colour.)
4. Discussion
Mussel adhesion is a dynamic process that depends critically on shielding DOPA at the plaque–substrate interface from oxidation. The mussel plaques of M. californianus retained soluble DOPA over 20 days, far longer than expected, suggesting a persistent protective mechanism. Mussels secrete H3O+ and antioxidants as a protective mechanism during plaque deposition, but the duration of this antioxidant activity had not been previously studied. We investigated age-related oxidation by tracking extractable protein concentration, DOPA content, and antioxidant activity in whole plaques over 20 days. We found a reservoir of antioxidant activity that also lasted over 20 days and that correlated with both cysteine and DOPA contents. We also located antioxidant activity at the adhesive plaque interface, further demonstrating the importance of antioxidant activity in mussel adhesion. These findings provide a new view of antioxidant activity in the long-term, underwater adhesion of M. californianus: rather than merely protecting DOPA during deposition, antioxidant activity maintains the mature byssus by shielding the plaque–adhesive interface from oxidation during flow-induced byssus deformation.
Although DOPA is essential for mussel adhesion and survival in the harsh intertidal zone [1,2], DOPA in solution oxidizes readily at seawater pH and becomes primarily cohesive [2,4,5,8,15]. Despite being surrounded by oxidizing seawater, plaque proteins in the byssus remained extractable (figure 2a) and retained 50% of their DOPA content over 20 days (figure 2b). This disparity in DOPA oxidation rates suggests an abiding protective mechanism for DOPA maintenance in the mussel byssus.
During plaque deposition, mussels secrete H3O+ and antioxidants into the distal depression of the foot to protect DOPA from oxidation [5,15]; but, when the foot retracts, it exposes the new thread and plaque to oxidizing seawater. Once the plaque pH equilibrates with the seawater pH, DOPA is more susceptible to oxidation. Although the pH equilibrates quickly [17], 30% of the initial reducing power in plaques remained after 20 days. This reducing power occurred at the plaque–substrate interface, but not at the top of the plaque or the thread exterior (figure 3b,c). Although further work is required to determine if antioxidant activity exists within the thread, these findings provide a new perspective for antioxidant activity in the long-term underwater adhesion of M. californianus; rather than merely protecting DOPA during deposition, antioxidant activity persists in the adhesive interface long after plaque maturation. Presumably, the reducing environment is adapted to maintaining the integrity of DOPA for as long as possible, however, the transient dip in DOPA content between 5–8 and 9–12 days (figure 2b) is not easily explainable. One testable hypothesis is that during the 5–8-day period there was a flush of metal-contaminated seawater that scavenged DOPA despite the reducing conditions. Perhaps, keeping interfacial proteins reduced allows mussel adhesive proteins to undergo de-adhesion/re-adhesion during repeated loading and withstand environmental fluctuations.
Protein antioxidant activity often relies on the reducing power of cysteine residues [22,23]. To gauge this dependence in the mussel plaques, we blocked (carboxymethylated) the thiols from acting as antioxidants in plaque extracts and tested for antioxidant activity. Indeed, cysteine residues were responsible for at least 50% of the antioxidant activity at pH 7 (figure 4). Our data also show that antioxidant activity is correlated (1 : 1) with DOPA content (figure 3). Considering that DOPA in mfp-3s exhibits two distinct redox populations [6], those with the higher oxidation potential may act as a reducing reservoir. If both cysteine and DOPA do indeed contribute to antioxidant activity, perhaps mussels adapt varying DOPA and cysteine redox potentials to tailor interfacial adhesive chemistry for specific surfaces. That mussels use DOPA for both structural and chemical roles demonstrates an economy of material and highlights DOPA's chemical diversity as well as its need for controlled environmental conditions.
Whereas living cells of plants, animals, fungi and some bacteria use reduced glutathione to actively (ATP dependently) prevent oxidative damage [24], the byssus lacks the ability to actively maintain a reducing environment. Extra-organismic structural materials such as the silk of Bombyx mori and the nacre of Pinctada fucata both include antioxidants to passively maintain reducing conditions [25,26]. Our results illustrate that the mussel byssus also passively maintains redox to protect the byssus from oxidative damage and suggest that plaque redox is non-equilibrium and under kinetic control.
In this study, we have shown that the adhesive face of byssal plaques is highly reducing and that extractable protein, DOPA content, and antioxidant activity last over 20 days. Antioxidant activity was correlated to both cysteine and DOPA contents, suggesting that mussels have adapted varying cysteine and DOPA redox potentials for favourable interfacial adhesive chemistry. These findings provide a new view for antioxidant activity in the long-term, underwater adhesion of M. californianus: rather than merely protecting DOPA during deposition, antioxidant activity persists in the adhesive interface long after plaque maturation. Perhaps, during plaque deformation, keeping interfacial proteins reduced allows mussel adhesive proteins to undergo de-adhesion/re-adhesion during repeated loading. This long-term maintenance suggests that plaque redox is non-equilibrium and under kinetic control over its functional lifetime.
Acknowledgements
We thank Dr Sascha Nicklish for contributing to the development of the in situ DPPH assay. We also thank Christophe Pierre for collecting and providing mussels for this research.
Endnote
Relating redox potential and pH: at the pH of seawater (pH 8.2), the E′o for quinone formation (reaction 1) is the difference of Nernst equations at the two pHs (E′o = Eo – 0.059 [ΔpH]), where Eo denotes the standard potential of the reducing half reaction. Accordingly, the E′o quinone reduction, +0.22 V (reaction −1), becomes +0.15 V at pH 8.2 (+0.22 V − 0.059[8.2–7] = +0.15 V) and by reversing the direction to oxidation, the Eo′ for quinone formation becomes −0.15 V at pH 8.2.
Authors' contributions
D.R.M. and J.H.W. conceived and designed the experimental approach. D.R.M. and J.E.S. preformed the experiments and analysed the data. All authors contributed to the writing and interpretation of the published findings.
Competing interests
The authors have no competing interests.
Funding
This work was supported, in whole or in part, by the National Institutes of Health grant number R01-DE018468 (J.H.W.) and the Materials Research Science and Engineering Centers programme of the National Science Foundation award number DMR 1121053.
References
- 1.Danner EW, Kan Y, Hammer MU, Israelachvili JN, Waite JH. 2012. Adhesion of mussel foot protein Mefp-5 to mica: an underwater superglue. Biochemistry 51, 6511–6518. ( 10.1021/bi3002538) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lee H, Scherer NF, Messersmith PB. 2006. Single-molecule mechanics of mussel adhesion. Proc. Natl Acad. Sci. USA 103, 12 999–13 003. ( 10.1073/pnas.0605552103) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Burzio LA, Burzio VA, Pardo J, Burzio LO. 2000. In vitro polymerization of mussel polyphenolic proteins catalyzed by mushroom tyrosinase. Comp. Biochem. Physiol. B 126, 383–389. ( 10.1016/S0305-0491(00)00188-7) [DOI] [PubMed] [Google Scholar]
- 4.McDowell LM, Burzio LA, Waite JH, Schaefer J. 1999. Rotational echo double resonance detection of cross-links formed in mussel byssus under high-flow stress. J. Biol Chem 274, 20 293–20 295. ( 10.1074/jbc.274.29.20293) [DOI] [PubMed] [Google Scholar]
- 5.Yu J, Wei W, Danner E, Ashley RK, Israelachvili JN, Waite JH. 2011. Mussel protein adhesion depends on interprotein thiol-mediated redox modulation. Nat. Chem. Biol. 7, 588–590. ( 10.1038/nchembio.630) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wei W, Yu J, Broomell C, Israelachvili JN, Waite JH. 2013. Hydrophobic enhancement of DOPA-mediated adhesion in a mussel foot protein. J. Am. Chem. Soc 135, 377–383. ( 10.1021/ja309590f) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yu J, et al. 2013. Adaptive hydrophobic and hydrophilic interactions of mussel foot proteins with organic thin films. Proc. Natl Acad. Sci. USA 110, 15 680–15 685. ( 10.1073/pnas.1315015110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fullenkamp DE, Barrett DG, Miller DR, Kurutz JW, Messersmith PB. 2014. pH-dependent cross-linking of catechols through oxidation via Fe3+ and potential implications for mussel adhesion. RSC Adv. 4, 25127 ( 10.1039/C4RA03178D) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Harrington MJ, Masic A, Holten-Andersen N, Waite JH, Fratzl P. 2010. Iron-clad fibers: a metal-based biological strategy for hard flexible coatings. Science 328, 216–220. ( 10.1126/science.1181044) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lu Q, Hwang DS, Liu Y, Zeng H. 2012. Molecular interactions of mussel protective coating protein, mcfp-1, from Mytilus californianus. Biomaterials 33, 1903–1911. ( 10.1016/j.biomaterials.2011.11.021) [DOI] [PubMed] [Google Scholar]
- 11.Weisser JT, Nilges MJ, Sever MJ, Wilker JJ. 2006. EPR investigation and spectral simulations of iron–catecholate complexes and iron–peptide models of marine adhesive cross-links. Inorg. Chem. 45, 7736–7747. ( 10.1021/ic060685p) [DOI] [PubMed] [Google Scholar]
- 12.Xu H, Nishida J, Ma W, Wu H, Kobayashi M, Otsuka H, Takahara A. 2012. Competition between oxidation and coordination in cross-linking of polystyrene copolymer containing catechol groups. ACS Macro Lett. 1, 457–460. ( 10.1021/mz200217d) [DOI] [PubMed] [Google Scholar]
- 13.Proudfoot G, Ritchie I. 1983. A cyclic voltammetric study of some 4-substituted benzene-1,2-diols. Aust. J. Chem. 36, 885–894. ( 10.1071/CH9830885) [DOI] [Google Scholar]
- 14.Segel IH. 1976. Biochemical calculations: how to solve mathematical problems in general biochemistry, 2nd edn New York, NY: John Wiley and Sons. [Google Scholar]
- 15.Zhao H, Waite JH. 2006. Linking adhesive and structural proteins in the attachment plaque of Mytilus californianus. J. Biol. Chem. 281, 26 150–26 158. ( 10.1074/jbc.M604357200) [DOI] [PubMed] [Google Scholar]
- 16.Nicklisch SCT, Das S, Martinez Rodriguez NR, Waite JH, Israelachvili JN. 2013. Antioxidant efficacy and adhesion rescue by a recombinant mussel foot protein-6. Biotechnol. Prog. 29, 1587–1593. ( 10.1002/btpr.1810) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Martinez-Rodriguez NR, Das S, Kaufman Y, Israelachvili JN, Waite JH. 2015. Interfacial pH during mussel adhesive plaque formation. Biofouling 31, 221–227. ( 10.1080/08927014.2015.1026337) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Waite JH. 1984. Determination of (catecholato)borate complexes using difference spectrophotometry. Anal. Chem. 56, 1935–1939. ( 10.1021/ac00275a040) [DOI] [Google Scholar]
- 19.Nicklisch SCT, Waite JH. 2014. Optimized DPPH assay in a detergent-based buffer system for measuring antioxidant activity of proteins. MethodsX 1, 233–238. ( 10.1016/j.mex.2014.10.004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Takebayashi J, Tai A, Gohda E, Yamamoto I. 2006. Characterization of the radical-scavenging reaction of 2-O-substituted ascorbic acid derivatives, AA-2G, AA-2P, and AA-2S: a kinetic and stoichiometric study. Biol. Pharm. Bull. 29, 766–771. ( 10.1248/bpb.29.766) [DOI] [PubMed] [Google Scholar]
- 21.Creighton TE, Chasman DI. 1997. Protein structure: a practical approach, vol. 26 Oxford, UK: IRL Press. [Google Scholar]
- 22.Banerjee R. 2012. Redox outside the box: linking extracellular redox remodeling with intracellular redox metabolism. J. Biol. Chem. 287, 4397–4402. ( 10.1074/jbc.R111.287995) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Paulsen CE, Carroll KS. 2013. Cysteine-mediated redox signaling: chemistry, biology, and tools for discovery. Chem. Rev. 113, 4633–4679. ( 10.1021/cr300163e) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tietze F. 1969. Enzymic method for quantitative determination of nanogram amounts of total and oxidized glutathione: applications to mammalian blood and other tissues. Anal. Biochem. 27, 502–522. ( 10.1016/0003-2697(69)90064-5) [DOI] [PubMed] [Google Scholar]
- 25.Kundu SC, Dash BC, Dash R, Kaplan DL. 2008. Natural protective glue protein, sericin bioengineered by silkworms: potential for biomedical and biotechnological applications. Prog. Polym. Sci. 33, 998–1012. ( 10.1016/j.progpolymsci.2008.08.002) [DOI] [Google Scholar]
- 26.Chaturvedi R, Singha PK, Dey S. 2013. Water soluble bioactives of nacre mediate antioxidant activity and osteoblast differentiation. PLoS ONE 8, e84584 ( 10.1371/journal.pone.0084584) [DOI] [PMC free article] [PubMed] [Google Scholar]