

Abstract
Breast cancer onset and disease progression have been linked to members of the TGFβ superfamily and their downstream signaling components, the Smads. Alterations in Smad3 signaling are associated with the dichotomous role of TGFβ in malignancy, mediating both tumor suppressant and pro-metastatic behaviors. Overexpression of cell cycle regulators, cyclins D and E, renders cyclin-dependent kinases (CDKs) 4/2 hyperactive. Noncanonical phosphorylation of Smad3 by CDK4/2 inhibits tumor suppressant actions of Smad3. We hypothesized that CDK inhibition (CDKi) would restore Smad3 action and help promote cancer cell regression. Treatment of triple-negative breast cancer (TNBC) cell lines (MDA-MB-231, MDA-MB-436, Hs578T) with CDK2i or CDK4i resulted in increased Smad3 activity and decreased cell migration. Transfection with a 5M Smad3 construct containing inhibitory mutations in 5 CDK phosphorylation sites also resulted in decreased TNBC cell migration and invasion. MDA-MB-231 cells treated with CDK2i or CDK4i resulted in decreased Smad3 protein phosphorylation at the CDK phosphorylation T179 site, decreased MMP2 and c-myc expression, and increased p15 and p21 expression. Using a novel transfected cell array, we found that CDK2i treatment decreased activity of the epithelial-to-mesenchymal transition related transcription factors Snail and Twist. In vivo studies in an MDA-MB-231 tumor model showed that individual and combination treatment with paclitaxel and CDK2i resulted in decreased tumor volume and Ki67 staining. Collectively, these data support further investigation of targeted CDK inhibitors as a promising therapeutic strategy for TNBC, a breast cancer subtype with limited treatment options.
Keywords: CDK, cyclin, paclitaxel, Smad3, triple negative breast cancer
Abbreviations
- BCSC
breast cancer stem cells
- CDK
cyclin dependent kinase
- CDKi
cyclin dependent kinase inhibitor
- CK
cytokeratin
- EGFR
epidermal growth factor receptor
- EMT
epithelial-mesenchymal transition
- ER
estrogen receptor
- HER2
human epidermal growth factor receptor 2
- Pin1
peptidyl-prolyl cis-trans isomerase NIMA-interacting 1
- PR
progesterone receptor
- TNBC
triple negative breast cancer
Introduction
TNBC, characterized by tumors that are negative for the estrogen receptor (ER), progesterone receptor (PR), and Human Epidermal Growth Factor Receptor 2 (HER2), represents approximately 15% of newly diagnosed invasive breast cancers and is associated with a higher incidence of relapse and death than other breast cancer subtypes.1,2 Treatment of TNBC represents a significant clinical challenge due to the limited molecular targets associated with this disease subtype. Patients with TNBC often have tumors with basal-like biology and an expression signature similar to that of the basal/myoepithelial cells of the breast, including cytokeratin (CK)5/6 and/or epidermal growth factor receptor (EGFR), as well as higher cyclin D and E expression.3,4 Cyclins D and E are overexpressed in more than 50% and 25% of breast cancers, respectively, and are considered oncogenes.5,6 Overexpression of cyclins, and the consequent impact on CDK activity, affects normal cell cycle control and contributes to disease progression and metastasis. Further understanding of the downstream impact of aberrant cyclin activity in TNBC may permit development of novel targeted therapies and improve outcomes for patients affected by this aggressive form of breast cancer.
Smad3 is a transcriptional regulator that canonically functions through phosphorylation at the C-terminus (Serine 423/425) upon ligand binding to TGFβ receptors.7 Alterations in Smad3 signaling have been implicated in the dichotomous role of TGFb in malignancy, enacting both tumor suppressant and tumor promoting behaviors in early- and later-stage breast carcinogenesis. We previously showed a significant correlation between decreased nuclear Smad3 and higher tumor grade, larger tumor size, and hormone receptor negativity.8 In human breast cancer cell lines, we also found that overexpression of Smad3 induced cell cycle arrest and transcription of p15.9 Matsuura et al. reported that Smad3 is a physiologic substrate for CDK4 and CDK2, and identified multiple CDK4/2 phosphorylation sites within the Smad3 protein, predominately in the linker region.10 Noncanonical CDK phosphorylation of these sites inhibited canonical Smad3 action, whereas mutation of the sites increased the transcriptional activity of Smad3, facilitated cell cycle arrest, and induced p15 transcription and c-myc repression.10 We confirmed these findings in breast cancer cells engineered to overexpress cyclins, showing that mutation or pharmacologic inhibition of CDK4/2 phosphorylation sites in the Smad3 protein resulted in restoration of transcription of cdk inhibitors p15 and p21 and repression of c-myc.11,12 Additional work by Matsuura et al. demonstrated that CDK-mediated phosphorylation of Smad3 is requisite for interaction with Smurf2 and SNAIL1, and promotion of cell migration, invasion, and epithelial-mesenchymal transition (EMT).13 Collectively, these studies indicate that CDK-induced non-canonical phosphorylation of Smad3 contributes to the loss of Smad3-mediated tumor suppression and gain of metastatic potential, and may underlie the observed oncogenic switch between TGFb-regulated tumor suppression to oncogenesis.
Based on these findings, we hypothesized that interventions that block non-canonical Smad3 phosphorylation to restore or maintain canonical Smad3 signaling and tumor-suppressive activity will slow breast cancer growth. Prior studies have shown that the CDK inhibitor (CDKi) flavopiridol in combination with trastuzumab, histone deacetylase inhibitors, and docetaxel lead to decreased solid tumor cell proliferation and tumor size.14 Furthermore, CDK4/6is have also demonstrated effectiveness against multiple myeloma and colon cancers in murine models.15,16 To date, clinical trials have not examined specific CDK2is and CDK4is in the context of TNBC and Smad3 signaling, highlighting a potentially novel and focused application for these agents. In the current work, we used human TNBC cell lines to examine the impact of CDK2i and CDK4i alone, and in combination with paclitaxel chemotherapy, on restoration of Smad3 signaling and reversion of oncogenic phenotypes in vitro and in vivo.
Results
Characterization of triple negative breast cancer cell lines
Table 1 describes the TNBC study cell lines.17–20 Immunoblotting showed differential expression of cell cycle proteins, with MDA-MB-231 cells expressing the highest levels of cyclin D, and MDA-MB-436 cells expressing the highest levels of cyclin E, (Fig. 1A). Relative expression of cyclins was lowest in Hs578T cells. Levels of CDK2 were similar across the 3 cell lines, while there was a lower level of CDK4 expression in Hs578T cells compared to the MDA-MB-231 and MDA-MB-436 cell lines. Smad 3 expression was highest in MDA-MB-436 cells when compared to the Hs578T and MDA-MB-231 cell lines, though this difference was not statistically significant.
Table 1.
Triple negative breast cancer cell lines
| Cell Line | Characteristics |
|---|---|
| Hs578T | Triple negative26 |
| Express TGFβ receptors, Smad327 | |
| |
Express cyclins D/E (Fig. 1A) |
| MDA-MB-231 | Triple negative27 |
| Basal-like subtype/highly metastatic27 | |
| Express TGFβ receptors, Smad327 | |
| |
Have high expression of cyclins D/E (Fig. 1A) |
| MDA-MB-436 | Triple negative28 |
| BRCA1 mutated28 | |
| Express TGFβ receptors, Smad329 | |
| High expression of cyclins D/E (Fig. 1A) |
Figure 1.

TNBC cells show increased Smad3 activity and decreased cell migration in response to CDKi treatment. (A) Immunoblotting confirmed expression of study proteins in TNBC cells. *P < 0.05 for indicated cell lines (B) Smad3 reporter activity was measured in study cells after increasing doses of CDK2i and CDK4i. Study cells were (C) treated with control DMSO (NT), CDK2i or CDK4i or (D) transfected with Vec, WT or 5M Smad3 and cell migration was assessed. (E) Representative images of Vec and 5M transfected cells are shown at 10× magnification.
CDK inhibitors increase Smad3 transcriptional activity in a dose-dependent manner
Overall, an increase in Smad3 transcriptional activity was observed in cells treated with the CDK inhibitors, indicating that, in the setting of elevated cyclin D and E, CDK4/2 inhibition augments canonical Smad3 activity (Fig. 1B). Treatment of Hs578T cells with relatively lower doses of the CDKis (Fig. 1B) resulted in the greatest increase in Smad3 activity compared with the other study cell lines, and this result may be associated with the lower cyclin/CDK expression levels found in the Hs578T cells (Fig. 1A). For the MDA-MB-231 and MDA-MB-436 cells, Smad3 reporter activity increased with a similar trend after treatment with either CDK2i or 4i. Based on these results, we used doses of 240 nM CDK2i and 400 nM CDK4i in subsequent studies.
Inhibition of CDK-mediated phosphorylation of Smad3 decreased migration of TNBC cells
For all cell lines, CDK inhibition resulted in decreased cell migration compared with untreated cells (Fig. 1C). CDK2 inhibition resulted in a greater decrease in MDA-MB-231 and MDA-MB-436 cell migration when compared to CDK4 inhibition, while the impact of both inhibitors was similar, though significant for CDK4i, in Hs578T cells. Next, we determined if overexpression of mutant 5M Smad3, resistant to CDK phosphorylation, would inhibit migration in a manner similar to pharmacological CDK inhibition. Study cells were transfected with Vec, WT, or 5M constructs and cell migration was assessed (Fig. 1D). Compared to Vec, transfection with WT and 5M Smad3 constructs resulted in decreased cell migration, with the greatest decrease found after transfection of MDA-MB-436 cells with the 5M construct. For MDA-MB-231 cells, transfection with the 5M construct resulted in a significantly greater decrease in migration when compared to transfection with the WT construct (Fig. 1D, E).
CDK inhibition results in decreased invasion and altered expression of metastasis- and cell cycle-related proteins in MDA-MB-231 cells
To further investigate the impact of CDK inhibition on Smad3 action we focused on the MDA-MB-231 cell line, capable of metastasis in vivo. We next examined the impact of overexpressing WT or 5M Smad3 on cell invasion. While both constructs significantly decreased the percentage of invading MDA-MB-231 cells, transfection of the 5M construct resulted in the greatest decrease in cell invasion (Fig. 2A). Previous studies by our group and others have revealed that, of the 5 noncanonical phosphorylation sites identified in Smad3, phosphorylation of the T179 site is critical to negative regulation of Smad3 action.11–13 As expected, treatment of MDA-MB-231 cells with CDK2i or CDK4i resulted in decreased phosphorylation at the T179 site, while total Smad3 levels remained relatively stable (Fig. 2B). Additionally, expression of MMP2 decreased with CDK2i and 4i (Fig. 2C). Further confirming restoration of canonical Smad3 action after treatment with CDK2i and CDK4i, expression of p15 and p21 increased, while expression of c-myc decreased (Fig. 2C).
Figure 2.
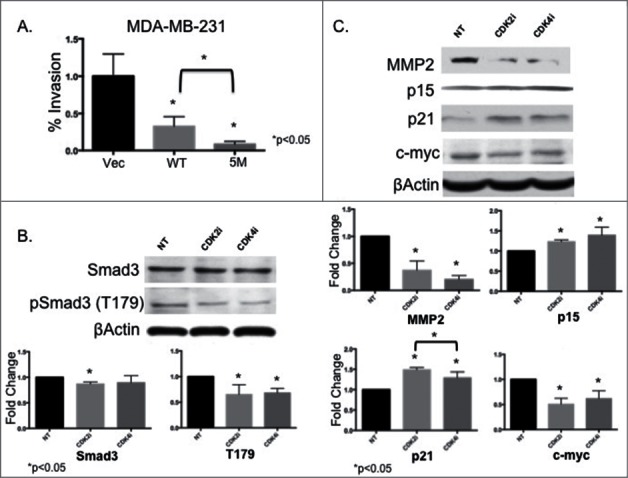
MDA-MB-231 cells expressing 5M Smad3 show decreased cell invasion and CDK inhibition results in decreased MMP2 expression, decreased CDK-mediated phosphorylation of Smad3 at the T179 site, and restoration of proteins related to cell cycle arrest. (A) MDA-MB-231 cells were transfected with Vec, WT, or 5M Smad3 and invasion was assessed (B and C) MDA-MB-231 cells were treated with control DMSO (NT), CDK2i, or CDK4i and immunoblotting was performed to assess expression of study proteins.
Treatment of MDA-MB-231 cells with paclitaxel/CDK2i results in apoptosis in vitro and decreased tumor size in vivo
Next we examined the effect of CDK2i and CDK4i alone and in combination with the chemotherapeutic paclitaxel on apoptosis (Fig. 3A). TUNEL staining of MDA-MB-231 cells showed that CDK2i, CDK4i, paclitaxel, and paclitaxel/CDK4i resulted in moderately increased apoptosis, while paclitaxel/CDK2i resulted in the greatest increase in apoptosis.
Figure 3.

Paclitaxel/CDK2i induced apoptosis in vitro and decreased tumor volume and Ki67 staining in vivo in a MDA-MB-231 xenograft model. (A) MDA-MB-231 cells were treated and analyzed for apoptosis using TUNEL staining. (B) Female athymic nu/nu mice were inoculated with MDA-MB-231 cells to establish tumors, and treated for 21 d Tumor volume was measured at indicated time points. The tumor volumes from treated groups were compared with the volume from the respective control treated group at Day 21, and significance was then determined; *P < 0.05, **P < 0.005. (C) Representative images from Ki67 stained xenografts from each treatment group at 20X magnification.
We then tested the impact of CDKis individually and in combination with paclitaxel in a xenograft model of TNBC. Compared to control, treatment with paclitaxel resulted in decreased tumor volume (Fig. 3B, C). Treatment with CDK2i alone also resulted in decreased tumor volume, and the effect was not significantly different from paclitaxel. Combination paclitaxel/CDK2i therapy resulted in the greatest decrease in tumor volume. In contrast, while treatment with CDK4i resulted in reduced tumor volume, the decrease was not significantly different from control. Treatment with paclitaxel alone, and in combination with CDK4i, resulted in a similar reduction in tumor growth compared to control treated animals. At the end of the treatment period, tumors were removed and stained for Ki67 (Fig. 3D). Compared to control treated animals, all study treatments except paclitaxel/CDK4i resulted in a similar significant decrease in tumor cell proliferation.
Characterization of signaling changes in MDA-MB-231 cells after CDK2 inhibition
Given the decrease in migration and invasion in vitro and cytoreductive impact of CDK2i treatment on TNBC in vivo, we applied a cell reporter array to further study mechanistic consequences of CDK2i.21,22 After treating MDA-MD-231 cells with control or CDK2i, activities of 11 TFs associated with oncogenesis were measured at several time points. Factors selected for study were based on association with proliferation, apoptosis, and metastasis (Table 2). After 4 hours of treatment, a significant increase in Smad3 reporter activity was detected (Fig. 4A). Increases in β-catenin, Snail, Sp1, Twist, YY1, and Zeb1 reporter activity were also identified at this early time point (Fig. 4A). At later time points (up to 6 d post treatment), Snail, Sp1, and Twist activity significantly decreased, while YY1 and Zeb1 activity plateaued (Fig. 4B). We also observed transient increases in p53 and Notch1 activity. This work supplements our earlier findings showing increased Smad3 activity upon treatment with CDK2i (Fig. 1).
Table 2.
List of reporters used in the array
| Reporter | Associated TF | Biological Function | Sequence | Reference for Reporter Sequence |
|---|---|---|---|---|
| βcat |
Catenin, Beta 1 |
Adhesion, Cell Cycle, Differentiation |
TAAGATCAAAGGGGGTAAGATCAAAGGGGGTAAAATCAAAGGGGGCCCCCTTTGATCTTACCCCCTTTGATCTTACCCCCTTTGATCTTAACCGGT |
TOP Flash sequence (Millipore) |
| Nanog |
Homeobox transcription factor nanog |
Stem cell proliferation, Self renewal |
CCATTGTCATGCTAATCCATTGTCATGCTAATCCATTGTCATGCTAAT |
Jauch et al 2008 (PMID 18177668) |
| NfkB |
Nuclear Factor kappa B |
Inflammation, Cell growth |
GGGAATTTCCGGGAATTTCCGGGAATTTCCGGGAATTTCCGGGAATTTCCGGGAATTTCC |
Panomics |
| Notch1 |
Notch |
Cell development, Proliferation |
TTTCCCACGTTTCCCACGTTTCCCACG |
TRANSFAC ID R27998 |
| p53 |
p53 |
Cell cycle, Apoptosis, DNA repair |
TACAGAACATGTCTAAGCATGCTGTGCCTTGCCTGGACTTGCCTGGCCTTGCCTTGGG |
Panomics |
| Smad3 |
Smad3 |
Cell cycle, Apoptosis, Metastasis |
TCGAGAGCCAGACAAAAAGCCAGACATTTAGCCAGACAC |
Panomics |
| Snail |
Zinc finger protein SNAI1 |
EMT |
CACCTGCACCTGCACCTGCACCTGCACCTGCACCTGCACCTGCACCTGCACCTGCACCTG |
TRANSFAC ID RE0073409 |
| Sp1 |
Transcription factor SP1 |
Apoptosis |
GTATTTCCCAGAAAAGGAACGTATTTCCCAGAAAAGGAACGTATTTCCCAGAAAAGGAAC |
Panomics |
| Twist |
Twist-related protein 1 |
EMT, Differentiation |
CATGTGCATGTGCATGTGCATGTGCATGTGCATGTGCATGTGCATGTGCATGTGCATGTG |
PMID 16585154 |
| YY1 |
Transcription Factor YY1 |
Differentiation, Embryogenesis, Proliferation |
CGCTCCCCGGCCATCTTGGCGGGTGGTCGCTCCCCGGCCATCTTGGCGGCTGGT |
Panomics |
| Zeb1 | ZEB1 | EMT | ACTCACCTGTGTACTCACCTGTGTACTCACCTGTGTACTCACCTGTGT | PMID 11713281 |
Source of Biological Function information: Online Mendelian Inheritance in Man database.23
Figure 4.

CDK2i treatment of MDA-MB-231 alters activity of transcription factors associated with oncogenic processes. MDA-MB-231 cells were transduced with TF reporters and treated with CDK2i. Bioluminescence was normalized to TA control at (A) 1, 2, and 4 hours (early timepoints) and (B) at 1, 3, and 6 d (late timepoints).
Discussion
Treatment of TNBC remains a clinical challenge. It is critical that we discover pathways underlying TNBC oncogenesis to identify potential targets and develop new therapies. Our previous work demonstrated the impact of cyclin/CDK-mediated Smad3 phosphorylation on oncogenic events in an engineered model of cyclin overexpressing breast cancer cells.11,12 We now report that, in more biologically relevant TNBC cell lines, inhibiting CDK-mediated phosphorylation of Smad3 through pharmacologic or genetic means supports canonical Smad3 activity and associated expression of cell cycle arrest proteins and inhibits EMT-associated cell processes of migration and invasion. Our in vitro results were recapitulated in vivo: CDK2i, paclitaxel, and paclitaxel/CDK2i significantly reduced tumor size and percent of Ki67-positive residual tumor cells. Lastly, treatment with CDK2i resulted in decreased transcriptional activity of EMT-associated factors including Snail and Twist, pre-clinical data that, together, helps to reveal mechanisms underlying TNBC oncogenesis and, potentially, a new therapeutic strategy.
TNBC may become more aggressive, in part, as cyclin expression increases and the cell cycle becomes more deregulated, leading to a switch from tumor-suppressing canonical Smad3 activity to tumor-promoting noncanonical CDK-mediated Smad3 phosphorylation.10 The mechanism by which this oncogenic switch occurs during cancer progression is likely reflective of dynamic changes evolving within tumor cells and the microenvironment over time. The rationale for CDKi therapy is linked to regulation of cell cycle progression and, to this end, we found a significant increase in p15 and p21 and decrease in c-myc protein expression after treatment with CDK2i and CDK4i (Fig. 3). Our data also suggests that CDK inhibition can impede metastatic phenotypes such as migration and invasion through blockade of noncanonical phosphorylation of Smad3 and the downstream effects on Smad3-mediated protein expression. MMP2 plays a critical role in EMT and was previously identified as a target of canonical Smad3 transcriptional activity in breast cancer, where TGFβ induced MMP2 expression in a Smad3/4-dependent manner.24 In normal and Ras-transformed rat gastric mucosa cell lines, inhibition of linker region Smad3 phosphorylation—such as that mediated by CDK2/4—was shown to result in decreased expression of MMP2.25 The influence of domain-specific phosphorylation of Smad3 on expression of MMP2, and the effect on migration and invasion, has not been previously studied in the context of breast cancer. We have demonstrated here in TNBC cells that inhibition of non-canonical phosphorylation of Smad3, through treatment with CDKis, led to a significant decrease in MMP2 (Fig. 2C).
Previous work has also shown that non-canonical phosphorylation of Smad3, particularly at the T179 site, leads to binding of the protein peptidyl-prolyl cis-trans isomerase NIMA-interacting 1 (Pin1) which in turn results in Smad3-Smurf2 interaction, ubiquitination, degradation, and loss of cell cycle control.13,26 Pin1 binding was also found to result in upregulation of N-cadherin and promotion of invasive and metastatic behaviors in a prostate cancer cell model.13 As shown, blockade of non-canonical Smad3 activity by CDKi would be expected to result in anti-metastatic phenotypes (Fig. 1). To this end, we had reported that transfection of a T179 site mutant construct resulted in increased Smad3 activity comparable to transfection of the 5M construct in the Hs578T triple negative cell line.12 In the current study, treatment with CDKi was also associated with decreased phosphorylation at the Smad3 T179 site (Fig. 2B), repression of MMP2 expression (Fig. 2C), and decreased TNBC cell migration (Fig. 1C). Expression of mutant 5M Smad3 was effective in reducing migration in the MDA-MB-231 and MDA-MB-436 cell lines, as well as invasion in MDA-MB-231 cells. Interestingly, for the Hs578T cell line, the anti-migratory impact of treatment with CDK2i or transfection with the 5M construct was not as prominent when compared to the other study cell lines. These findings underscore the heterogeneity and complexity of TNBC. To this end, it is possible that the disruption of pro-migratory PIN1 binding may be differentially affected by CDKi therapy, resulting in the less pronounced anti-migratory effects found in the Hs578T cells after CDK2i treatment, when compared with the reduction in cell migration found in the MDA-MB-231 and MDA-MB-436 cell lines. Furthermore, additional factors contributing to cell migration have also shown disparate effects in TNBC, with the Rho family GTPase Cdc42 found to negatively regulate cell migration in MDA-MB-231 cells while having a pro-migratory effect in Hs578T cells.27 In recognizing the complexity of Smad3 action, impacted by both canonical and noncanonical phosphorylation, the specific phospho-Smad3 status may also have a role in the metastatic phenotypes of TNBC. As such, increased levels of Smad3 phosphorylated in the linker region (pSmad3L) have been detected in invasive human colorectal and hepatocellular cancers when compared to non-cancerous tissues.28–30 It follows that increased pSmad3L levels may then facilitate pro-migratory PIN1 binding. Accordingly, investigation into the prognostic and predictive relevance of pSmad3C (Smad3 phosphorylated at the C-terminus) versus pSmad3L in TNBC is warranted.
To further examine the impact of CDKi on EMT related events we used a novel transcription factor (TF) reporter cell array21,22,31 and found that CDK2i treatment of TNBC cells led to an early increase in activity of β-catenin, Smad3, Snail, Sp1, Twist, YY1, and Zeb1 (Fig. 4). The association between canonical Smad3 signaling and transcriptional drivers of EMT, including Snail/Twist/Zeb1 has been well documented.32 Restoration of canonical Smad3 signaling by CDK2i may trigger cross-talk between TGFβ/Smad3 signaling and other oncogenic signaling pathways, such as RAS-related signaling, leading to activation of EMT factors. Accordingly, RAF-MAPK signaling induced EMT only in cooperation with TGFβ signaling in mammary epithelial cells in vivo.33 At later timepoints, Smad3 and β-catenin activity normalized; Snail, Sp1, and Twist activity significantly decreased; and Notch1, YY1 and Zeb1 activity plateaued at levels comparable to early timepoints. Temporal changes in EMT factor activity in response to CDK2i may reflect efficacy of this therapy in promoting canonical, tumor-suppressive, Smad3 signaling; as noted, decreased activity of Snail and Twist found at the 24 hour time point correlated to the decrease in migration found in study cells after 24 hours of treatment with CDK2i. Additionally, ubiquitin ligases can preferentially bind to activated Smad complexes resulting in a subsequent decrease in canonical Smad3 signaling, potentially contributing to the later onset decreased levels of Snail and Twist activity.34 As stated, potential blockade at the T179 site by CDK2i may also offset Pin1 binding and result in decreased metastatic phenotypes. Thus, although activity levels of Zeb1 and Notch1 were also increased after prolonged CDK2i treatment, taking into consideration the complex interplay of mechanistic events that contribute to a changing phenotype, the impact of CDK2i was sufficient to alter the balance toward an anti-migratory phenotype in TNBC study cells. Correspondingly, Siletz et al. recently reported on the critical nature of sequential changes in TF activity within a dynamic model of an EMT network that captures the dedifferentiation program.31 Taken together, our work suggests that Smad3 may be an important hub within the TF network, functioning both canonically and non-canonically, to affect downstream activity of factors such as Snail and Twist, with CDK2i acting to mitigate phenotypic changes associated with EMT.
CDK inhibitors have previously shown significantly greater cytotoxic potency compared to receptor tyrosine kinase inhibitors in a clinically diverse panel of breast cancer cell lines, demonstrating that breast cancer cells may be particularly sensitive to disruption in CDK activity.35 Interestingly, in MDA-MB-231 and MDA-MB-436 cell, which displayed relatively higher levels of cyclin E than Hs578T cells, CDK2i was more effective than CDK4i in reducing cell migration (Fig. 1). Our in vivo studies showed similarly higher efficacy of CDK2i vs. CDK4i in reduction of TNBC tumor volume and cell proliferation (Fig. 3). This suggests a correlation between cyclin E expression and efficacy of CDK inhibition, and that CDKis may be a promising therapeutic approach for cyclin E overexpressing TNBC. Previous reports have demonstrated the predominant action of cyclin E activity in TNBC36,37 and immunohistochemical analysis of cyclins in human breast cancer samples showed that cyclin E expression correlated with ER/PR-negative breast cancer and increased intensity of Ki67 staining.36 Additionally, expression of cyclin E was found to correlate with basal type and poor prognosis in a breast cancer tissue microarray study.37 Furthermore, the ability of CDK2 to bind to cyclin D (in the presence of p21), as well as cyclin E, may result in increased control of the cell cycle by this CDK in TNBC.38 Thus, for the MDA-MB-231 TNBC model that expresses increased levels of cyclins D and E, disrupting CDK2 activity may be more impactful than disrupting CDK4 activity.
Taxanes are potent anti-tumor agents widely implemented in the care of patients with breast cancer;39 however, acquired resistance to taxanes has prompted investigation into combination drug studies.39 A study of MCF7 cells determined that flavopiridol, a pan-CDK inhibitor against CDKs-1,2,4,6,7, augmented paclitaxel-mediated apoptosis when administered after paclitaxel through enhanced activation of caspases.40 Pretreatment with flavopiridol resulted in G1/G2 cell cycle arrest, limiting effectiveness of paclitaxel, which acts predominately in the M phase. Thus, the impact of drug combinations may be sequence dependent. In the current study of TNBC xenografts, CDKis, paclitaxel, and combination paclitaxel/CDK2i were effective in decreasing the percent of Ki67+ cells, but a subset of predominantly Ki67+ cells remained after treatment with paclitaxel/CDK4i (Fig. 5). These residual Ki67+ cells may be indicative of a chemoresistant TNBC stem cell (BCSC) population, as breast cancer cells treated with pacIitaxel can develop populations of resistant BCSCs.41 Furthermore, a paclitaxel-resistant stem-like variant of the MDA-MB-231 cell line had increased expression of p21.42 As mentioned, in the presence of p21, CDK2 can form active complexes with cyclin D in addition to cyclin E38 which may render CDK2i treatment more impactful in this scenario, possibly by promoting apoptosis through continual E2F activation in the setting of pRb deregulation.43 Correspondingly, CDK4i would be expected to be less effective after treatment with paclitaxel—with its associated elevated p21 expression—compared to either paclitaxel or CDK4i treatment alone. In addition to the Ki67+ cells with higher p21 expression, another subset of cells may co-exist, cycling outside of the point of paclitaxel action in the M phase. This cell subset could be impacted by CDK4i treatment in G1/G0 phase, thus protecting these specific cells from the action of paclitaxel upon further dosing. Prior work in MDA-MB-231 cells showed that combination therapy with doxorubicin and a CDK4/6 inhibitor compromised the efficacy of doxorubicin.44 This decreased efficacy was secondary to cell cycle-specific phase sequestration of cancer cells, dependent on Rb functional status, and resulted in persistence of CDK4/6i-mediated cancer cell viability.44 Additionally, in cell models of fibrosarcoma and melanoma, induction of p21, p16, or treatment with a CDK4/6 inhibitor along with MEK/ERK (MAPK pathway) activation resulted in overexpression of cyclin D, and in conjunction with functional MTOR (mechanistic target of rapamycin), these cellular events also led to cancer cell senescence.45,46 Taken together, this prior work helps contextualize our findings after paclitaxel/CDK4i treatment when compared to the other study treatments. Furthermore, these findings support the rational implementation of combination therapies that have multiple targets of action in order to achieve superior therapeutic outcomes.
Figure 5.
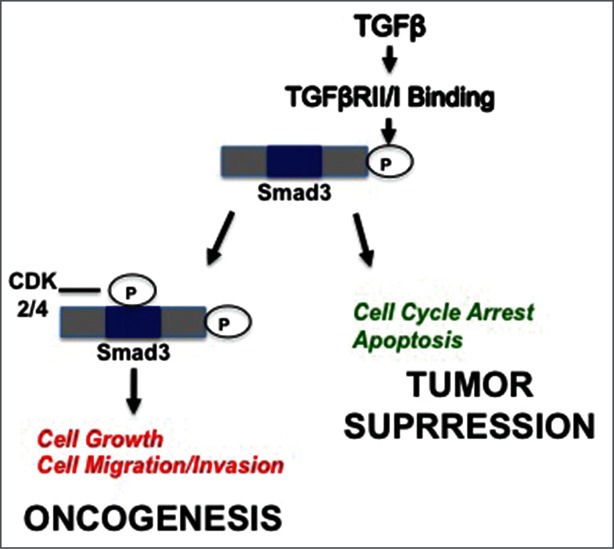
Proposed model for impact of CDK2/4 on dichotomous TGFβ/Smad3 signaling in breast cancer. Canonical Smad3 signaling, where phosphorylation occurs at the C-terminus, results in tumor suppressive events, including cell cycle arrest and apoptosis. Cyclin overexpression in breast cancer cells leads to hyperactive CDK activity that results in CDK-mediated noncanonical phosphorylation of Smad3 in the linker region and downstream oncogenic events such as cell proliferation and migration/invasion. Inhibiting CDK activity through use of pharmacological CDK inhibitors may prevent noncanonical phosphorylation of Smad3 and favor tumor suppressive Smad signaling.
While change in tumor size remains a critical factor when determining effectiveness of neoadjuvant therapy prior to surgery,47 other indicators of tumor response, such as post treatment tumor cellularity, are also prognostic. Evaluation of response becomes more complicated when tumor size does not correlate to histopathologic changes occurring within the tumor. Our data revealed a discrepancy between impact of therapy on tumor size when compared with Ki67 staining; specifically, CDK4i-treated tumors showed a high desmoplastic response indicative of tumor fibrosis and decreased Ki67 staining despite a non-significant reduction in tumor volume, whereas paclitaxel/CDK4i-treated tumors showed an intermediate desmoplastic response with higher Ki67 staining compared to treatment with paclitaxel or CDK4i alone. These findings underscore the importance of evaluating several markers of therapeutic response including tumor volume, cellularity, and histologic appearance, as well as expression of tumor markers such as Ki67 during pathologic analysis.48 The measure of residual tumor burden, which factors in pathological measurements of tumor size and cellularity, as well as nodal metastasis, was a significant predictor of distant relapse-free survival in breast cancers treated with neoadjuvant chemotherapy.49 Thus, accounting for both the change in tumor size and cellularity may give more precise pathologic response information than tumor size alone when evaluating response to therapy.50 As such, the potential impact of CDK4i therapy on reduction of tumor cellularity will motivate future studies focusing on alternative chemotherapies in combination with CDK4i for the treatment of TNBC.
Collectively, these studies demonstrate the effectiveness of CDK inhibitors to increase Smad3 activity, decrease cell migration and invasion, decrease tumor growth in a xenograft mouse model of TNBC, and alter transcription factor profiles in TNBC cells. While pan–anti-TGFβ signaling agents have shown some efficacy in treating metastatic breast cancer,51,52 implementing CDKi therapy may provide a more rational means to target the pro-oncogenic aspects of TGFβ signaling, providing more enhanced anticancer effects (Fig. 5). Consequently, phosphorylation of active nuclear Smad3 would shift away from non-canonical, oncogenic phosphorylation of the linker region and toward the more tumor suppressive C-terminal phosphorylation.25 Our preclinical data identifies potential cyclin/CDK and TGFb related biomarkers relevant to TNBC and a possible novel therapeutic drug combination that may help improve outcomes for patients who develop this aggressive breast cancer subtype.
Materials and Methods
Cell culture and antibodies
Human MDA-MB-231, MDA-MB-436, and Hs578T cells were obtained from ATCC (Manassas, VA) and maintained according to supplier recommendations. Antibodies: cyclin D1, GAPDH, cyclin E, p21, CDK2: Santa Cruz Biotechnology (Santa Cruz, CA); Smad3, CDK4, p15, MMP2: Cell Signaling Technology (Danvers, MA); β-actin: Sigma (St. Louis, MO); c-myc: Epitomics (Burlingame, CA); phospho-Smad3 T179: Abcam (Cambridge, MA). CDK4 inhibitor II (IC50 200 nmol/L) and CDK2 inhibitor (IC50 60 nmol/L) Calbiochem (La Jolla, CA). Each inhibitor was resuspended in DMSO, and DMSO was control treatment. Based on IC50 and dose-ranging luciferase assay results, treatments were: 0.1% DMSO, 400 nM CDK4i, and 240 nM CDK2i.
Immunoblotting
For MMP2, p15, p21, and c-myc immunoblots, cells were treated for 48 hours with CDKis. For Smad3 and Smad3-T179 immunoblots, cells were treated for 2 hours with CDKis, followed by 10 ng/mL TGFb for 1 hour. Following treatment, protein was extracted and immunoblotting was performed as previously described.12 Densitometry was completed using ImageJ software and values for treated samples were normalized to controls.
Luciferase assay
A construct consisting of a Smad3-responsive promoter upstream of a firefly luciferase reporter (CAGA-luc) and a control reporter (pRenilla) was transfected into study cells, with 200 ng of Smad3 expression plasmid, and treated with CDKi for 48 hours. Cells were processed and luminescence measured as previously described.12
Migration and invasion assays
For the migration assay, 10% FBS was used as a chemoattractant and added to the wells of a 24-well plate. Transwell inserts of 8-μm polyethylene terephthalate (PET) membranes (BD Biosciences) were placed in each well. Suspensions of serum-starved cells treated for 24 hours with CDKis or transfected with CS2+ vector control (Vec), wild-type Smad3 (WT), or 5M Smad3 (5M) were added to inserts and incubated for 24 hours. The 5M Smad3 expression plasmid, (a kind gift from Dr. Fang Liu, Rutgers, Piscataway, NJ) has been described previously, and contains multiple CDK phosphorylation site mutations (T8/T179/S204/S208/S213) that result in the inhibition of CDK4/2 phosphorylation.10 For invasion assays, reconstituted basement membrane Matrigel inserts (BD Biosciences) and cells were incubated for 48 hours. After scrubbing the inserts, they were transferred into 24-well plates containing 0.5% crystal violet to stain and fix cells that migrated to the bottom of membrane. Stained, migrated cells were counted using 10X magnification.
Cell block preparation for in situ TUNEL assay
MDA-MB-231 breast cancer cells were treated for 48 hours with control DMSO, 400 nM CDK4i, 240 nM CDK2i, 1 nM paclitaxel alone or in combination. Supernatant was collected to isolate floating apoptotic cells with a minimum of 10 million cells per sample. Cells were spun down, 3 drops of plasma and thrombin were added to pellet, followed by centrifugation, and placed into a mesh bag, immersed in formalin for 30 minutes, processed and paraffin embedded.
Histology, immunohistochemistry and TUNEL assay
Sections were cut to 4-μm thickness, deparaffinized in xylene and rehydrated through graded alcohols to deionized water. Antigen retrieval and endogenous peroxidase block were carried out by standard procedure. IHC examination of mouse xenografts was performed on FFPE sections using Dako Mouse EnVisionTM+ HRP method. Ki67 [(1:100) (DAKO M7240) Glostrup, Denmark] immunostaining was performed using DAKO auto-stainer. Apoptotic cells were detected in FFPE cell blocks using ApopTag® plus peroxidase in situ apoptosis kit (#S7101, Millipore, Billerica, MA).
Slide evaluation
Slides were evaluated with NanoZoomer 2.0-HT: C9600-13 scanner (Hamamatsu Photonics, Iwata City, Japan) at 20X magnification. Mayachitra (Santa Barbara, CA) imago-1 software was used to count total number of nuclei (range 15,000–100,000 cells) in acquired TIFF images. Three sections from each paraffin cell block were stained at intervals of 50 μm depth. Manual identification of apoptotic bodies at 20X objective was based on (a) marked condensation of chromatin and cytoplasm (apoptotic cells); (b) cytoplasmic fragments with or without condensed chromatin (apoptotic bodies); and (c) intra- and extracellular chromatin fragments (micronuclei) [17]. An apoptotic index (%) was calculated as follows: apoptotic bodies/total number of cells per slide X 100. For Ki67 staining, 6 peripheral/edge regions were sampled within a fixed field of 0.427 mm2 in each tumor sample. Areas of necrosis were discarded and V.P. was blinded to experimental treatment. ImageJ software was used to quantify Ki67 immunoreactivity. Ki67 Labeling Index (LI) was calculated as a percentage of positively stained cells per total number of tumor cells considered for evaluation.
In vivo studies
MDA-MB-231 cells (1 × 106) suspended in 100 μL of chilled Matrigel (BD Biosciences) were injected bilaterally into lactiferous ducts of 4th mammary gland of 5- to 6-week-old female athymic nu/nu mice (Charles River). Two weeks post inoculation, mice were randomized into 6 treatment groups(8 mice per group): control DMSO, CDK2i, CDK4i, paclitaxel, paclitaxel/CDK2i, and paclitaxel/CDK4i. Mice were treated using intra-peritoneal injection: control or 1 day of paclitaxel treatment followed by control or 2 d of CDKi. Study groups were treated 3 times weekly for 4 weeks, or when tumor burden reached more than 1.5 cm2. Tumors were measured with digital calipers; tumor volumes were calculated using equation VTumor = (w2 × l )/2.53 Mice were weighed twice weekly. The tumor volumes from treated groups were compared with the volume from the respective control treated group at Day 21, and significance was then determined. At the end 4 weeks, mice were sacrificed and xenografts removed for IHC analysis. All animal experiments were conducted under protocols approved by Animal Care and Use Committee.
Formation of transduced cell arrays
To form an array, MDA-MB-231 cells were transduced with lentivirus encoding TA-FLuc or one of the TF reporter genes, as previously described,21,22,31 (25,000 PP/cell), by centrifugation, resuspension in Matrigel (BD Biosciences), and seeding into wells of a black 384-well plate (Greiner BioSciences, Monroe, NC). Cells were treated with 0.1% DMSO control or 240 nM CDK2i at indicated time points. For each treatment group, 11 TF reporters (TFr) were studied, in addition to control TA-Fluc reporter. Non-transduced cells were also used to control for background imaging. For all treatment groups, each TFr was present in each plate in quadruplicate.
Bioluminescent imaging to measure FLuc activity
To assess FLuc activity, bioluminescence imaging was performed at indicated time points. One mM d-luciferin (Perkin Elmer, Waltham, MA) was added to wells and plates were incubated at 37°C for 30 minutes, followed by imaging with In Vivo Imaging System (Caliper). Data were analyzed using R.54 Statistical methodology22 was updated accordingly: data below the background that contained non-infected cells were removed (confidence interval [CI] = 99.5%) and missing data were replaced with the average value of the same treated wells. Subsequently, data were further normalized by the average bioluminescence intensity of the control wells, TA-Fluc, for the same time and treatment and logarithmically transformed (in based 2). Normalized and transformed intensities were shifted to the same initial time value by multiplying ratio of initial measured activity to average of all initial measured activities for a given TFr such that the initial normalized value was the same across all conditions for the same TFr. R package limma55 was employed to determine differentially activated TFr versus initial time and experimental control. False discovery rate (fdr) was used to correct for multiple comparisons. Data were plotted using the ggplots package.56
Statistical analysis
Results are presented as mean values ± SE from representative experiments performed in triplicate and were analyzed by unpaired, 2 tailed Student's t test. p < 0.05 was considered statistically significant. Power analysis was performed to determine the number of animals (each with 2 xenografts) needed in each treatment group using sample size formula for comparison of means and PASS sample size software (Power Analysis and Sample Size for Windows 2000, NCSS). For a CV of 20%, 8 animals (16 tumors) per group were utilized.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Dr. Stacey Tobin for her editorial assistance, Dr. Stanislav Zelivianski for technical assistance with immunoblotting, Dr. Alfred Rademaker and Dr. Samira Azarin for assistance with statistical analysis in design of the in vivo work, and Dr. Fang Liu for her generous gift of the Smad3 mutant constructs.
Funding
JSJ is a Lynn Sage Scholar supported by NIH K22 CA138776 and R01GM097220, Central Surgical Association, Society of Surgical Oncology, Saslow Family and A Sister's Hope. ET is a Dr. John N. Nicholson Fellow, Chicago Biomedical Consortium Scholar supported by Searle Funds at Chicago Community Trust, NIH/NCI T32CA09560 and Kirchstein-NRSA Fellowship F31 CA168106-02.
References
- 1. Guarneri V, Broglio K, Kau SW, Cristofanilli M, Buzdar AU, Valero V, Buchholz T, Meric F, Middleton L, Hortobagyi GN, et al. Prognostic value of pathologic complete response after primary chemotherapy in relation to hormone receptor status and other factors. J Clin Oncol 2006; 24:1037-44; PMID:16505422; http://dx.doi.org/ 10.1200/JCO.2005.02.6914 [DOI] [PubMed] [Google Scholar]
- 2. Foulkes WD, Smith IE, Reis-Filho JS. Triple-negative breast cancer. N Engl J Med 2010; 363:1938-48; PMID:21067385; http://dx.doi.org/ 10.1056/NEJMra1001389 [DOI] [PubMed] [Google Scholar]
- 3. Fadare O, Tavassoli FA. Clinical and pathologic aspects of basal-like breast cancers. Nat Clin Pract Oncol 2008; 5:149-59; PMID:18212769; http://dx.doi.org/ 10.1038/ncponc1038 [DOI] [PubMed] [Google Scholar]
- 4. Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA, et al. Molecular portraits of human breast tumours. Nature 2000; 406:747-52; PMID:10963602; http://dx.doi.org/ 10.1038/35021093 [DOI] [PubMed] [Google Scholar]
- 5. Velasco-Velazquez MA, Li Z, Casimiro M, Loro E, Homsi N, Pestell RG. Examining the role of cyclin D1 in breast cancer. Future Oncol 2011; 7:753-65; PMID:21675838; http://dx.doi.org/ 10.2217/fon.11.56 [DOI] [PubMed] [Google Scholar]
- 6. Keyomarsi K, Tucker SL, Buchholz TA, Callister M, Ding Y, Hortobagyi GN, Bedrosian I, Knickerbocker C, Toyofuku W, Lowe M, et al. Cyclin E and survival in patients with breast cancer. N Engl J Med 2002; 347:1566-75; PMID:12432043; http://dx.doi.org/ 10.1056/NEJMoa021153 [DOI] [PubMed] [Google Scholar]
- 7. Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell 2003; 113:685-700; PMID:12809600; http://dx.doi.org/ 10.1016/S0092-8674(03)00432-X [DOI] [PubMed] [Google Scholar]
- 8. Jeruss JS, Sturgis CD, Rademaker AW, Woodruff TK. Down-regulation of activin, activin receptors, and Smads in high-grade breast cancer. Cancer Res 2003; 63:3783-90; PMID:12839974 [PubMed] [Google Scholar]
- 9. Burdette JE, Jeruss JS, Kurley SJ, Lee EJ, Woodruff TK. Activin A mediates growth inhibition and cell cycle arrest through Smads in human breast cancer cells. Cancer Res 2005; 65:7968-75; PMID:16140969 [DOI] [PubMed] [Google Scholar]
- 10. Matsuura I, Denissova NG, Wang G, He D, Long J, Liu F. Cyclin-dependent kinases regulate the antiproliferative function of Smads. Nature 2004; 430:226-31; PMID:15241418; http://dx.doi.org/ 10.1038/nature02650 [DOI] [PubMed] [Google Scholar]
- 11. Cooley A, Zelivianski S, Jeruss JS. Impact of cyclin E overexpression on Smad3 activity in breast cancer cell lines. Cell Cycle 2010; 9:4900-7; PMID:21150326; http://dx.doi.org/ 10.4161/cc.9.24.14158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zelivianski S, Cooley A, Kall R, Jeruss JS. Cyclin-dependent kinase 4-mediated phosphorylation inhibits Smad3 activity in cyclin d-overexpressing breast cancer cells. Mol Cancer Res 2010; 8:1375-87; PMID:20736297; http://dx.doi.org/ 10.1158/1541-7786.MCR-09-0537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Matsuura I, Chiang KN, Lai CY, He D, Wang G, Ramkumar R, Uchida T, Ryo A, Lu K, Liu F. Pin1 promotes transforming growth factor-beta-induced migration and invasion. J Biol Chem 2010; 285:1754-64; PMID:19920136; http://dx.doi.org/ 10.1074/jbc.M109.063826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fornier MN, Rathkopf D, Shah M, Patil S, O’Reilly E, Tse AN, Hudis C, Lefkowitz R, Kelsen DP, Schwartz GK. Phase I dose-finding study of weekly docetaxel followed by flavopiridol for patients with advanced solid tumors. Clin Cancer Res 2007; 13:5841-6; PMID:17908977; http://dx.doi.org/ 10.1158/1078-0432.CCR-07-1218 [DOI] [PubMed] [Google Scholar]
- 15. Baughn LB, Di Liberto M, Wu K, Toogood PL, Louie T, Gottschalk R, Niesvizky R, Cho H, Ely S, Moore MA, et al. A novel orally active small molecule potently induces G1 arrest in primary myeloma cells and prevents tumor growth by specific inhibition of cyclin-dependent kinase 46. Cancer Res 2006; 66:7661-7; PMID:16885367; http://dx.doi.org/ 10.1158/0008-5472.CAN-06-1098 [DOI] [PubMed] [Google Scholar]
- 16. Fry DW, Harvey PJ, Keller PR, Elliott WL, Meade M, Trachet E, Albassam M, Zheng X, Leopold WR, Pryer NK, et al. Specific inhibition of cyclin-dependent kinase 46 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol Cancer Ther 2004; 3:1427-38; PMID:15542782 [PubMed] [Google Scholar]
- 17. Hughes L, Malone C, Chumsri S, Burger AM, McDonnell S. Characterisation of breast cancer cell lines and establishment of a novel isogenic subclone to study migration, invasion and tumourigenicity. Clin Exp Metastasis 2008; 25:549-57; PMID:18386134; http://dx.doi.org/ 10.1007/s10585-008-9169-z [DOI] [PubMed] [Google Scholar]
- 18. Kalkhoven E, Roelen BA, de Winter JP, Mummery CL, van den Eijnden-van Raaij AJ, van der Saag PT, van der Burg B. Resistance to transforming growth factor beta and activin due to reduced receptor expression in human breast tumor cell lines. Cell Growth Differ 1995; 6:1151-61; PMID:8519692 [PubMed] [Google Scholar]
- 19. Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, Pietenpol JA. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest 2011; 121:2750-67; PMID:21633166; http://dx.doi.org/ 10.1172/JCI45014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Brown KA, Aakre ME, Gorska AE, Price JO, Eltom SE, Pietenpol JA, Moses HL. Induction by transforming growth factor-beta1 of epithelial to mesenchymal transition is a rare event in vitro. Breast Cancer Res 2004; 6:R215-31; PMID:15084245; http://dx.doi.org/ 10.1186/bcr778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bellis AD, Penalver-Bernabe B, Weiss MS, Yarrington ME, Barbolina MV, Pannier AK, Jeruss JS, Broadbelt LJ, Shea LD. Cellular arrays for large-scale analysis of transcription factor activity. Biotechnol Bioeng 2011; 108:395-403; PMID:20812256; http://dx.doi.org/ 10.1002/bit.22916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Weiss MS, Penalver Bernabe B, Bellis AD, Broadbelt LJ, Jeruss JS, Shea LD. Dynamic, large-scale profiling of transcription factor activity from live cells in 3D culture. PLoS One 2010; 5:e14026; PMID:21103341; http://dx.doi.org/ 10.1371/journal.pone.0014026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Online Mendelian Inheritance in Man, OMIM®. McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University; Baltimore, MD 2014; http://omim.org/ [Google Scholar]
- 24. Wiercinska E, Naber HP, Pardali E, van der Pluijm G, van Dam H, Ten Dijke P. The TGF-betaSmad pathway induces breast cancer cell invasion through the up-regulation of matrix metalloproteinase 2 and 9 in a spheroid invasion model system. Breast Cancer Res Treat 2011; 128:657-66; PMID:20821046; http://dx.doi.org/ 10.1007/s10549-010-1147-x [DOI] [PubMed] [Google Scholar]
- 25. Sekimoto G, Matsuzaki K, Yoshida K, Mori S, Murata M, Seki T, Matsui H, Fujisawa J, Okazaki K. Reversible smad-dependent signaling between tumor suppression and oncogenesis. Cancer Res 2007; 67:5090-6; PMID:17545585; http://dx.doi.org/ 10.1158/0008-5472.CAN-06-4629 [DOI] [PubMed] [Google Scholar]
- 26. Nakano A, Koinuma D, Miyazawa K, Uchida T, Saitoh M, Kawabata M, Hanai J, Akiyama H, Abe M, Miyazono K, et al. Pin1 down-regulates transforming growth factor-beta (TGF-beta) signaling by inducing degradation of Smad proteins. J Biol Chem 2009; 284:6109-15; PMID:19122240; http://dx.doi.org/ 10.1074/jbc.M804659200 [DOI] [PubMed] [Google Scholar]
- 27. Zuo Y, Wu Y, Chakraborty C. Cdc42 negatively regulates intrinsic migration of highly aggressive breast cancer cells. J Cell Physiol 2012; 227:1399-407; PMID:21618528; http://dx.doi.org/ 10.1002/jcp.22853 [DOI] [PubMed] [Google Scholar]
- 28. Matsuzaki K, Murata M, Yoshida K, Sekimoto G, Uemura Y, Sakaida N, Kaibori M, Kamiyama Y, Nishizawa M, Fujisawa J, et al. Chronic inflammation associated with hepatitis C virus infection perturbs hepatic transforming growth factor beta signaling, promoting cirrhosis and hepatocellular carcinoma. Hepatology 2007; 46:48-57; PMID:17596875; http://dx.doi.org/ 10.1002/hep.21672 [DOI] [PubMed] [Google Scholar]
- 29. Yamagata H, Matsuzaki K, Mori S, Yoshida K, Tahashi Y, Furukawa F, Sekimoto G, Watanabe T, Uemura Y, Sakaida N, et al. Acceleration of Smad2 and Smad3 phosphorylation via c-Jun NH(2)-terminal kinase during human colorectal carcinogenesis. Cancer Res 2005; 65:157-65; PMID:15665291 [PubMed] [Google Scholar]
- 30. Matsuzaki K, Okazaki K. Transforming growth factor-beta during carcinogenesis: the shift from epithelial to mesenchymal signaling. J Gastroenterol 2006; 41:295-303; PMID:16741607; http://dx.doi.org/ 10.1007/s00535-006-1795-0 [DOI] [PubMed] [Google Scholar]
- 31. Siletz A, Schnabel M, Kniazeva E, Schumacher AJ, Shin S, Jeruss JS, Shea LD. Dynamic transcription factor networks in epithelial-mesenchymal transition in breast cancer models. PLoS One 2013; 8:e57180; PMID:23593114; http://dx.doi.org/ 10.1371/journal.pone.0057180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Xu J, Lamouille S, Derynck R. TGF-beta-induced epithelial to mesenchymal transition. Cell Res 2009; 19:156-72; PMID:19153598; http://dx.doi.org/ 10.1038/cr.2009.5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Grunert S, Jechlinger M, Beug H. Diverse cellular and molecular mechanisms contribute to epithelial plasticity and metastasis. Nat Rev Mol Cell Biol 2003; 4:657-65; PMID:12923528; http://dx.doi.org/ 10.1038/nrm1175 [DOI] [PubMed] [Google Scholar]
- 34. Ross S, Hill CS. How the Smads regulate transcription. Int J Biochem Cell Biol 2008; 40:383-408; PMID:18061509; http://dx.doi.org/ 10.1016/j.biocel.2007.09.006 [DOI] [PubMed] [Google Scholar]
- 35. Nagaria TS, Williams JL, Leduc C, Squire JA, Greer PA, Sangrar W. Flavopiridol synergizes with sorafenib to induce cytotoxicity and potentiate antitumorigenic activity in EGFRHER-2 and mutant RASRAF breast cancer model systems. Neoplasia 2013; 15:939-51; PMID:23908594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bostrom P, Soderstrom M, Palokangas T, Vahlberg T, Collan Y, Carpen O, Hirsimaki P. Analysis of cyclins A, B1, D1 and E in breast cancer in relation to tumour grade and other prognostic factors. BMC Res Notes 2009; 2:140; PMID:19615042; http://dx.doi.org/ 10.1186/1756-0500-2-140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Voduc D, Nielsen TO, Cheang MC, Foulkes WD. The combination of high cyclin E and Skp2 expression in breast cancer is associated with a poor prognosis and the basal phenotype. Hum Pathol 2008; 39:1431-7; PMID:18620730; http://dx.doi.org/ 10.1016/j.humpath.2008.03.004 [DOI] [PubMed] [Google Scholar]
- 38. Jahn SC, Law ME, Corsino PE, Rowe TC, Davis BJ, Law BK. Assembly, activation, and substrate specificity of cyclin D1Cdk2 complexes. Biochemistry 2013; PMID:23627734; http://dx.doi.org/ 10.1021/bi400047u [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gradishar WJ. Taxanes for the treatment of metastatic breast cancer. Breast Cancer (Auckl) 2012; 6:159-71; PMID:23133315; http://dx.doi.org/ 10.4137/BCBCR.S8205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Motwani M, Delohery TM, Schwartz GK. Sequential dependent enhancement of caspase activation and apoptosis by flavopiridol on paclitaxel-treated human gastric and breast cancer cells. Clin Cancer Res 1999; 5:1876-83; PMID:10430095 [PubMed] [Google Scholar]
- 41. Mao J, Song B, Shi Y, Wang B, Fan S, Yu X, Tang J, Li L. ShRNA targeting Notch1 sensitizes breast cancer stem cell to paclitaxel. Int J Biochem Cell Biol 2013; 45:1064-73; PMID:23500524; http://dx.doi.org/ 10.1016/j.biocel.2013.02.022 [DOI] [PubMed] [Google Scholar]
- 42. Liu P, Kumar IS, Brown S, Kannappan V, Tawari PE, Tang JZ, Jiang W, Armesilla AL, Darling JL, Wang W. Disulfiram targets cancer stem-like cells and reverses resistance and cross-resistance in acquired paclitaxel-resistant triple-negative breast cancer cells. Br J Cancer 2013; 109:1876-85; PMID:24008666; http://dx.doi.org/ 10.1038/bjc.2013.534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chen YN, Sharma SK, Ramsey TM, Jiang L, Martin MS, Baker K, Adams PD, Bair KW, Kaelin WG., Jr. Selective killing of transformed cells by cyclincyclin-dependent kinase 2 antagonists. Proc Natl Acad Sci U S A 1999; 96:4325-9; PMID:10200261; http://dx.doi.org/ 10.1073/pnas.96.8.4325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. McClendon AK, Dean JL, Rivadeneira DB, Yu JE, Reed CA, Gao E, Farber JL, Force T, Koch WJ, Knudsen ES. CDK46 inhibition antagonizes the cytotoxic response to anthracycline therapy. Cell Cycle 2012; 11:2747-55; PMID:22751436; http://dx.doi.org/ 10.4161/cc.21127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Leontieva OV, Blagosklonny MV. CDK46-inhibiting drug substitutes for p21 and p16 in senescence: duration of cell cycle arrest and MTOR activity determine geroconversion. Cell Cycle 2013; 12:3063-9; PMID:23974099; http://dx.doi.org/ 10.4161/cc.26130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Leontieva OV, Demidenko ZN, Blagosklonny MV. MEK drives cyclin D1 hyperelevation during geroconversion. Cell Death Differ 2013; 20:1241-9; PMID:23852369; http://dx.doi.org/ 10.1038/cdd.2013.86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. McMasters KM, Hunt KK. Neoadjuvant chemotherapy, locally advanced breast cancer, and quality of life. J Clin Oncol 1999; 17:441-4; PMID:10080583 [DOI] [PubMed] [Google Scholar]
- 48. Sahoo S, Lester SC. Pathology of breast carcinomas after neoadjuvant chemotherapy: an overview with recommendations on specimen processing and reporting. Arch Pathol Lab Med 2009; 133:633-42; PMID:19391665; http://dx.doi.org/ 10.1043/1543-2165-133.4.633 [DOI] [PubMed] [Google Scholar]
- 49. Symmans WF, Peintinger F, Hatzis C, Rajan R, Kuerer H, Valero V, Assad L, Poniecka A, Hennessy B, Green M, et al. Measurement of residual breast cancer burden to predict survival after neoadjuvant chemotherapy. J Clin Oncol 2007; 25:4414-22; PMID:17785706; http://dx.doi.org/ 10.1200/JCO.2007.10.6823 [DOI] [PubMed] [Google Scholar]
- 50. Rajan R, Poniecka A, Smith TL, Yang Y, Frye D, Pusztai L, Fiterman DJ, Gal-Gombos E, Whitman G, Rouzier R, et al. Change in tumor cellularity of breast carcinoma after neoadjuvant chemotherapy as a variable in the pathologic assessment of response. Cancer 2004; 100:1365-73; PMID:15042669; http://dx.doi.org/ 10.1002/cncr.20134 [DOI] [PubMed] [Google Scholar]
- 51. Kang Y, He W, Tulley S, Gupta GP, Serganova I, Chen CR, Manova-Todorova K, Blasberg R, Gerald WL, Massague J. Breast cancer bone metastasis mediated by the Smad tumor suppressor pathway. Proc Natl Acad Sci U S A 2005; 102:13909-14; PMID:16172383; http://dx.doi.org/ 10.1073/pnas.0506517102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bandyopadhyay A, Agyin JK, Wang L, Tang Y, Lei X, Story BM, Cornell JE, Pollock BH, Mundy GR, Sun LZ. Inhibition of pulmonary and skeletal metastasis by a transforming growth factor-beta type I receptor kinase inhibitor. Cancer Res 2006; 66:6714-21; PMID:16818646; http://dx.doi.org/ 10.1158/0008-5472.CAN-05-3565 [DOI] [PubMed] [Google Scholar]
- 53. Euhus DM, Hudd C, LaRegina MC, Johnson FE. Tumor measurement in the nude mouse. J Surg Oncol 1986; 31:229-34; PMID:3724177; http://dx.doi.org/ 10.1002/jso.2930310402 [DOI] [PubMed] [Google Scholar]
- 54. R Development Core Team 2005. R Foundation for Statistical Computing R: A Language and Environment for Statistical Computing. Vienna, Austria, 2012 [Google Scholar]
- 55. Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol 2004; 3:1-25 Article3; PMID:16646809 [DOI] [PubMed] [Google Scholar]
- 56. Wickham H. ggplot2. Elegant Graphics for Data Analysis. New York, NY: Springer Science + Business Media, LLC, 2009 [Google Scholar]