

Abstract
Following fertilization, oviparous embryos undergo rapid, mostly transcriptionally silent cleavage divisions until the mid-blastula transition (MBT), when large-scale developmental changes occur, including zygotic genome activation (ZGA) and cell cycle remodeling, via lengthening and checkpoint acquisition. Despite their concomitant appearance, whether these changes are co-regulated is unclear. Three models have been proposed to account for the timing of (ZGA). One model implicates a threshold nuclear to cytoplasmic (N:C) ratio, another stresses the importance cell cycle elongation, while the third model invokes a timer mechanism. We show that precocious Chk1 activity in pre-MBT zebrafish embryos elongates cleavage cycles, thereby slowing the increase in the N:C ratio. We find that cell cycle elongation does not lead to transcriptional activation. Rather, ZGA slows in parallel with the N:C ratio. We show further that the DNA damage checkpoint program is maternally supplied and independent of ZGA. Although pre-MBT embryos detect damage and activate Chk2 after induction of DNA double-strand breaks, the Chk1 arm of the DNA damage response is not activated, and the checkpoint is nonfunctional. Our results are consistent with the N:C ratio model for ZGA. Moreover, the ability of precocious Chk1 activity to delay pre-MBT cell cycles indicate that lack of Chk1 activity limits checkpoint function during cleavage cycles. We propose that Chk1 gain-of-function at the MBT underlies cell cycle remodeling, whereas ZGA is regulated independently by the N:C ratio.
Keywords: cell cycle checkpoints, embryogenesis, mid-blastula transition, zygotic genome activation
Abbreviations
- MBT
Mid-blastula Transition
- ZGA
Zygotic genome activation
Introduction
Immediately following fertilization, most metazoan embryos undergo synchronous cleavage divisions that lack gap phases and cell cycle checkpoints. During this period, most zygotic genes are transcriptionally silent; rather, embryos rely on maternally loaded mRNAs for development. These abbreviated cycles persist until the mid-blastula transition (MBT), when several large-scale changes occur together.
Zygotic genome activation (ZGA) is one hallmark of the MBT. With a few exceptions1-7 transcription of the vast majority of zygotic genes starts at the MBT. How the onset of ZGA is regulated is a long-standing question, and several models have been proposed. In one model, activation of zygotic transcription depends on a threshold nuclear to cytoplasmic (N:C) ratio that is achieved through the reductive cleavage divisions leading up to the MBT, when cells divide but do not grow. Addition of exogenous DNA to increase the N:C ratio in pre-MBT Xenopus embryos can induce premature onset of zygotic transcription.8 These findings suggest that transcription is regulated by an unknown cytoplasmic repressor that is titrated as the N:C ratio increases.9 A second model suggests that cell cycle elongation is required for ZGA,9 as rapid cell cycles may not support transcription, particularly of long genes.6, 10-13 Thus, although it may not directly trigger ZGA, cell cycle elongation at the MBT may create a permissive environment for increased transcriptional activity. A third model postulates that a cell cycle-independent timer governs ZGA, either through accumulation of transcription machinery components, loss of a transcriptional repressor, or degradation of maternal transcripts.1,11,14
Massive cell cycle remodeling is a second hallmark of the MBT, as embryonic cleavage divisions transform into typical somatic cell cycles: losing cell division synchrony, elongating the cell cycle dramatically, and adding gap phases that are lacking during pre-MBT cell cycles. Both activation of Chk1, independent of damage,15,16 and degradation of the Cdk1-activating phosphatase Cdc2517-19 contribute to these changes. Another major component of cell cycle remodeling at the MBT is acquisition of DNA damage checkpoints. Pre-MBT embryos neither delay their cell cycles nor initiate DNA repair and apoptotic pathways in response to DNA damage.20,21 Mechanisms underlying this aspect of cell cycle remodeling at the MBT are poorly understood.
Despite the striking synchrony of ZGA and DNA damage checkpoint acquisition, how these events are coordinated is unclear. One model hypothesizes that checkpoint function is directly coupled to ZGA: pre-MBT checkpoint signaling pathways might lack essential components provided only by the zygotic genome. Adding exogenous DNA to pre-MBT Xenopus embryos leads to precocious checkpoint function,22 but this effect could be indirect if the N:C ratio controls zygotic transcription.
By experimentally manipulating Chk1 activity before the MBT in zebrafish embryos, we interrogate its role in cell cycle remodeling at the MBT and distinguish between models for ZGA. We find that zygotic transcription increases in parallel with the N:C ratio and not in response to premature cell cycle elongation. We also show that the DNA damage checkpoint program is independent of zygotic transcription, and that checkpoint function is limited by the lack of Chk1 activity prior to the MBT. Overall, we conclude that while happening simultaneously, cell cycle remodeling and zygotic genome activation are regulated independently at the MBT.
Results
Precocious cell cycle elongation prior to the MBT does not lead to zygotic genome activation
To determine whether cell cycle elongation leads to ZGA, we first tested whether premature Chk1 activity could lengthen pre-MBT cell cycles. We expressed exogenous wildtype GFP-tagged zebrafish Chk1 (Chk1-GFP) or a constitutively active, phosphomimetic form (Chk1-4E-GFP)23 in pre-MBT zebrafish embryos. Pre-MBT cell cycles progressively lengthened to an average of 21.0 min (Chk1-GFP) or 26.1 min (Chk1-4E-GFP) between the 6th and 10th cleavages, compared to 16 min in control embryos (Fig. 1). We also tested 2 Chk1 truncation mutants. ΔC-Chk1-GFP contains residues 1-99 of zebrafish Chk1, corresponding to the N-terminal kinase domain, which by itself is catalytically inactive.24-27 ΔN-Chk1-GFP contains residues 215-410 of zebrafish Chk1, which corresponds to the C-terminal regulatory domain.25,27 Neither truncation mutant affected cleavage cycle lengths, indicating that the cell cycle lengthening depends on Chk1 kinase activity (Fig 1B).
Figure 1.
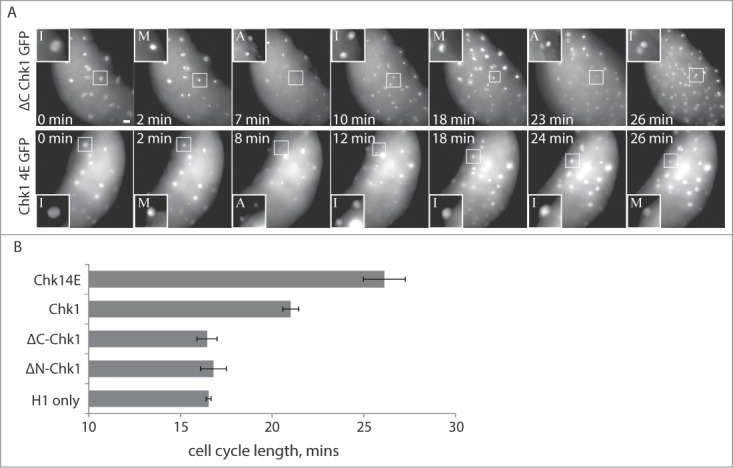
Expression of a constitutively active Chk1 kinase elongates pre-MBT cell cycles. (A and B) Embryos were injected with Alexa 594-Histone H1 protein and mRNA encoding one of the indicated Chk1 constructs (wtChk1, Chk14E-GFP, ΔC-Chk1-GFP, or ΔN-Chk1-GFP), then imaged live during cleavage-stage cell cycles. Image (A) show cell cycle progression based on Histone H1, starting at ∼2.25 HPF (t = 0). Scale bar 20 μm. Insets are displayed with higher contrast settings to show the changing morphology of a single nucleus at different cell cycle stages: I, interphase; M, prometaphase/metaphase; A, anaphase. The time between M-phases is 16 min for ΔC-Chk1-GFP and 24 min for Chk1-4E-GFP. Although not shown, the second mitotic nucleus in the Chk1-4E-GFP embryo goes into anaphase 6 min later. Cell cycle lengths (B) were measured as time between M-phases, based on Histone H1 morphology, and averaged over cleavages 6-10 for multiple embryos (n ≥ 9).
Cell cycle elongation caused by exogenous Chk1 allowed us to test models for how the timing of ZGA is controlled. If ZGA depends directly on cell cycle elongation, which occurs prematurely in Chk1-4E embryos, we expect transcription at a lower N:C ratio compared to controls. Similarly, if ZGA is controlled by a cell cycle-independent timing mechanism, we expect transcription at a lower N:C ratio when cell cycles are elongated in Chk1-4E embryos, compared to controls, because the N:C ratio increases more slowly. In contrast, if ZGA is controlled by the N:C ratio, we expect control and Chk1-4E embryos to exhibit a similar increase in transcriptional activity with respect to the N:C ratio.
To examine the relationship between N:C ratio and transcription, we measured both simultaneously in individual fixed embryos. We monitored transcriptional activation using a phospho-specific antibody to stain for phospho-Rbp1 Ser2/5 (pRbp1), which is specific for phosphoepitopes present on active, elongating RNA polymerase II20 in pre-MBT zebrafish and Xenopus embryos.5,28,29 This assay allows us to evaluate global transcriptional activity in combination with N:C ratio on an individual cell basis.
To confirm that pRbp1 staining correlates with transcription, we examined expression of nanor and apoeb, 2 zygotically expressed genes that only appear after the MBT, at 3.75 HPF (Fig 2A).21 Injection with α-amanitin, an inhibitor of RNA polymerase II, abolished transcript expression at 3.75 HPF. Consistent with the transcript levels, pRbp1 staining is low at 2.5 HPF and high at 3.5 HPF in both control and Chk1-4E embryos (Fig 2B). We also tracked changes in the N:C ratio by measuring nuclei densities at 15-minute intervals between 2.25 and 3.5 HPF. The nuclei density increased more slowly in Chk1-4E embryos compared to control embryos (Fig 2C), as expected for elongated cell cycles. We find that pRbp1 staining increases with nuclei density (and therefore, N:C ratio) in Chk1-4E embryos similarly as in control embryos, and the 2 groups are indistinguishable from each other (Fig 2D). In addition, pRbp1 staining did not increase prematurely at 2.5 HPF in Chk1-4E embryos, though cell cycles are elongated (Fig 2B). These data demonstrate that ZGA is coupled to the N:C ratio rather than cell cycle elongation, and is not controlled by a timing mechanism.
Figure 2.

For figure legend, see page 3831.Figure 2 (See previous page). Transcriptional activity is coupled to the N:C ratio. (A) Expression of the zygotic genes apoeb and lrat were monitored by RT-PCR at the times indicated, with or without Chk1-4E-GFP expression. α-amanitin injected embryos serve as a transcriptionally silent post-MBT control. β-actin is the internal control. (B) Embryos with our without Chk1-4E-GFP expression were fixed and stained for phosphorylated RNA Polymerase II (pRbp1) and DNA at the indicated times. pRbp1 staining was quantified and averaged over multiple embryos (n ≥ 10). Error bars indicate s.e.m., *P < 0.05. (C and D) Control and Chk1-4E-GFP embryos were fixed and stained for DNA and pRbp1 at 15-minute intervals between 2.25 and 3.5 HPF. Representative images show nuclei density at the first and last timepoints (C), and the plot shows average nuclei density (n ≥ 37) at each timepoint, pooled from 4 independent experiments. Chk1-4E-GFP embryos have fewer nuclei as a result of elongated cell cycles. pRbp1 staining was calculated for embryos grouped by nuclei density (D). Representative images are shown for each group, and the plot shows average pRbp1 staining intensity for each group, calculated as a fraction of the ≥60 control group (n ≥ 9 for Chk14E with >50 nuclei, n ≥ 13 for all other groups). Error bars indicate s.e.m. All scale bars 20 μm.
Acquisition of DNA damage checkpoints is independent of zygotic transcription
Chk1 is typically activated in response to DNA damage or replication stress, and phosphorylation of Chk1 substrates leads to cell cycle delay and DNA repair.30 The ability of precocious Chk1 activity to lengthen pre-MBT cell cycles (Fig. 1) indicates that downstream checkpoint signaling is intact and suggests that the lack of Chk1 activity limits checkpoint function prior to the MBT. Alternatively, other components could be provided via zygotic transcription, which would coordinate checkpoint acquisition and ZGA.
To test whether checkpoint acquisition depends on zygotic transcription at the MBT, we inhibited transcription by injecting one-cell stage embryos with α-amanitin, an inhibitor of RNA polymerase II.31,32 To confirm that the treatment prevented activation of zygotic transcription, we stained for pRbp1 and monitored expression of the zygotic genes nanor and lrat. Post-MBT embryos treated with α-amanitin have decreased Rbp1 staining compared to uninjected control embryos and lack nanor and lrat, indicating successful transcription inhibition (Fig. 3A, B).
Figure 3.

DNA damage checkpoint acquisition at the MBT does not depend on zygotic transcription. (A) 1-cell stage embryos were injected with α-amanitin as indicated, incubated until ∼3.75 HPF, then fixed and stained for pRbp1 and DNA. pRbp1 staining intensity was quantified and averaged over multiple embryos (n ≥ 9). Error bars indicate s.e.m. Scale bars 10 μm. (B) Expression of the zygotic genes nanor and lrat was monitored by RT-PCR at the times indicated in control embryos or embryos injected with α-amanitin. β-actin is the internal control; — indicates no sample loaded. A representative gel is show, and similar results were obtained in 3 independent experiments. (C) 1-cell stage embryos were injected with α-amanitin as indicated, treated with 250 mM HU at 3.25 HPF for 45 min, fixed at 4 HPF, and stained for pH3 and DNA. The % of nuclei positive for pH3 was calculated and averaged over multiple embryos (n ≥ 21 for each condition, pooled from 3 independent experiments). Error bars indicate s.e.m. Scale bars 20 μm.
To test for a DNA damage response in the absence of zygotic transcription, embryos were injected with α-amanitin and then treated with hydroxyurea (HU) to induce DNA damage. Hydroxyurea is a ribonucleotide reductase inhibitor which causes stalled replication forks that eventually lead to fork collapse and DNA single-strand breaks.33 After HU treatment, embryos were fixed and stained for phospho-Ser10 histone H3 (pH3), a well-established marker for mitosis,34 to track cell cycle arrest after DNA damage. Under normal conditions, post-MBT embryos treated with HU have a robust checkpoint response: the mitotic index decreases from ∼20% to 5%, demonstrating successful cell cycle delay and inhibition of mitotic entry after sustaining DNA damage (Fig. 3C, left 4 panels). Inhibition of zygotic transcription did not affect the cell cycle response to HU, as the mitotic index was not affected by α-amanitin (Fig. 3C, right 4 panels). Our results demonstrate that pre-MBT embryos have a maternally supplied checkpoint program, as checkpoint acquisition does not depend on zygotic transcription.
Pre-MBT embryos have one of 2 DNA damage signaling pathways intact
Our results show that checkpoint signaling downstream of Chk1 is intact in pre-MBT embryos, since premature Chk1 activation leads to cell cycle delay, and further that all components of the checkpoint program are maternally provided. We next asked how DNA damage signaling changes at the MBT. One of the earliest cellular responses to DNA damage is phosphorylation of the histone variant H2AX, which creates foci at sites of DNA damage that act as docking sites for additional DNA damage response proteins.35 Given its essential role in DNA damage response initiation, we monitored γH2AX by immunofluorescence in pre- and post-MBT embryos in response to ionizing radiation (IR), using a phospho-specific antibody. Treatment with 10 Gy ionizing radiation causes severe DNA damage, as indicated by abnormal nuclei, but does not lead to cell cycle delay in pre-MBT embryos (Fig 4A), as expected since these embryos lack cell cycle checkpoints. Despite the lack of cell cycle delay, γH2AX levels increased dramatically in response to IR in pre-MBT embryos (Fig. 4B, left panels), demonstrating that these embryos are indeed capable of detecting DNA damage and can activate the first major step in the checkpoint signaling pathway. Treatment with IR also induced an increase in γH2AX in post-MBT embryos, as expected for cells with fully functional damage checkpoints (Fig. 4B, right panels).
Figure 4.

Pre-MBT embryos can initiate a DNA damage response. (A) Embryos were injected with Alexa 594-Histone H1 protein and treated with 10 Gy IR as indicated, then imaged live during cleavage-stage cell cycles. Images (A) show cell cycle progression based on Histone H1, starting at ∼2.5 HPF (t = 0), with insets as in Figure. 1A. Insets are shown with higher contrast settings so nuclei morphology are clear. Cell cycle lengths were measured as time between M-phases, based on Histone H1 morphology, and averaged over cleavages 5-9 for multiple embryos (n ≥ 9). (B) Pre-MBT (1.5 HPF) or post-MBT (4 HPF) embryos were treated with 10 Gy IR then fixed and stained for γH2AX and DNA. γH2AX staining intensity was quantified and averaged over multiple embryos (n ≥ 10) for each condition. Similar results were obtained in 4 independent experiments; a representative plot is shown. Error bars indicate s.e.m., *P < 0.05. All scale bars 20 μm. Arrowheads show examples of nuclear fragmentation and lagging chromosomes from DNA damage in the irradiated embryo.
Downstream of γH2AX, the cellular response to DNA damage can be divided into 2 main signaling cascades that lead to phosphorylation and activation of the Chk1 or Chk2 checkpoint kinases. Both pathways converge on Cdc25, a positive regulator of cell cycle progression that is inhibited by phosphorylation by Chk1 or Chk2. To directly test whether Chk1 and Chk2 signaling pathways are active prior to the MBT, we examined phosphorylation of each after DNA damage. We used phospho-specific antibodies against human Chk2-Thr68 (pChk2) or human Chk1-Ser345 (pChk1), well established markers for Chk2 or Chk1 activation that are also used in zebrafish.36,37 Embryos were treated with IR to induce DNA damage, then fixed and stained with the phospho-specific antibodies. Chk2 is robustly phosphorylated in response to IR in both pre-MBT and post-MBT embryos (Fig 5A, C), indicating that the Chk2 pathway is intact before the MBT. Chk1 is also robustly phosphorylated in response to IR in post-MBT embryos, but Chk1 phosphorylation increases only slightly in pre-MBT embryos (Fig 5B, C), although Chk1 protein is present at both stages (Fig 5D). The failure to fully activate Chk1 is consistent with a previous finding in Xenopus embryos with aphidicolin, a DNA damage-inducing replication inhibitor.15 Taken together, our results demonstrate a key difference in DNA damage signaling in pre-MBT versus post-MBT embryos. The Chk1 pathway is active only after the MBT, whereas Chk2 signaling is intact even prior to the MBT, although activation of this pathway alone does not lead to cell cycle delay. These results, together with our finding that premature Chk1 activation leads to cell cycle delay before the MBT (Fig 1), indicate that the lack of Chk1 activity prior to the MBT limits checkpoint function.
Figure 5.
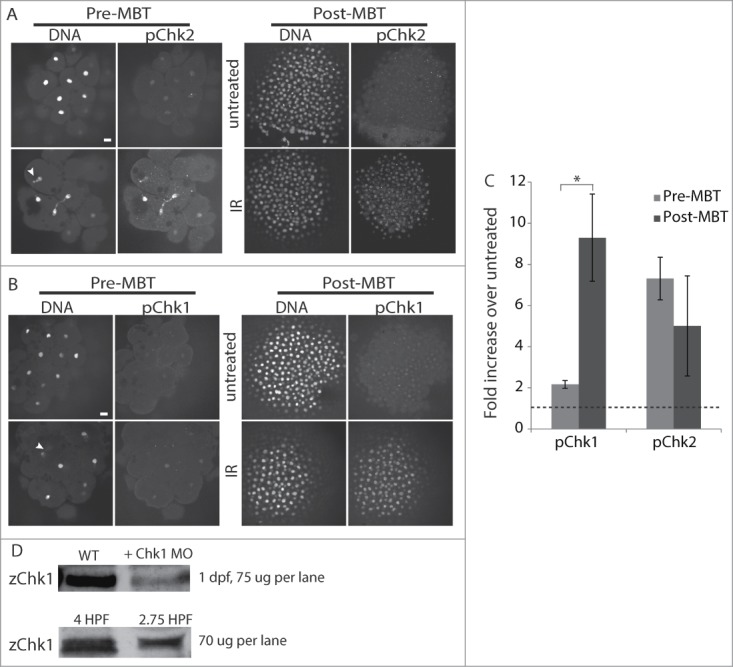
Pre-MBT embryos can activate Chk2 but not Chk1. (A–C) Pre-MBT (1.5 HPF) or post-MBT (4 HPF) embryos were treated with 10 Gy IR as indicated, then fixed and stained for pChk2 (A) or pChk1 (B) and DNA. Arrowheads show examples of nuclear fragmentation and lagging chromosomes from DNA damage. pChk1 and pChk2 staining were quantified, averaged over multiple embryos (n ≥ 8, pooled from multiple experiments), and plotted (C) as the fold increase of IR treated over untreated. The dashed line represents no increase over untreated control. Error bars indicate SD, *P < 0.05, scale bars 20 μm. (D) Western blots for Chk1 using zebrafish-specific anti-Chk1 antibody. Top blot: zChk1 polyclonal antibody was validated first in embryos 1 day post fertilization. Lanes show 75 ug of total protein from control embryos and 75 ug of total protein from embryos injected with a Chk1 morpholino to confirm the correct band. Bottom blot: 70 ug of total protein was loaded into each lane for 4 and 2.75 HPF embryos.
Discussion and Conclusions
Our findings address how embryos orchestrate the maternal to zygotic transition, specifically ZGA and cell cycle remodeling at the MBT. Despite their simultaneous emergence, we show that these major events are governed by independent mechanisms. Our data are consistent with the model that zygotic transcription is coupled to the N:C ratio. Premature Chk1 activation in cleavage-stage embryos lengthens the cell cycles and therefore slows the increase in N:C ratio. In these embryos, transcriptional activity increases in proportion to the N:C ratio.
Drosophila Chk1 mutants do not slow their cell cycles and do not undergo ZGA.38 However, Drosophila Chk1/Chk2 double mutants maintain rapid cell cycles like Chk1 single mutants yet still activate transcription of several zygotic genes, suggesting that neither Chk1 activation nor cell cycle elongation is required for ZGA in Drosophila.39 On the other hand, evidence from other systems indicates that cell cycle elongation is involved in ZGA, based on the observation that the production of gene products is limited by the time it takes to transcribe and process RNA.40 The rapid cell cycles of pre-MBT embryos might not support this process, as mitosis is not compatible with transcription.10 Consistent with this idea, overexpression of replication factors in Xenopus embryos leads to continuation of rapid cell cycles and delays the expression of a large number of zygotic genes,13 while precocious RNA synthesis occurs when cell cycles are artificially lengthened in Xenopus pre-MBT embryos treated with cycloheximide, an inhibitor of protein synthesis.9 In addition, the majority of zygotic genes expressed before the MBT in zebrafish are short, underscoring the idea that cell cycle elongation may be required to create a permissive environment for the expression of longer gene products.6 Although it may contribute to ZGA, our results show that cell cycle elongation is not sufficient for ZGA, as premature cell cycle lengthening does not lead to transcriptional activation at a lower N:C ratio.
Instead, our findings are consistent with previous studies that suggest that the N:C ratio governs ZGA. Work in Xenopus embryos demonstrated that transcription in pre-MBT embryos can be triggered precociously either in polyspermic embryos or by the addition of exogenous DNA to reach an N:C ratio characteristic of the MBT.8 Precocious transcription also occurs in polyploid cells, with a high N:C ratio, of zebrafish embryos with a mutation that prevents chromosome segregation but leaves cleavage intact.28 How transcriptional activation depends on the N:C ratio remains unclear. A cytoplasmic ZGA repressor may inhibit zygotic transcription until it is titrated out by a critical amount of DNA, and candidate transcriptional repressors have been identified in Xenopus and Drosophila. 41,42 Alternatively, the N:C ratio could indirectly control ZGA if it affects other MBT events not addressed in the present study, such as maternal transcript degradation and chromatin modifications, which have also been suggested to regulate ZGA.43,44 The influence of the N:C ratio on these events remains a question for future work.
An implication of our data is that transcriptional activity does not dramatically increase at a specific N:C ratio, which suggests that ZGA is not a single, abrupt event. Rather, transcriptional competence gradually increases, even throughout the cleavage divisions. Indeed, analyses of RNA synthesis and large-scale gene expression have found zygotic gene expression in Drosophila, Xenopus and zebrafish well before the MBT.1, 4-6, 45,46 Additionally, gene expression profiling data from haploid and diploid Drosophila embryos suggest more complex regulation of zygotic transcription, with distinct subsets of zygotic transcripts that depend either on time or on the N:C ratio.1
Our studies also provide insight into cell cycle remodeling at the MBT. We show that checkpoint acquisition can be uncoupled from zygotic transcription, and that checkpoint components are maternally supplied. Rather, differences in Chk1 activation underlie checkpoint function in pre- vs. post-MBT embryos. We demonstrate that pre-MBT embryos can detect DNA damage and activate Chk2 but not Chk1. Although there are mixed results on the ability of Chk2 to support a checkpoint without Chk1,47-49 our findings indicate that the lack of Chk1 activity accounts for the absence of DNA damage checkpoints during cleavage cycles. In addition, we show that premature Chk1 activation lengthens cell cycles, likely through inhibition of Cdc25,16 consistent with previous findings in Xenopus.15,16 Together, these results indicate that signaling downstream of Chk1 is intact before the MBT, but the checkpoint is not functional because Chk1 is not activated after DNA damage.
The principal goal of cleavage-stage cell cycles is to rapidly amass enough cells for later developmental stages. Uninterrupted cell cycle progression takes priority in pre-MBT stage embryos, even in the face of DNA damage. Absence of Chk1 activity is a strategy for embryos to avoid cell cycle delays prior to the MBT. Furthermore, embryos hijack the ability of Chk1 to delay cell cycle progression by transiently activating Chk1 at the MBT, which contributes to cell cycle elongation.16,50 We suggest a unifying model for cell cycle remodeling in which Chk1 gain-of-function dictates timing of both cell cycle elongation and DNA damage checkpoint acquisition in early embryogenesis. Chk1 gain-of-function at the MBT has been attributed to replication stalling and the phosphorylation of Claspin, an adaptor kinase that promotes Chk1 activation,51,52 which are both sensitive to the N:C ratio,22,53 but further work is required for a full understanding of this process.
Methods
Fish husbandry
Embryos were collected from natural mating and incubated in E3 buffer (5 mM NaCl, 0.17 mM KCl, 0.33 mM CaCl2, 0.33 mM MgSO4) at 28˚C. All experiments were carried out in the TLF strain.
Cell cycle length measurements using Histone H1-594
Histone H1 from calf thymus (Sigma) was conjugated to Alexa-Fluor 594 (Invitrogen) following the manufacturer's instructions. 1-cell stage embryos were injected with Alexa 594-histone H1 and incubated in E3 buffer at 28°C until live imaging. For live imaging, embryos were dechorionated with 1 mg/mL pronase in E3 buffer for 10 min, washed 2x in E3 buffer, then mounted on a 4-well fluorodish (Grenier Bio-One) in 0.4% agarose dissolved in E3 buffer.. Images were acquired using a 20x 0.7 NA objective on an inverted fluorescence microscope (DM6000, Leica Microsystems) equipped with an automated XYZ stage and a charge-coupled device camera (Orca-AG, Hamamatsu Photonics), controlled by Metamorph Software (MDS Analytical Technologies). Embryos were imaged every 1-2 minutes by fluorescence. At each time point a z series of 10 images was collected at 10 μm intervals. Max intensity projections are shown. Cell cycle lengths were measured by manually tracking nuclear morphology visualized by Alexa 594-histone H1, using ImageJ and Metamorph software. For cell cycle length measurements in embryos treated with IR, embryos were irradiated immediately before mounting and imaging.
DNA damage and embryo drug treatments
For pH3 staining, post-MBT embryos were treated with 250 mM HU dissolved in E3 buffer for at 3.25 HPF for 45 min, then fixed. For γH2AX, pChk1 and pChk2 staining embryos were treated with 10 Gy ionizing radiation with a Gammacell 40 irradiator (Nordion International), using cesium-137 as the radiation source. Pre-MBT embryos either fixed immediately after irradiation or after 7 min as above, and interphase nuclei were analyzed. Post-MBT embryos were all fixed immediately after irradiation. To inhibit transcription, 2 nL of 1 mg/mL α-amanitin dissolved in ddH2O was injected into the cell of 1-cell stage embryos. Embryos were incubated in E3 buffer at 28˚C until drug treatment with HU or RNA extraction.
Reverse transcription-polymerase chain reaction
RNA was isolated using Trizol (Invitrogen) from embryos at 2, 3.5, 3.75 and 4 HPF following the manufacturer's instructions, using 50 embryos were used for each condition and timepoint. 3.75 HPF embryos were used for testing transcription activity in Chk1-4E embryos as transcripts did not appear until then. Single-stranded cDNA was synthesized from total RNA extracted, using the SuperScript III First Strand Synthesis System (Invitrogen) following the following the manufacturer's instructions, using random hexamer primers. β-actin was used as the loading control. Primers used for PCR reaction were as follows:
nanor
Fwd: CAGCGAGCAGCGTTTACAGCGG
Rev: GAGGGAATCACCGCTCTGGTCTG
lrat
Fwd: ACGGGTCCAATATTTTGCTG
Rev: AGRCATCCACAAGATGAAGG
apoeb
Fwd: AGCAGAATGCAGATGACGTG
Rev: TCAGAGAGGTGCGTAGGTT
β-actin
Fwd: ACGCTTCTGGTCGTACTA
Rev: GATCTTGATCTTCATGGT
Immunofluorescence
Embryos were fixed with 4% paraformaldehyde in PBST (PBS with 0.1% Tween-20) overnight at 4°C, then manually dechorionated and dehydrated in 100% methanol overnight at −20°C. Embryos were rehydrated the next day sequentially with 75%, 50% and then 25% methanol in PBST (5 min each), then permeabilized with 100% acetone at −20°C for 7 min, then blocked with buffer containing 20% Heat-inactivated FBS, 20% Blocking reagent (Roche) and 1% DMSO in PBST for 1 hr at room temperature. Antibodies were diluted in blocking buffer, applied to embryos and incubated overnight at 4°C. Embryos were then washed 4 times (30 min each) with PBST, incubated with secondary antibody in blocking buffer, washed another 3 times, stained for 5 min with SYTOX Green (Invitrogen), then washed once with PBST. Embryos were mounted on fluorodishes (World Precision) in 4% methylcellulose dissolved in E3 buffer.
Primary antibodies were: mouse monoclonal against phospho-Ser10 Histone H3 (1:1000, Millipore cat #05-806); rabbit polyclonal against RNA Polymerase II subunit B1 CTD phospho-Ser2/5 (pRbp1) (1:1000, Cell Signaling cat #4735); rabbit polyclonal anti-γH2AX (1:1000, gift from Dr. James Amatruda, University of Texas Southwestern); rabbit monoclonal against human Chk1 phospho-Ser345 (1:500, Cell Signaling cat #2348); rabbit polyclonal against human Chk2 phospho-Thr68 (1:250, Cell Signaling cat #2661). Secondary antibodies were Alexa-Fluor 594-conjugated anti-mouse or anti-rabbit (1:200, Invitrogen).
All fixed embryos were imaged with a spinning disk confocal: a microscope (DM4000, Leica) with a 20 × 0.7 NA objective or a 63 × 1.3 NA glycerol objective, an XY piezo-z stage (Applied Scientific Instrumentation), a spinning disk (Yokogawa), an electron multiplier charge-coupled device camera (ImageEM, Hamamatsu Photonics), and an LMM5 laser merge module equipped with 488 and 593 nm lasers (Spectral Applied Research) controlled by Metamorph software. To quantify γH2AX, pRbp1, pChk1, or pChk2 staining, nuclei were defined based on DNA staining, and phospho-antibody staining intensity was averaged over all nuclei in each field after subtracting background based on cytoplasmic intensity using ImageJ software. % mitotic cells was quantified by calculating pH3 positive nuclei as a fraction of total nuclei (by DNA stain) in a field.
Nuclei density measurements
Control and Chk14E-injected embryos were fixed at 15-minute intervals between 2.25 and 3.5 HPF and stained with STYOX Green to label nuclei. Embryos were imaged as described above, and a z-series of 10 images was collected at 10 μm intervals for each embryo. We used ImageJ Object 3D Counter to count the number of nuclei within an isolated field measuring 157 × 157 μm across 3 z-slices for individual embryos.
mRNA constructs and injections
Wildtype zebrafish Chk1 cDNA was purchased from ATCC (Cat no. 5410666) and cloned into a GFP-pCS2+ mRNA expression vector. The constitutively active, phosphomimetic zChk123 was created by mutating 4 residues (S256E, S280E, T292E, S301E) in zebrafish Chk1 in a 492 basepair gBlock gene fragment (IDT). This fragment replaced residues 193-358 of wildtype zChk1 in GFP-pCS2+. A cDNA encoding the N-terminal kinase domain of Chk1 (amino acids 1-99) was amplified from wildtype zChk1 and cloned into GFP-pCS2+ to create ΔC-Chk1-GFP construct. Another cDNA encoding a C-terminal domain of Chk1 (amino acids 215-410) was amplified from wildtype zChk1 and cloned into GFP-pCS2+ to create the ΔN-Chk1-GFP construct. The Ambion mMessage mMachine SP6 in vitro transcription kit was used to make Chk1-GFP and Chk1-4E-GFP, ΔC-Chk1-GFP, and ΔN-Chk1-GFP mRNA. All constructs were injected into the cell of the 1-cell stage embryo. mRNAs were diluted in 0.2M KCl. Each embryo was injected with a 6.75 ng of mRNA. For live imaging, Histone H1 protein was also added to the injection mix. Expression of Chk1 constructs was monitored by GFP expression.
Morpholino injections
The morpholino oligonucleotide (MO) for Chk1 was synthesized by Gene Tools™ LLC, resuspended in sterile water at a concentration of 1mM and delivered into zebrafish embryos at the one-cell stage by microinjection. Chk1 MO sequence is: aggcacagccattatgcaatcttcg.54 Working concentration of the MO was 0.75 mM
Chk1 Immunoblotting
Frozen embryo samples were thawed on ice, diluted in 6x Laemmli sample buffer and loaded on 4-20% Ready Gel Tris-HCl Gels (Bio-Rad). 70 ug of protein was loaded onto each lane. Samples were electrophoresed at 100 mA, transferred to Hybond ECL nitrocellulose membrane (GE Healthcare), and blocked for 1 hour in PBS plus 0.1% (v/v) Tween-20 and 3% Blocking Agent (ECL Advance; GE Healthcare) at 25°C. Zebrafish-specific anti-Chk1 antibody (60B253; Antagene; 1:500 dilution) was used to detect Chk1. Horseradish peroxidase-conjugated secondary antibody (Amershame Biosciences; 1:100,000 dilution) were detected with chemiluminescence (ECL Advance; Amersham Biosciences).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Author Contributions
MZ and MAL conceived of the study, and participated in its design and coordination. MZ performed all the experiments, analyzed the data and performed statistical analysis. PK helped analyze gH2AX staining experiments. MZ, MAL and MCM drafted the manuscript. All authors read and approved the final manuscript.
Acknowledgments
We thank James Amatruda for supplying the γH2AX antibody. We also thank Jie He and the Zebrafish Core facility for helping with zebrafish husbandry. Additionally, we thank Peter Klein, Jennifer Skirkanich and Elliott Abrams for guidance and advice.
Funding
This work was supported by the NIH (T32HD007516 to MZ and GM083988 MAL), a Searle Scholar award to MAL, and the Penn Genome Frontiers Institute and a grant with the Pennsylvania Department of Health. The Department of Health specifically disclaims responsibility for any analyses, interpretations, or conclusions.
References
- 1. Lu X, Li JM, Elemento O, Tavazoie S, Wieschaus EF. Coupling of zygotic transcription to mitotic control at the Drosophila mid-blastula transition. Development 2009; 136:2101-10; PMID:19465600; http://dx.doi.org/ 10.1242/dev.034421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. De Renzis S, Elemento O, Tavazoie S, Wieschaus EF. Unmasking activation of the zygotic genome using chromosomal deletions in the Drosophila embryo. PLoS Biol 2007; 5:e117; PMID:17456005; http://dx.doi.org/ 10.1371/journal.pbio.0050117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Liang H-L, Nien C-Y, Liu H-Y, Metzstein MM, Kirov N, Rushlow C. The zinc-finger protein Zelda is a key activator of the early zygotic genome in Drosophila. Nature 2008; 456:400-3; PMID:18931655; http://dx.doi.org/ 10.1038/nature07388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Skirkanich J, Luxardi G, Yang J, Kodjabachian L, Klein PS. An essential role for transcription before the MBT in Xenopus laevis. Dev Biol 2011; 357:478-91; PMID:21741375; http://dx.doi.org/ 10.1016/j.ydbio.2011.06.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Blythe SA, Cha SW, Tadjuidje E, Heasman J, Klein PS. β-catenin primes organizer gene expression by recruiting a histone H3 arginine 8 methyltransferase, Prmt2. Dev Cell 2010; 19:220-231; PMID:20708585; http://dx.doi.org/ 10.1016/j.devcel.2010.07.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Heyn P, Kircher M, Dahl A, Kelso J, Tomancak P, Kalinka AT, Neugebauer KM. The earliest transcribed zygotic genes are short, newly evolved, and different across species. Cell Rep 2014; 6:285-92; PMID:24440719; http://dx.doi.org/ 10.1016/j.celrep.2013.12.030 [DOI] [PubMed] [Google Scholar]
- 7. Lindeman LC, Andersen IS, Reiner AH, Li N, Aanes H, Østrup O, Winata C, Mathavan S, Müller F, Aleström P, Collas P. Prepatterning of developmental gene expression by modified histones before zygotic genome activation. Dev Cell 2011; 21:993-1004; PMID:22137762; http://dx.doi.org/ 10.1016/j.devcel.2011.10.008 [DOI] [PubMed] [Google Scholar]
- 8. Newport J, Kirschner M. A major developmental transition in early Xenopus embryos: II. Control of the onset of transcription. Cell 1982; 30:687-96; PMID:7139712; http://dx.doi.org/ 10.1016/0092-8674(82)90273-2 [DOI] [PubMed] [Google Scholar]
- 9. Kimelman D, Kirschner M, Scherson T. The events of the midblastula transition in Xenopus are regulated by changes in the cell cycle. Cell 1987; 48:399-407; PMID:3802197; http://dx.doi.org/ 10.1016/0092-8674(87)90191-7 [DOI] [PubMed] [Google Scholar]
- 10. Shermoen AW, O’Farrell PH. Progression of the cell cycle through mitosis leads to abortion of nascent transcripts. Cell 1991; 67:303-310; PMID:1680567; http://dx.doi.org/ 10.1016/0092-8674(91)90182-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tadros W, Lipshitz HD. The maternal-to-zygotic transition: a play in two acts. Development 2009; 136:3033-42; PMID:19700615; http://dx.doi.org/ 10.1242/dev.033183 [DOI] [PubMed] [Google Scholar]
- 12. McHale P, Mizutani CM, Kosman D, MacKay DL, Belu M, Hermann A, McGinnis W, Bier E, Hwa T. Gene length may contribute to graded transcriptional responses in the Drosophila embryo. Dev Biol 2011; 360:230-40; PMID:21920356;. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Collart C, Allen GE, Bradshaw CR, Smith JC, Zegerman P. Titration of four replication factors is essential for the Xenopus laevis midblastula transition. Science 2013; 341:893-6; PMID:23907533; http://dx.doi.org/ 10.1126/science.1241530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tadros W, Goldman AL, Babak T, Menzies F, Vardy L, Orr-Weaver T, Hughes TR, Westwood JT, Smibert C a, Lipshitz HD. SMAUG is a major regulator of maternal mRNA destabilization in Drosophila and its translation is activated by the PAN GU kinase. Dev Cell 2007; 12:143-55; PMID:17199047; http://dx.doi.org/ 10.1016/j.devcel.2006.10.005 [DOI] [PubMed] [Google Scholar]
- 15. Kappas NC, Savage P, Chen KC, Walls AT, Sible JC. Dissection of the XChk1 signaling pathway in Xenopus laevis embryos. Mol Biol Cell 2000; 11:3101-8; PMID:10982403; http://dx.doi.org/ 10.1091/mbc.11.9.3101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shimuta K, Nakajo N, Uto K, Hayano Y, Okazaki K, Sagata N. Chk1 is activated transiently and targets Cdc25A for degradation at the Xenopus midblastula transition. EMBO J 2002; 21:3694-703; PMID:12110582; http://dx.doi.org/ 10.1093/emboj/cdf357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sung H-W, Spangenberg S, Vogt N, Großhans J. Number of nuclear divisions in the Drosophila blastoderm controlled by onset of zygotic transcription. Curr Biol 2013; 23:133-8; PMID:23290555; http://dx.doi.org/ 10.1016/j.cub.2012.12.013 [DOI] [PubMed] [Google Scholar]
- 18. Farrell J. a, O’Farrell PH. Mechanism and regulation of cdc25/twine protein destruction in embryonic cell-cycle remodeling. Curr Biol 2013; 23:118-26; PMID:23290551; http://dx.doi.org/ 10.1016/j.cub.2012.11.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Di Talia S, She R, Blythe S a, Lu X, Zhang QF, Wieschaus EF. Posttranslational control of Cdc25 degradation terminates Drosophila's early cell-cycle program. Curr Biol 2013; 23:127-32; PMID:23290553; http://dx.doi.org/ 10.1016/j.cub.2012.11.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ikegami R., Rivera-Bennetts A., Brooker D., Yager T. (University of T . Effect of inhibitors of DNA replication on early zebrafish embryos: evidence for coordinate activation of multiple intrinsic cell-cycle checkpoints at the mid-blastula transition. Zygote 1997:153-175; PMID:9276512; . [DOI] [PubMed] [Google Scholar]
- 21. Hensey C, Gautier J. A developmental timer that regulates apoptosis at the onset of gastrulation. Mech Dev 1997; 69:183-195; PMID:9486540; http://dx.doi.org/ 10.1016/S0925-4773(97)00191-3 [DOI] [PubMed] [Google Scholar]
- 22. Conn CW, Lewellyn AL, Maller JL. The DNA damage checkpoint in embryonic cell cycles is dependent on the DNA-to-cytoplasmic ratio. Dev Cell 2004; 7:275-81; PMID:15296723; http://dx.doi.org/ 10.1016/j.devcel.2004.07.003 [DOI] [PubMed] [Google Scholar]
- 23. Katsuragi Y, Sagata N. Regulation of Chk1 Kinase by Autoinhibition and ATR-mediated Phosphorylation. Mol Biol Cell 2004; 15(April):1680-1689; PMID:14767054; http://dx.doi.org/ 10.1091/mbc.E03-12-0874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kosoy A, Connell MJO. Regulation of Chk1 by Its C-terminal Domain. Mol Biol Cell 2008; 19(November):4546-4553; PMID:18716058; http://dx.doi.org/ 10.1091/mbc.E08-04-0444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nakajo N, Oe T, Uto K, Sagata N. Involvement of Chk1 kinase in prophase I arrest of Xenopus oocytes. Dev Biol 1999; 207:432-44; PMID:10068474; http://dx.doi.org/ 10.1006/dbio.1998.9178 [DOI] [PubMed] [Google Scholar]
- 26. Caparelli ML, O’Connell MJ. Regulatory motifs in Chk1. Cell Cycle 2013; 12:916-22; PMID:23422000; http://dx.doi.org/ 10.4161/cc.23881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Oe T, Nakajo N, Katsuragi Y, Okazaki K, Sagata N. Cytoplasmic occurrence of the Chk1/Cdc25 pathway and regulation of Chk1 in Xenopus oocytes. Dev Biol 2001; 229:250-61; PMID:11133168; http://dx.doi.org/ 10.1006/dbio.2000.9968 [DOI] [PubMed] [Google Scholar]
- 28. Dekens MPS. The maternal-effect gene futile cycle is essential for pronuclear congression and mitotic spindle assembly in the zebrafish zygote. Development 2003; 130:3907-3916; PMID:12874114; http://dx.doi.org/ 10.1242/dev.00606 [DOI] [PubMed] [Google Scholar]
- 29. Dalle Nogare DE, Pauerstein PT, Lane ME. G2 acquisition by transcription-independent mechanism at the zebrafish midblastula transition. Dev Biol 2009; 326:131-42; PMID:19063878; http://dx.doi.org/ 10.1016/j.ydbio.2008.11.002 [DOI] [PubMed] [Google Scholar]
- 30. Stracker TH, Usui T, Petrini JHJ. Taking the time to make important decisions: the checkpoint effector kinases Chk1 and Chk2 and the DNA damage response. DNA Repair (Amst) 2009; 8:1047-54; PMID:19473886; http://dx.doi.org/ 10.1016/j.dnarep.2009.04.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nogare DED, Arguello A, Sazer S, Lane ME. Zebrafish cdc25a is expressed during early development and limiting for post-blastoderm cell cycle progression. Dev Dyn 2007; 236:3427-35; PMID:17969147; http://dx.doi.org/ 10.1002/dvdy.21363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Meinecke B, Meinecke-Tillmann S (Justus-L-UG). Effects of α-amanitin on nuclear maturation of porcine oocytes in vitro. J Reprod Fertil 1993:195-201; PMID:8345464; http://dx.doi.org/ 10.1530/jrf.0.0980195 [DOI] [PubMed] [Google Scholar]
- 33. Shechter D, Costanzo V, Gautier J. Regulation of DNA replication by ATR: signaling in response to DNA intermediates. DNA Repair (Amst) 2004; 3:901-8; PMID:15279775; http://dx.doi.org/ 10.1016/j.dnarep.2004.03.020 [DOI] [PubMed] [Google Scholar]
- 34. Paulson JR, Taylor SS. Phosphorylation of histones 1 and 3 and nonhistone high mobility group 14 by an endogenous kinase in HeLa metaphase chromosomes. J Biol Chem 1982; 257:6064-6072; PMID:6281254; . [PubMed] [Google Scholar]
- 35. Paull TT, Rogakou EP, Yamazaki V, Kirchgessner CU, Gellert M, Bonner WM. A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Curr Biol 2000; 10:886-895; PMID:10959836; http://dx.doi.org/ 10.1016/S0960-9822(00)00610-2 [DOI] [PubMed] [Google Scholar]
- 36. Gao N, Davuluri G, Gong W, Seiler C, Lorent K, Furth EE, Kaestner KH, Pack M. The nuclear pore complex protein Elys is required for genome stability in mouse intestinal epithelial progenitor cells. Gastroenterology 2011; 140:1547-55.e10; PMID:21315719; http://dx.doi.org/ 10.1053/j.gastro.2011.01.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhao H, Piwnica-Worms H. ATR-Mediated Checkpoint Pathways Regulate Phosphorylation and Activation of Human Chk1. Mol Cell Biol 2001; 21:4129-4139; PMID:11390642; http://dx.doi.org/ 10.1128/MCB.21.13.4129-4139.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sibon OC, Stevenson V. a, Theurkauf WE. DNA-replication checkpoint control at the Drosophila midblastula transition. Nature 1997; 388:93-7; PMID:9214509; http://dx.doi.org/ 10.1038/40439 [DOI] [PubMed] [Google Scholar]
- 39. Takada S, Kwak S, Koppetsch BS, Theurkauf WE. grp (chk1) replication-checkpoint mutations and DNA damage trigger a Chk2-dependent block at the Drosophila midblastula transition. Development 2007; 134:1737-44; PMID:17409117; http://dx.doi.org/ 10.1242/dev.02831 [DOI] [PubMed] [Google Scholar]
- 40. Swinburne IA, Silver PA. Intron delays and transcriptional timing during development. Dev Cell 2008:324-330; PMID:18331713; http://dx.doi.org/ 10.1016/j.devcel.2008.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Stancheva I, Meehan RR. Xenopus embryos Transient depletion of xDnmt1 leads to premature gene activation in Xenopus embryos. 2000:313-327. [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 42. Pritchard DK, Schubiger G. Activation of transcription in Drosophila embryos is a gradual process mediated by the nucleocytoplasmic ratio. Genes Dev 1996; 10:1131-1142; PMID:8654928; http://dx.doi.org/ 10.1101/gad.10.9.1131 [DOI] [PubMed] [Google Scholar]
- 43. Benoit B, He CH, Zhang F, Votruba SM, Tadros W, Westwood JT, Smibert C a, Lipshitz HD, Theurkauf WE. An essential role for the RNA-binding protein Smaug during the Drosophila maternal-to-zygotic transition. Development 2009; 136:923-32; PMID:19234062; http://dx.doi.org/ 10.1242/dev.031815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pérez-Montero S, Carbonell A, Morán T, Vaquero A, Azorín F. The embryonic linker histone H1 variant of Drosophila, dBigH1, regulates zygotic genome activation. Dev Cell 2013; 26:578-90; http://dx.doi.org/ 10.1016/j.devcel.2013.08.011 [DOI] [PubMed] [Google Scholar]
- 45. Collart C, Owens NDL, Bhaw-Rosun L, Cooper B, De Domenico E, Patrushev I, Sesay AK, Smith JN, Smith JC, Gilchrist MJ. High-resolution analysis of gene activity during the Xenopus mid-blastula transition. Development 2014; 141:1927-39; PMID:24757007; http://dx.doi.org/ 10.1242/dev.102012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mathavan S, Lee SGP, Mak A, Miller LD, Murthy KRK, Govindarajan KR, Tong Y, Wu YL, Lam SH, Yang H, et al. Transcriptome analysis of zebrafish embryogenesis using microarrays. PLoS Genet 2005; 1:260-76; PMID:16132083; http://dx.doi.org/ 10.1371/journal.pgen.0010029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Xu B, Kim S, Lim D, Kastan MB. Two Molecularly Distinct G 2 / M Checkpoints Are Induced by Ionizing Irradiation Two Molecularly Distinct G 2 / M Checkpoints Are Induced by Ionizing Irradiation. Mol Cell Biol 2002; 22:1049-1059; PMID:11809797; http://dx.doi.org/ 10.1128/MCB.22.4.1049-1059.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Brown EJ, Baltimore D. Essential and dispensable roles of ATR in cell cycle arrest and genome maintenance. Genes Dev 2003; 17:615-28; PMID:12629044; http://dx.doi.org/ 10.1101/gad.1067403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Cimprich K a, Cortez D. ATR: an essential regulator of genome integrity. Nat Rev Mol Cell Biol 2008; 9:616-27; PMID:18594563; http://dx.doi.org/ 10.1038/nrm2450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Gotoh T, Kishimoto T, Sible JC. Phosphorylation of Claspin is triggered by the nucleocytoplasmic ratio at the Xenopus laevis midblastula transition. Dev Biol 2011; 353:302-8; PMID:21396931; http://dx.doi.org/ 10.1016/j.ydbio.2011.03.002 [DOI] [PubMed] [Google Scholar]
- 51. Gotoh T, Ohsumi K, Matsui T, Takisawa H, Kishimoto T. Inactivation of the checkpoint kinase Cds1 is dependent on cyclin B-Cdc2 kinase activation at the meiotic G(2)/M-phase transition in Xenopus oocytes. J Cell Sci 2001; 114(Pt 18):3397-406; PMID:11591827; . [DOI] [PubMed] [Google Scholar]
- 52. Newport J, Dasso M. On the coupling between DNA replication and mitosis. J Cell Sci Suppl 1989; 12:149-60; PMID:2517560; http://dx.doi.org/ 10.1242/jcs.1989.Supplement_12.13 [DOI] [PubMed] [Google Scholar]
- 53. Pogoriler J, Du W. Chk1 Activation and the Nuclear / Cytoplasmic Ratio. Dev Cell 2004:147-148; PMID:15296709; http://dx.doi.org/ 10.1016/j.devcel.2004.07.015 [DOI] [PubMed] [Google Scholar]
- 54. Sidi S, Sanda T, Kennedy RD, Hagen AT, Jette C a, Hoffmans R, Pascual J, Imamura S, Kishi S, Amatruda JF, et al. Chk1 suppresses a caspase-2 apoptotic response to DNA damage that bypasses p53, Bcl-2, and caspase-3. Cell 2008; 133:864-77; PMID:18510930; http://dx.doi.org/ 10.1016/j.cell.2008.03.037 [DOI] [PMC free article] [PubMed] [Google Scholar]