

Abstract
In the last decade taxane-based therapy has emerged as a standard of care for hormone-refractory prostate cancer. Nevertheless, a significant fraction of tumors show no appreciable response to the treatment, while the others develop resistance and recur. Despite years of intense research, the mechanisms of taxane resistance in prostate cancer and other malignancies are poorly understood and remain a topic of intense investigation. We have used improved mutagenesis via random insertion of a strong promoter to search for events, which enable survival of prostate cancer cells after Taxol exposure. High-throughput mapping of the integration sites pointed to the PRKAR2A gene, which codes for a type II-α regulatory subunit of protein kinase A, as a candidate modulator of drug response. Both full-length and N-terminally truncated forms of the PRKAR2A gene product markedly increased survival of prostate cancer cells lines treated with Taxol and Taxotere. Suppression of protein kinase A enzymatic activity is the likely mechanism of action of the overexpressed proteins. Accordingly, protein kinase A inhibitor PKI (6–22) amide reduced toxicity of Taxol to prostate cancer cells. Our findings support the role of protein kinase A and its constituent proteins in cell response to chemotherapy.
Keywords: docetaxel, doxorubicin, forward genetics, insertional mutagenesis, paclitaxel, prostate cancer, protein kinase A, vincristine
Abbreviations
- miRNA
micro RNA
- PKA
Protein kinase A (aka cAMP-Dependent Protein Kinase)
- PPKAR2A
cAMP-Dependent Protein Kinase Regulatory Subunit RII Alpha
- shRNA
short hairpin RNA
Introduction
Taxanes remain among the most commonly used chemotherapeutic agents. Taxanes have a unique mechanism of action: they stabilize microtubules, essentially “freezing” mitosis and some forms of intracellular transport.1 Higher mitotic rates and inefficiency of protective cell cycle checkpoints may contribute to the sensitivity of cancer cells to such treatments. Paclitaxel (Taxol) and docetaxel (Taxotere) have shown efficacy against a wide range of malignancies. For example, docetaxel was the first chemotherapeutic agent that significantly extend the life of patients with hormone-refractory metastatic disease.2,3
Unfortunately, a significant number of tumors show no response, and most of those that respond initially, eventually develop resistance to the drug. In prostate cancer, taxanes fail to achieve sustained response in approximately half the cases, and when the response is achieved, its median duration is limited to 6–9 months.4 This situation provides the impetus for intense research into the mechanisms of cancer cell resistance to taxanes.
The best characterized mechanism of Taxol and Taxotere resistance is efflux of the drugs. This could be achieved via overexpression of molecular pumps, most notably—of p-glycoprotein (aka MDR1 or ABCB1).5,6 However, the available data on the elevated expression of these pumps cannot explain the high incidence of resistance. In fact, there are conflicting reports on whether MDR1 expression increases or declines during the natural progression of prostate cancer.7,8 Even when expression of transporters is elevated, it is not a proof of their involvement in resistance. Indeed, the therapeutic compounds, which could be effluxed by such pumps, demonstrate increased toxicity, but hardly gain any additional clinical efficacy in the presence of efflux inhibitors.9,10 Furthermore, the new generation of taxanes may overcome the issues of efflux, but fail to avoid eventual emergence of resistant cancers, while causing severe side effects.11
Another family of resistance mechanisms may arise from the alterations in tubulins, including changes in relative abundance of various tubulin classes as well as mutations in the corresponding genes. While we are not aware of drug-resistant mutants of tubulin emerging in prostate cancer, overexpression of class III β-tubulin in this disease is statistically associated with poor response to docetaxel.12 The interpretation, however, is somewhat complicated by the fact that high expression of this protein is associated with transdifferentiation and altered proliferation of prostate cancer cells; the overexpressing tumors show significantly more aggressive features prior to treatment, and a response to therapy is observed in some overexpressing cases.12
Programmed cell death is one of the outcomes of taxane exposure, so one might expect that anti-apoptotic Bcl-2 family members13 would provide some degree of protection. Indeed, elevated activity of Bcl-2 has been reported to reduce acute toxicity of the drug in some cell lines.14 However, clinical relevance of this mechanism in prostate cancer is still uncertain, and it has also been reported that Taxanes efficiently suppress the function of the endogenous Bcl-2.15 Moreover, one would expect that castration-resistant prostate cancer, which is the form commonly treated with taxanes, has already been selected for decreased sensitivity to apoptosis. Of note, inhibition of Bcl-2 caused severe side effects, but failed to increase the efficacy of docetaxel.16 Indeed, the prostate cancer patients with elevated Bcl-2 expression were reported to benefit the most from Taxol treatment.17
It is important to note that the current knowledge of taxane-resistance mechanisms originates predominantly from in vitro experiments on cell lines. Some of these experiments are limited to measuring cell number or a surrogate (e.g. activity of certain enzymes) after a continuous short-term exposure to the drug. Therefore, such observations are insufficient to predict long-term clonogenic survival of cells. Other experiments select for resistant clones and score cell survival upon continuous long-term exposure to very high doses of the compound. Under this setup, the only mutations that could render effective protection are those that provide absolute insensitivity to the drug. Not surprisingly, such a selection commonly results in overexpression of transporters18 and mutations in tubulin, the latter of which decrease tubulin polymerization and make cells dependent on the presence of the drug.19 Interestingly, the inhibitory action of taxanes on cancer cells may differ significantly at different doses. Higher doses are likely to induce an absolute block of mitosis, while the moderate ones would permit some “slippage” of affected cells into G1. Importantly, higher doses are not necessarily better in inducing cells death.20 Depending on the genetic makeup and the environment, individual cells differ in the propensity to arrest and survive in G2, to “slip” into G1, to sustain and tolerate the damage incurred during “slippage,” and to resume proliferation after withdrawal of the drug.21,22 These differences may be obscured in long-term continuous high-dose exposure experiments, but are likely to affect the effectiveness of real-life taxane treatment, which typically consists of a series of several injections spaced by 7 or more days and followed by complete drug withdrawal. To this end, we have set forth to implement a selection scheme, which could identify the genes whose products mediate such differences.
Results
In order to identify new mechanisms of Taxol resistance, we relied on insertional mutagenesis with a construct (Fig. 1) that randomly inserts a strong regulated promoter throughout the genome. The promoter drives transcription into the adjacent host DNA, generating a hybrid mRNA. Depending on the site and orientation of the insert, an integration event may lead to overexpression of the nearby gene or its partial products. After a mutant with the phenotype of interest is selected, the affected gene remains marked by the insert and can be readily identified.23 Appropriate vector design favors splicing of vector- and host-derived sequences, enhancing the yield of phenotypically discernable mutants. This approach was successfully used to interrogate mammalian signaling pathways before,24–26 but suffers from the need to isolate and separately characterize many individual clones of the presumed mutants.
Figure 1.
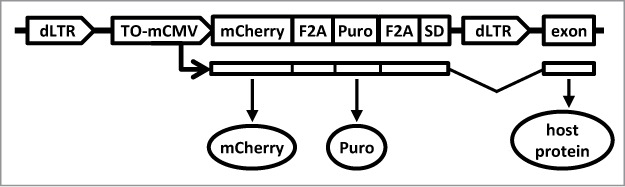
The structure of the insertional mutagen. The HIV1-based construct is shown as a provirus integrated collinearly with a host gene. The provirus is flanked by promoter-deficient LTRs (“dLTR”). The internal promoter (“TO-mCMV”) is composed of tet-operators and a minimal CMV promoter. In the presence of an active tetracycline trasnactivator protein, it drives expression of the red fluorescent protein mCherry (“mCherry”) and puromycin N-acetyl-transferase (“puro”). Utilization of a splice-donor site (“SD”) permits fusion with downstream exons. The proteins encoded by the hybrid transcript are produced as separate peptides due to the presence of picornavirus F2A sequences between the respective coding regions.
Importantly, because of the random nature of insertional mutagenesis, the junction between vector- and host-derived sequences is different for each individual mutant, presenting a unique “tag.” Theoretically, such tags can be readily recovered, either from the DNA or of the RNA of the mutagenized cells. Furthermore, while the sequence of a tag reveals the site of integration, the changes in the relative abundance of individual tags could be used to judge the relative fitness to the corresponding mutants. For example, by comparing pools of tags from the same mutagenized population with and without drug selection, one should be able to predict, which insertional events enhance resistance. Such enrichment analysis should be feasible without isolation of individual clones. This should be especially helpful when stringency of selection is relatively low and a large number of surviving clones is expected.
The insertional mutagen used in this study (Fig. 1) is constructed on HIV-1 backbone, with self-inactivating LTRs and an internal tetracycline-regulated promoter, which controls expression of a puromycin resistance marker and a fluorescent protein. The transcript, which is driven by the regulated promoter, also contains an unpaired splice donor site. As was shown before,27 this design permits read-through transcription past the LTR and “trapping” of downstream exons of a host genes. Inclusion of F2A sequence ensures that a peptide produced from the host portion of the hybrid RNA would be detached from vector-encoded markers during translation.28
We have chosen LNCaP cells as the model system, since the cell line was established from a relatively early state metastatic disease and, in comparison with more evolved tumors, might be open to acquiring a more diverse set of protective alterations. These cells are also free from contaminating viruses, which might complicate the experiments in some other prostate cancer models.29,30
In order to support the function of the regulated promoter, we used a derivative of these cells (LNGK9), which expresses tetracycline-regulated transactivator protein.31 A pool of these cells was infected with the insertional mutagen (∼25% infection rate) and subjected a one-time treatment with 11nM Taxol. After 5 days, the cells were transferred to a Taxol-free medium and allowed to recover. The recovered cells were expanded, and Taxol treatment was repeated. DNA and RNA were extracted from the cells after recovery from the second treatment. The host-vector fusion products were recovered and sequenced. While the RNA- and the DNA-based mapping approaches have their advantages and limitations, both revealed that a mutant with an insert in the first intron of PRKAR2A gene (Fig. 2A) is prevalent in the selected population.
Figure 2.

PRKAR2A affects Taxol resistance. (A) Schematic illustration of the insert in the PRKAR2A gene in Taxol-resistant cells. The sequences recovered through DNA- and RNA-based mapping of insertional events are shown below and above the drawing respectively. (B) Full-length and truncated PRKAR2A gene products protect LNGK9 cells from Taxol. The cells harboring PRKAR2A variants or an empty vector were treated with 11nM Taxol for 5 days, and allowed 1 week to recover. Cell numbers were assessed by methylene blue staining and expressed as fold increase over vector –infected control. The graph shows cumulative results of 4 independent experiments. Each box shows the range from the first to third quartiles and is divided by the median. The error bars depict the range. The geometric mean of each data set is denoted by a black diamond. (C) Representative image of cell staining following Taxol treatment and recovery for LNGK9 cells expressing PRKAR2A variants or the empty vector. Twenty thousand cells were plated per well of a 6-well plate, treated and stained as in (B). (D) DU145 cells were treated with 13nM Taxol for 5 days, and allowed 5 d to recover before cell numbers were assessed by methylene blue staining. The fold change imparted by PRKAR2A expression versus control cells expressing the empty vector was graphed. (E) LNGK9 cells were treated with 10nM Taxol for 5 days, allowed 1 week to recover and then challenged a second time by 11nM Taxol for 5 days. Following another recovery period of one week cell number was assessed by methylene blue staining, and the fold change imparted by PRKAR2A expression vs. empty vector-transduced LNGK9 cells that were not pre-selected on Taxol was graphed for each treatment. In panels D and E, the average results of 3 independent experiments are shown and the error bars depict standard deviations.
As discussed elsewhere,27 the presence of an insert in the proximity of a known gene is not enough to prove the relevance of the gene to the altered phenotype of the mutant clone. Thus, we proceeded to functional confirmation of the candidate Taxol-resistance gene. We established LNGK9 cells transduced with a full-length PRKAR2A expression construct, a construct expressing truncated PRKAR2A (missing the N-terminus of the protein, which is encoded by the first exon), and the respective empty-vector control. Both PRKAR2A variants greatly increased the number of colonies that were formed by LNGK9 cells upon recovery from Taxol exposure (Fig. 2B, C). This effect was not limited to LNGK9 and was seen in DU145 cells as well (Fig. 2D).
Of note, only a small minority (typically, <1%) of PRKAR2A-expressing cells would form colonies after the withdrawal of the drugs in our experimental conditions. We considered 2 explanations for this phenomenon. First, in order to provide protection from the direct effect of the drug, a transgene may need cooperation from additional genetic events or be expressed only at a particular level. Second, a transgene may not protect a cell from the primary impact of the drug, but may increase the chances of survival and recovery of the drug-affected cells. The first scenario implies that a PRKAR2A-transduced population, which had recovered from one round of Taxol exposure, should be greatly enriched for well-protected cells, and would resist a subsequent Taxol challenge en mass. If the second scenario applies, one would expect the recovered populations to show only a modest, if any, additional increase in resistance, with most of the cells still remaining vulnerable to the drug. Indeed, the latter was observed (Fig. 2E), suggesting that most of the transgene-bearing cells remain vulnerable to the drug despite surviving a prior treatment. Moreover, the PRKAR2A-expressing cells showed as strong a cell cycle arrest as the control LNGK9 cells following Taxol treatment (Fig. 3A and data not shown). This is also consistent with the cells experiencing the same primary effects of the drug. Furthermore, although PRKAR2A was previously implicated in intracellular transport of p-glycoprotein,32 we did not detect any significant difference between PRKAR2A-expressing and control cells in their ability to accumulate or efflux fluorescently-labeled Taxol (Fig. 3B and data not shown).
Figure 3.

PRKAR2A expression does not alter cell cycle progression or drug efflux. (A) Cell cycle profiling of PRKAR2A-expressing LNGK9 cells treated with 11nm Taxol for 48 hours. The cells were stained with propidium iodide and subpopulations are shown next to untreated controls. (B) LNGK9 cells were treated with Bodipy-labeled Taxol for 45 minutes, then washed and incubated for 5 h to allow for Taxol efflux. Residual fluorescence was measured for each of the PRKAR2A variants. The histograms display unstained cells in white, unwashed stained cells in black, and those which were washed in gray.
The effect of the transgenes was not limited to Taxol. Strong protection was seen against Taxotere (Fig. 4A), and, to a smaller degree, against vincristine and doxorubicin (Fig. 4B-C). No significant protection was observed in irradiated or cisplatin-treated cells (Fig. 4D-E), confirming that PRKAR2A is not a generic suppressor of cell death.
Figure 4.
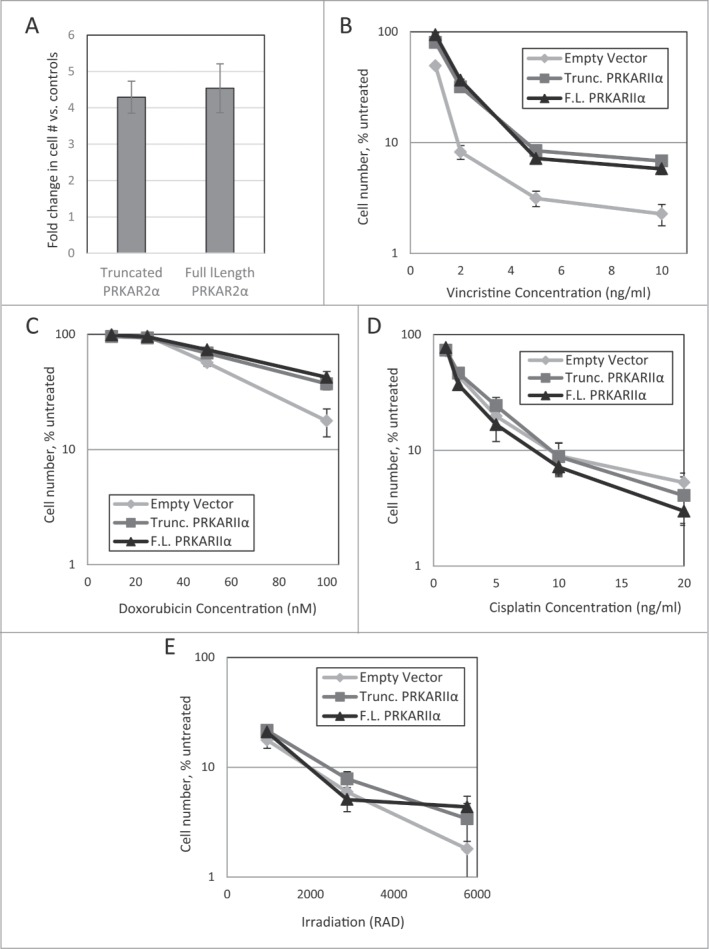
Effect of PRKAR2A expression on response to cytotoxic impacts. LNGK9 cell variants were treated with (A) 1nM Taxotere (5-day treatment, 1 week recovery), (B) vincristine (5 day treatment, 4 day recovery), (C) doxorubicin (2 day treatment, 4 day recovery), (D) cisplatin (1 day treatment, 3-day recovery), or (E) gamma-irradiation (3 day recovery), after which, cell number was assessed by methylene blue, and the change imparted by PRKAR2A variants was graphed versus untreated control cells. The average results of 3 independent experiments are shown and the error bars depict standard deviations.
The regulatory subunit of PKA serves to tether the enzyme to specific locations within the cell, as well as to keep the catalytic subunit inactive until an appropriate signal is received. The truncated form of PRKAR2A lacks the domain, which is usually associated with the tethering function, but retains the domain, which interacts with the catalytic subunit. Considering that the full-length and the truncated PRKAR2A variants have a similar effect on Taxol resistance, we hypothesized that the phenomenon is mediated by inhibition of PKA activity. Indeed, we found that low doses of a PKA inhibitor significantly increased recovery of cells following drug treatment (Fig. 5).
Figure 5.
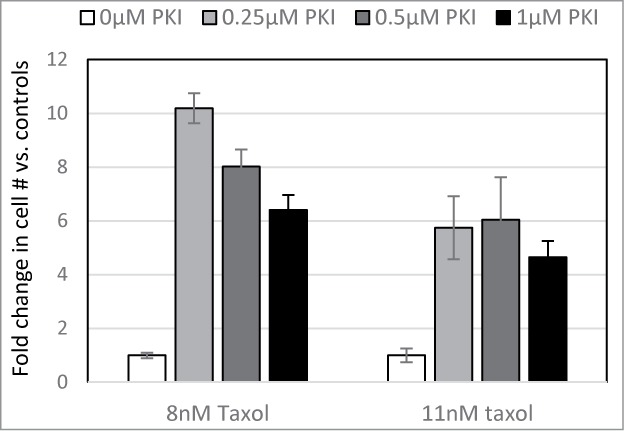
PKA inhibitor confers Taxol resistance. LNGK9 cells were treated with 0, 0.25, 0.5, or 1μM PKI in combination with 0, 8, or 11nM Taxol for 5 days. Following a 1 week recovery period, cell numbers were assessed by methylene blue staining and graphed as a fold change vs. untreated cells. The average results of 3 independent experiments are shown and the error bars depict standard deviations.
Discussion
The mechanisms of Taxol resistance have long been investigated using tools of forward genetics (e.g. refs.18,33,34), with newer attempts often reiterating older findings (e.g. the protective role of MDR1).18 Our experimental system is distinct on 2 accounts. First, it is based on prostate cancer cells. While some mechanisms of drug response may be shared between cancers of various tissue origins, this does not always have to be the case. Second, our system was deliberately set up to permit identification of events that improve recovery of cells upon drug withdrawal. In this regard, our system is similar to the one that was used recently to select for shRNAs that protect HeLa cells from a short-term paclitaxel treatment.35,36 This approach contrasts with many other studies, which have selected cells for the ability to grow continuously in the presence of taxanes.
In addition, our study has benefitted from the use of high-throughput sequencing, which greatly simplifies mapping of the integration sites. Both DNA- and RNA-based detection of vector-host fusion products were successful, and the presence of a mutant with an insert in the PRKAR2A gene was revealed by both techniques (Fig. 3). It should be noted that the DNA-based approach has a drawback of frequently encountering repetitive sequences, which cannot be unambiguously mapped to the genome (data not shown). On the other hand, it may reflect the composition of the pool of mutants better than the RNA-based approach, which is inherently dependent on the relative levels of expression of the fusion products. Of course, both approaches might encounter difficulties in mapping some individual inserts due to unfavorable position of specific restriction sites in genomic DNA (too close or too far from the insert) or due to the nucleotide composition of the adjacent host DNA, which may be unfavorable for efficient amplification.
The major bottleneck of the technology, however, remains functional confirmation of candidates. Recurrent presence of inserts in the same gene among the cells with a common phenotype may reflect an integration bias rather than a biological role of the gene in question.37 The bias of viral integration may depend on the species of the virus, and DNA sequence and the chromatin state of the target. At present, no statistical model can fully account for this phenomenon. As a result, the relevance of the identified genes has to be established from functional tests, rather than mere statistical arguments. Perhaps, the bulk of false-positives could be excluded by conducting a promoter-dependence test along with high-throughput insert mapping. For example, the same pool of cells could be treated in parallel in conditions that permit or restrict the function of the inserted promoter (e.g. in the presence and in the absence of tetracycline analogs in our experimental system). In this case, promoter-dependent mutants will be enriched only in “promoter on” condition, while spontaneous mutants will be enriched in both. Indeed, the relative abundance of PRKAR2A mutant was lower when the cells after the first Taxol selection were re-challenged with the drug in the presence of doxycycline (data not shown). We are currently exploring the other candidate Taxol-resistance genes, insertions into which display a similar pattern of behavior.
While overexpression of full-length or truncated PRKAR2A increases long-term survival of treated prostate cancer cells, the precise mechanism of this phenomenon remains to be elucidated. The N-terminal domain of PRKAR2A, which mediates dimerization and binding to protein kinase A anchoring proteins (AKAPs) and some other interactors,38 is dispensable for the protective effect. This suggests that the increased long-term survival in our experiments was mediated by the inhibitory interaction of PRKAR2A with a catalytic subunit of PKA. Under this scenario, the overexpressed subunit acts a de facto dominant-negative peptide, causing loss of the enzyme's function. This hypothesis is supported by the observation that a low dose of a PKA inhibitor also increases the survival of treated cells (Fig. 5).
Previously, the role of PKA in the response to taxane treatment was viewed primarily in the context of regulating the levels and activities of p-glycoprotein32 and the proteins of Bcl2 family.39,40 Consequently, anti-PKA therapy (based on antisense targeting of regulatory subunit PRKAR1A) was explored in combination with docetaxel for drug-resistant malignancies. Considerable side effects were seen in a Phase I trial, but not a single objective response was achieved.41 A direct comparison of these studies with our results is complicated by the differences in the roles of distinct PKA isoforms and their individual subunits in various cell compartments and in different cell types, and by possible artifacts of the inhibitors that are used to probe these systems (e.g. the off-target effects of anti-sense oligonucleotides). On the other hand, even a mild elevation in cAMP levels is promoting cell death in many cancer cell lines,42,43 suggesting a death-enhancing role for PKA.44 While apoptosis is not the only type of death promoted by taxanes,45 PKA has been reported to play a direct role in Taxol-induced apoptosis in a breast cancer cell line MDA-MB-435.46 It is possible that the same mechanisms also account for the role of PRKAR2A in taxane response in prostate cancer cells. Taken together, our findings and the published data suggest that the net results of modulating PKA activity in taxane response may vary, depending on whether p-glycoprotein is expressed or is inducible in a given cell, and on which specific PKA subunits are being targeted. This has to be considered in planning any future attempts to modulate PKA activity in taxane-treated patients.
Multiple prior studies were focused on discovering genes whose inhibition would sensitize cells to Taxol.47–50 On the other hand, our current report and the observations of others suggest that taxane resistance could also arise from inhibition of various proteins51–54 or miRNAs.55 We would like to speculate that a comprehensive screen for loss-of-function events, which protect from taxane toxicity, would likely reveal numerous genes in addition to the 2 (USP15 and SEPR10) that were previously found via shRNA library selection.35,36 The significance of such findings is not limited to understanding the routes to taxane resistance in cancer. There is also a considerable interest in developing strategies to protect normal cells from the toxicity of Taxol.56–58 It remains to be seen whether PKA inhibitors could be used in such applications. It is tempting to speculate that such agents may be of use in the cases when a tumor itself is expressing p-glycoprotein in a PKA-dependent manner. Hence, the same compound would protect most normal cells, while decreasing drug resistance of the tumor. In this regard, it is noteworthy that the loss of some PKA subunits is not only tolerated, but appears to be life-extending in animal models.59
It remains to be elucidated, which of the many possible PKA targets are responsible for sensitivity of prostate cancer cells to taxanes. The ongoing investigation of numerous additional genes, which were revealed as candidate determinants of Taxol resistance in our genetic screen (data not shown), might be able to shed a new light of this problem.
Materials and Methods
Cell culture and viral transduction
Tet-activator expressing LNCaP cells (LNGK931) and DU145 cells (ATCC) were maintained in Dulbecco's Modified Eagle Medium (DMEM) (Life Technologies) supplemented with 10% fetal bovine serum and 100 U/ml penicillin/streptomycin. Viral transduction was performed as previously.27
Drug treatment
The cells were plated at ∼20% confluence one day before being subjected to the indicated concentrations of Taxol (Sigma Aldrich), Taxotere (Selleck Chem), vincristine (Sigma Aldrich), cisplatin (Sigma Aldrich), or doxorubicin (Sigma Aldrich). When indicated, a protein kinase A inhibitor (PKI (6–22) amide60; Santa Cruz Biotechnology) was applied 1 hour before addition of the drugs. Following treatment, the cells were washed and allowed to recover in fresh growth media. At this time, the cells were fixed and their numbers were assessed using a methylene blue staining and extraction method.61 The values were normalized with relation to untreated controls and graphed as described in figure legends. In the course of this study, no difference in plating efficiency or growth rate was detected between the cells harboring empty vector and either of the PKRAR2A-expressing constructs (data not shown).
Gamma-irradiation
LNGK9 cells were plated at ∼20% confluence one day before being subjected to the indicated doses of radiation. Three days later, the cells were fixed, their numbers were assessed using a methylene blue staining and extraction method,61 and the values were graphed relatively to untreated controls.
Taxol efflux assay
The cells were treated with 5μM Taxol conjugated to Bodipy 564/570 (Life Technologies) for 45 minutes. The cells were then washed twice with PBS and incubated in fresh growth media for 5 hours before residual fluorescence was measured using flow cytometry. For flow cytometry, cells were collected, washed, fixed, stained, and analyzed on an LSRII flow cytometer (BD PharMingen) according to manufacturer's protocol.
Cell cycle analysis
Propidium iodide staining was performed as previously.62 Stained cells were analyzed on an LSRII flow cytometer (BD PharMingen), and the data were processed using WinList 8.0.
PRKAR2A plasmid construction
PRKAR2A gene was amplified from a PRKAR2A vector (Addgene) using the full length PRKAR2A forward primer, CTTATTGACTAGTGCCACCATGGGTAGCCACATCCAGATCCCG, and the PRKAR2A reverse primer, CCCAAGACAATTGCTACTGCCCGAGGTTGCC, or the truncated PRKAR2A forward primer, GCGTCAGACTAGTGCCACCATGGTCTGTGCTGAGACCTATAAC, with the aforementioned reverse primer. The resulting products were then digested with SpeI (New England Biolabs) and MunI (New England Biolabs). The inserts were ligated into pLM-CMV-mAktER-neo vector (a gift of Dr. P. Chumakov), which had been digested with SpeI and EcoRI (New England Biolabs).
Oligonucleotides and adapters used for insert mapping
Gex1MSup oligonucleotide (AGCAGAAGACGGCATACGAGAGGGCTCGAGCGGCGGCGGCAGGT) was annealed with Ad-Tsp-P-b (AATTACCTGCCGCC-NH2) or Ad-Mbo-P-b (GATCACCTGCCGCC) to form the Tsp509I- and MboI-specific adapter respectively. Bio-Fwd4788 (Bio-TEG-TCCCTCAGACCCTTTTAGTCA) and Bio-FwdT7 (Bio-TEG-AGTAATACGACTCACTATAGGGAGA) PCR primers were biotin-labeled to allow affinity purification. Additional PCR primers used were Gex1MS (AGCAGAAGAGGGCATACGAGA), Gex2M-SD (AATGATACGGCGACCACCGAGATCTTCCGCATCGCTGTCTGCGAG) and Gex2M-F4774 (AATGATACGGCGACCACCGAGAGTCAGTGTGGAAAATCTCTA). Primers GexSeqG (TCTGCGAGGGCCAGCTGTTG) and F4774 (TCCCTCAGACCCTTTTAGTCA) were used in sequencing reactions. The sites for Bio-Fwd4788, Bio-TEG-FwdT7, Gex2M-SD, Gex2M-F4774, F4788 and GexSeqG were pre-engineered into the insertional mutagen (Fig. 1).
Recovery and sequencing of vector-host RNA junctions
Total RNA from each sample (∼2 × 106 cells) was extracted with Trizol (Thermo-Fisher) using the manufacturer's protocol, and the polyadenylated RNA fraction was isolated using Dynabeads (Thermo-Fisher). The cDNA was synthesized using reagents and protocol from SuperScript double –stranded cDNA synthesis kit (Thermo-Fisher) and purified by Qiaquick PCR purification kit (Qiagen). Each ds cDNA sample was split into 2: one was treated with MboI, the other with Tsp509I (New England Biolabs). After purification using Qiaquick PCR purification kit, the digested DNA was ligated with the corresponding MboI- or Tsp509I-specific adapter using T4 DNA ligase (New England Biolabs). The reactions were stopped by heat inactivation (65°C for 15 min), then treated with SalI (to remove inter-spliced vector mRNA products) and ExoI nuclease (to remove non-ligated adaptors), mixed together and purified by Qiaquick PCR purification kit. Adapter-ligated cDNAs were amplified by Phusion HF Titanium DNA polymerase (Thermo-Fisher) and Bio-TEG-FwdT7 and Gex1MS primers. Amplified cDNA products were purified by Qiaquick PCR purification kit and MyOne Streptavidin Magnetic Beads (Thermo-Fisher) using manufacturers’ protocols. The adaptor-ligated cDNAs were eluted from magnetic beads at 95°C and amplified in the second round of nested PCR with Gex1MS and Gex2M-SD primers and Phusion HF DNA polymerase. The PCR products with sizes 140–300 bp were purified from an agarose gel by Qiaquick Gel Extraction Kit (Qiagen) and sequenced in HiSeq2000 (Illumina) single read flow cells with GexSeqG sequencing primer.
Recovery and sequencing of vector-host DNA junctions
Genomic DNA from each sample (∼2 × 106 cells) was extracted with DNeasy blood and tissue kit (Qiagen) and was split in 2 aliquots: one treated with MboI, the other—with Tsp509I. Each MboI- or Tsp509I-digested genomic DNA was purified using Qiaquick purification kit and ligated with the corresponding MboI- or Tsp509I-specific adapter using T4 DNA ligase. The reactions were stopped by heating (65°C for 15 min), then treated with ExoI nuclease (to remove non-ligated adaptors), mixed together and purified by Qiaquick PCR purification kit. Adapter-ligated DNA was amplified by Phusion HF Titanium DNA polymerase using Bio-TEG-F4788 and Gex1MS primers. The PCR products were purified by Qiaquick PCR purification kit and MyOne Streptavidin Magnetic Beads (Thermo-Fisher) using manufacturers’ protocols. The DNA was eluted from magnetic beads at 95°C and amplified in the second round of nested PCR with Gex1MS and Gex2M-F4774 primers and Phusion HF DNA polymerase. The PCR products with sizes 140–300 bp were purified from an agarose gel by Qiaquick Gel Extraction Kit and sequenced in HiSeq2000 single read flow cells with F4774 sequencing primer.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Drs. C.Porter and A.Gudkov for sharing cells and reagents.
Funding
The work was supported by grants from the Roswell Park Alliance Foundation and from the NIH IMAT program (R21CA137708) to E.S.K.
References
- 1. Higa GM. The microtubule as a breast cancer target. Breast Cancer (Tokyo, Japan) 2011; 18:103-19; PMID:20862571; http://dx.doi.org/ 10.1007/s12282-010-0224-7 [DOI] [PubMed] [Google Scholar]
- 2. Petrylak DP, Tangen CM, Hussain MH, Lara PN, Jr., Jones JA, Taplin ME, Burch PA, Berry D, Moinpour C, Kohli M, et al. Docetaxel and estramustine compared with mitoxantrone and prednisone for advanced refractory prostate cancer. N Engl J Med 2004; 351:1513-20; PMID:15470214; http://dx.doi.org/ 10.1056/NEJMoa041318 [DOI] [PubMed] [Google Scholar]
- 3. Tannock IF, de Wit R, Berry WR, Horti J, Pluzanska A, Chi KN, Oudard S, Theodore C, James ND, Turesson I, et al. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N Engl J Med 2004; 351:1502-12; PMID:15470213; http://dx.doi.org/ 10.1056/NEJMoa040720 [DOI] [PubMed] [Google Scholar]
- 4. Mathew P, Dipaola R. Taxane refractory prostate cancer. J Urol 2007; 178:S36-41; PMID:17644120; http://dx.doi.org/ 10.1016/j.juro.2007.04.032 [DOI] [PubMed] [Google Scholar]
- 5. Safa AR, Stern RK, Choi K, Agresti M, Tamai I, Mehta ND, Roninson IB. Molecular basis of preferential resistance to colchicine in multidrug-resistant human cells conferred by Gly-185–Val-185 substitution in P-glycoprotein. Proc Natl Acad Sci U S A 1990; 87:7225-9; PMID:1976255; http://dx.doi.org/ 10.1073/pnas.87.18.7225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ise W, Heuser M, Sanders K, Beck J, Gekeler V. P-glycoprotein-associated resistance to taxol and taxotere and its reversal by dexniguldipine-HCl, dexverapamil-HCl, or cyclosporin A. Int J Oncol 1996; 8:951-6; PMID:21544450 [DOI] [PubMed] [Google Scholar]
- 7. Bhangal G, Halford S, Wang J, Roylance R, Shah R, Waxman J. Expression of the multidrug resistance gene in human prostate cancer. Urol Oncol 2000; 5:118-21; PMID:10765019; http://dx.doi.org/ 10.1016/S1078-1439(99)00055-1 [DOI] [PubMed] [Google Scholar]
- 8. Henrique R, Oliveira AI, Costa VL, Baptista T, Martins AT, Morais A, Oliveira J, Jeronimo C. Epigenetic regulation of MDR1 gene through post-translational histone modifications in prostate cancer. BMC Genomics 2013; 14:898; PMID:24344919; http://dx.doi.org/ 10.1186/1471-2164-14-898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rago RP, Einstein A, Jr., Lush R, Beer TM, Ko YJ, Henner WD, Bubley G, Merica EA, Garg V, Ette E, et al. Safety and efficacy of the MDR inhibitor Incel (biricodar, VX-710) in combination with mitoxantrone and prednisone in hormone-refractory prostate cancer. Cancer Chemother Pharmacol 2003; 51:297-305; PMID:12721757 [DOI] [PubMed] [Google Scholar]
- 10. Yu M, Ocana A, Tannock IF. Reversal of ATP-binding cassette drug transporter activity to modulate chemoresistance: why has it failed to provide clinical benefit? Cancer Metastasis Rev 2013; 32:211-27; PMID:23093326; http://dx.doi.org/ 10.1007/s10555-012-9402-8 [DOI] [PubMed] [Google Scholar]
- 11. de Bono JS, Oudard S, Ozguroglu M, Hansen S, Machiels JP, Kocak I, Gravis G, Bodrogi I, Mackenzie MJ, Shen L, et al. Prednisone plus cabazitaxel or mitoxantrone for metastatic castration-resistant prostate cancer progressing after docetaxel treatment: a randomised open-label trial. Lancet 2010; 376:1147-54; PMID:20888992; http://dx.doi.org/ 10.1016/S0140-6736(10)61389-X [DOI] [PubMed] [Google Scholar]
- 12. Ploussard G, Terry S, Maille P, Allory Y, Sirab N, Kheuang L, Soyeux P, Nicolaiew N, Coppolani E, Paule B, et al. Class III beta-tubulin expression predicts prostate tumor aggressiveness and patient response to docetaxel-based chemotherapy. Cancer Res 2010; 70:9253-64; PMID:21045157; http://dx.doi.org/ 10.1158/0008-5472.CAN-10-1447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Braun F, de Carne Trecesson S, Bertin-Ciftci J, Juin P. Protect and serve: Bcl-2 proteins as guardians and rulers of cancer cell survival. Cell Cycle 2013; 12:2937-47; PMID:23974114; http://dx.doi.org/ 10.4161/cc.25972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Srivastava RK, Sasaki CY, Hardwick JM, Longo DL. Bcl-2-mediated drug resistance: inhibition of apoptosis by blocking nuclear factor of activated T lymphocytes (NFAT)-induced Fas ligand transcription. J Exp Med 1999; 190:253-65; PMID:10432288; http://dx.doi.org/ 10.1084/jem.190.2.253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Haldar S, Chintapalli J, Croce CM. Taxol induces bcl-2 phosphorylation and death of prostate cancer cells. Cancer Res 1996; 56:1253-5; PMID:8640809 [PubMed] [Google Scholar]
- 16. Sternberg CN, Dumez H, Van Poppel H, Skoneczna I, Sella A, Daugaard G, Gil T, Graham J, Carpentier P, Calabro F, et al. Docetaxel plus oblimersen sodium (Bcl-2 antisense oligonucleotide): an EORTC multicenter, randomized phase II study in patients with castration-resistant prostate cancer. Ann Oncol 2009; 20:1264-9; PMID:19297314; http://dx.doi.org/ 10.1093/annonc/mdn784 [DOI] [PubMed] [Google Scholar]
- 17. Yoshino T, Shiina H, Urakami S, Kikuno N, Yoneda T, Shigeno K, Igawa M. Bcl-2 expression as a predictive marker of hormone-refractory prostate cancer treated with taxane-based chemotherapy. Clin Cancer Res 2006; 12:6116-24; PMID:17062688; http://dx.doi.org/ 10.1158/1078-0432.CCR-06-0147 [DOI] [PubMed] [Google Scholar]
- 18. Chen L, Stuart L, Ohsumi TK, Burgess S, Varshney GK, Dastur A, Borowsky M, Benes C, Lacy-Hulbert A, Schmidt EV. Transposon activation mutagenesis as a screening tool for identifying resistance to cancer therapeutics. BMC Cancer 2013; 13:93; PMID:23442791; http://dx.doi.org/ 10.1186/1471-2407-13-93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Schibler MJ, Cabral F. Taxol-dependent mutants of Chinese hamster ovary cells with alterations in alpha- and beta-tubulin. J Cell Biol 1986; 102:1522-31; PMID:2870070; http://dx.doi.org/ 10.1083/jcb.102.4.1522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Das GC, Holiday D, Gallardo R, Haas C. Taxol-induced cell cycle arrest and apoptosis: dose-response relationship in lung cancer cells of different wild-type p53 status and under isogenic condition. Cancer Lett 2001; 165:147-53; PMID:11275363; http://dx.doi.org/ 10.1016/S0304-3835(01)00404-9 [DOI] [PubMed] [Google Scholar]
- 21. Giovinazzi S, Bellapu D, Morozov VM, Ishov AM. Targeting mitotic exit with hyperthermia or APC/C inhibition to increase paclitaxel efficacy. Cell Cycle 2013; 12:2598-607; PMID:23907120; http://dx.doi.org/ 10.4161/cc.25591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhu Y, Zhou Y, Shi J. Post-slippage multinucleation renders cytotoxic variation in anti-mitotic drugs that target the microtubules or mitotic spindle. Cell Cycle 2014; 13:1756-64; PMID:24694730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kandel ES, Stark GR. Forward genetics in mammalian cells. In: Sehgal PB, Levy DE, Hirano T, eds. Signal Transducers and Activators of Transcription (STATs): Activation and Biology. The Netherlands: Kluwer Academic Publishers, 2003 [Google Scholar]
- 24. Kandel ES, Lu T, Wan Y, Agarwal MK, Jackson MW, Stark GR. Mutagenesis by reversible promoter insertion to study the activation of NF-kappaB. Proc Natl Acad Sci U S A 2005; 102:6425-30; PMID:15851657; http://dx.doi.org/ 10.1073/pnas.0502463102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dasgupta M, Agarwal MK, Varley P, Lu T, Stark GR, Kandel ES. Transposon-based mutagenesis identifies short RIP1 as an activator of NFkappaB. Cell Cycle 2008; 7:2249-56; PMID:18635966; http://dx.doi.org/ 10.4161/cc.7.14.6310 [DOI] [PubMed] [Google Scholar]
- 26. Lu T, Jackson MW, Singhi AD, Kandel ES, Yang M, Zhang Y, Gudkov AV, Stark GR. Validation-based insertional mutagenesis identifies lysine demethylase FBXL11 as a negative regulator of NFkappaB. Proc Natl Acad Sci U S A 2009; 106:16339-44; PMID:19805303; http://dx.doi.org/ 10.1073/pnas.0908560106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Singhal R, Deng X, Chenchik AA, Kandel ES. Long-distance effects of insertional mutagenesis. PLoS One 2011; 6:e15832; PMID:21246045; http://dx.doi.org/ 10.1371/journal.pone.0015832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lengler J, Holzmuller H, Salmons B, Gunzburg WH, Renner M. FMDV-2A sequence and protein arrangement contribute to functionality of CYP2B1-reporter fusion protein. Anal Biochem 2005; 343:116-24; PMID:15955524; http://dx.doi.org/ 10.1016/j.ab.2005.05.004 [DOI] [PubMed] [Google Scholar]
- 29. Dong B, Silverman RH, Kandel ES. A natural human retrovirus efficiently complements vectors based on murine leukemia virus. PLoS One 2008; 3:e3144; PMID:18769545; http://dx.doi.org/ 10.1371/journal.pone.0003144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yang J, Battacharya P, Singhal R, Kandel ES. Xenotropic murine leukemia virus-related virus (XMRV) in prostate cancer cells likely represents a laboratory artifact. Oncotarget 2011; 2:358-62; PMID:21642749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gschwend JE, Fair WR, Powell CT. Evaluation of the tetracycline-repressible transactivator system for inducible gene expression in human prostate cancer cell lines. Prostate 1997; 33:166-76; PMID:9365544; http://dx.doi.org/ 10.1002/(SICI)1097-0045(19971101)33:3%3c166::AID-PROS4%3e3.0.CO;2-H [DOI] [PubMed] [Google Scholar]
- 32. Wojtal KA, de Vries E, Hoekstra D, van Ijzendoorn SC. Efficient trafficking of MDR1/P-glycoprotein to apical canalicular plasma membranes in HepG2 cells requires PKA-RIIalpha anchoring and glucosylceramide. Mol Biol Cell 2006; 17:3638-50; PMID:16723498; http://dx.doi.org/ 10.1091/mbc.E06-03-0230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. De S, Cipriano R, Jackson MW, Stark GR. Overexpression of kinesins mediates docetaxel resistance in breast cancer cells. Cancer Res 2009; 69:8035-42; PMID:19789344; http://dx.doi.org/ 10.1158/0008-5472.CAN-09-1224 [DOI] [PubMed] [Google Scholar]
- 34. Xu X, Leo C, Jang Y, Chan E, Padilla D, Huang BC, Lin T, Gururaja T, Hitoshi Y, Lorens JB, et al. Dominant effector genetics in mammalian cells. Nat Genet 2001; 27:23-9; PMID:11137994; http://dx.doi.org/ 10.1038/83717 [DOI] [PubMed] [Google Scholar]
- 35. Xu M, Takanashi M, Oikawa K, Tanaka M, Nishi H, Isaka K, Kudo M, Kuroda M. USP15 plays an essential role for caspase-3 activation during Paclitaxel-induced apoptosis. Biochem Biophys Res Commun 2009; 388:366-71; PMID:19665996; http://dx.doi.org/ 10.1016/j.bbrc.2009.08.015 [DOI] [PubMed] [Google Scholar]
- 36. Xu M, Takanashi M, Oikawa K, Nishi H, Isaka K, Yoshimoto T, Ohyashiki J, Kuroda M. Identification of a novel role of Septin 10 in paclitaxel-resistance in cancers through a functional genomics screen. Cancer Sci 2012; 103:821-7; PMID:22320903; http://dx.doi.org/ 10.1111/j.1349-7006.2012.02221.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Biffi A, Bartolomae CC, Cesana D, Cartier N, Aubourg P, Ranzani M, Cesani M, Benedicenti F, Plati T, Rubagotti E, et al. Lentiviral vector common integration sites in preclinical models and a clinical trial reflect a benign integration bias and not oncogenic selection. Blood 2011; 117:5332-9; PMID:21403130; http://dx.doi.org/ 10.1182/blood-2010-09-306761 [DOI] [PubMed] [Google Scholar]
- 38. Newlon MG, Roy M, Hausken ZE, Scott JD, Jennings PA. The A-kinase anchoring domain of type IIalpha cAMP-dependent protein kinase is highly helical. J Biol Chem 1997; 272:23637-44; PMID:9295304; http://dx.doi.org/ 10.1074/jbc.272.38.23637 [DOI] [PubMed] [Google Scholar]
- 39. Rohlff C, Glazer RI. Regulation of the MDR1 promoter by cyclic AMP-dependent protein kinase and transcription factor Sp1. Int J Oncol 1998; 12:383-6; PMID:9458366 [DOI] [PubMed] [Google Scholar]
- 40. Srivastava RK, Srivastava AR, Korsmeyer SJ, Nesterova M, Cho-Chung YS, Longo DL. Involvement of microtubules in the regulation of Bcl2 phosphorylation and apoptosis through cyclic AMP-dependent protein kinase. Mol Cell Biol 1998; 18:3509-17; PMID:9584191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Goel S, Desai K, Macapinlac M, Wadler S, Goldberg G, Fields A, Einstein M, Volterra F, Wong B, Martin R, et al. A phase I safety and dose escalation trial of docetaxel combined with GEM231, a second generation antisense oligonucleotide targeting protein kinase A R1alpha in patients with advanced solid cancers. Invest New Drugs 2006; 24:125-34; PMID:16683205; http://dx.doi.org/ 10.1007/s10637-006-2378-x [DOI] [PubMed] [Google Scholar]
- 42. McEwan DG, Brunton VG, Baillie GS, Leslie NR, Houslay MD, Frame MC. Chemoresistant KM12C colon cancer cells are addicted to low cyclic AMP levels in a phosphodiesterase 4-regulated compartment via effects on phosphoinositide 3-kinase. Cancer Res 2007; 67:5248-57; PMID:17545604; http://dx.doi.org/ 10.1158/0008-5472.CAN-07-0097 [DOI] [PubMed] [Google Scholar]
- 43. Zhang L, Insel PA. The pro-apoptotic protein Bim is a convergence point for cAMP/protein kinase A- and glucocorticoid-promoted apoptosis of lymphoid cells. J Biol Chem 2004; 279:20858-65; PMID:14996839; http://dx.doi.org/ 10.1074/jbc.M310643200 [DOI] [PubMed] [Google Scholar]
- 44. Coley AM, Moujalled D, Puthalakath H. The PKA paradox: is Bim the answer? Cell Cycle 2011; 10:729-30; PMID:21311230; http://dx.doi.org/ 10.4161/cc.10.5.14869 [DOI] [PubMed] [Google Scholar]
- 45. Blagosklonny MV, Robey RW, Sheikh MS, Fojo T. Paclitaxel-Induced FasL-Independent Apoptosis and Slow (Non-Apoptotic) Cell Death. Cancer Biol Ther 2002; 1:113-7; PMID:12170770; http://dx.doi.org/ 10.4161/cbt.53 [DOI] [PubMed] [Google Scholar]
- 46. Reshkin SJ, Bellizzi A, Cardone RA, Tommasino M, Casavola V, Paradiso A. Paclitaxel induces apoptosis via protein kinase A- and p38 mitogen-activated protein-dependent inhibition of the Na+/H+ exchanger (NHE) NHE isoform 1 in human breast cancer cells. Clin Cancer Res 2003; 9:2366-73; PMID:12796407 [PubMed] [Google Scholar]
- 47. MacKeigan JP, Murphy LO, Blenis J. Sensitized RNAi screen of human kinases and phosphatases identifies new regulators of apoptosis and chemoresistance. Nat Cell Biol 2005; 7:591-600; PMID:15864305; http://dx.doi.org/ 10.1038/ncb1258 [DOI] [PubMed] [Google Scholar]
- 48. Peterson D, Lee J, Lei XC, Forrest WF, Davis DP, Jackson PK, Belmont LD. A chemosensitization screen identifies TP53RK, a kinase that restrains apoptosis after mitotic stress. Cancer Res 2010; 70:6325-35; PMID:20647325; http://dx.doi.org/ 10.1158/0008-5472.CAN-10-0015 [DOI] [PubMed] [Google Scholar]
- 49. Swanton C, Marani M, Pardo O, Warne PH, Kelly G, Sahai E, Elustondo F, Chang J, Temple J, Ahmed AA, et al. Regulators of mitotic arrest and ceramide metabolism are determinants of sensitivity to paclitaxel and other chemotherapeutic drugs. Cancer Cell 2007; 11:498-512; PMID:17560332; http://dx.doi.org/ 10.1016/j.ccr.2007.04.011 [DOI] [PubMed] [Google Scholar]
- 50. Whitehurst AW, Bodemann BO, Cardenas J, Ferguson D, Girard L, Peyton M, Minna JD, Michnoff C, Hao W, Roth MG, et al. Synthetic lethal screen identification of chemosensitizer loci in cancer cells. Nature 2007; 446:815-9; PMID:17429401; http://dx.doi.org/ 10.1038/nature05697 [DOI] [PubMed] [Google Scholar]
- 51. Coley HM, Hatzimichael E, Blagden S, McNeish I, Thompson A, Crook T, Syed N. Polo Like Kinase 2 Tumour Suppressor and cancer biomarker: new perspectives on drug sensitivity/resistance in ovarian cancer. Oncotarget 2012; 3:78-83; PMID:22289679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Syed N, Coley HM, Sehouli J, Koensgen D, Mustea A, Szlosarek P, McNeish I, Blagden SP, Schmid P, Lovell DP, et al. Polo-like kinase Plk2 is an epigenetic determinant of chemosensitivity and clinical outcomes in ovarian cancer. Cancer Res 2011; 71:3317-27; PMID:21402713; http://dx.doi.org/ 10.1158/0008-5472.CAN-10-2048 [DOI] [PubMed] [Google Scholar]
- 53. Lovat F, Ishii H, Schiappacassi M, Fassan M, Barbareschi M, Galligioni E, Gasparini P, Baldassarre G, Croce CM, Vecchione A. LZTS1 downregulation confers paclitaxel resistance and is associated with worse prognosis in breast cancer. Oncotarget 2014; 5:970-7; PMID:24448468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Nakouzi NA, Cotteret S, Commo F, Gaudin C, Rajpar S, Dessen P, Vielh P, Fizazi K, Chauchereau A. Targeting CDC25C, PLK1 and CHEK1 to overcome Docetaxel resistance induced by loss of LZTS1 in prostate cancer. Oncotarget 2014; 5:667-78; PMID:24525428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Chen DQ, Pan BZ, Huang JY, Zhang K, Cui SY, De W, Wang R, Chen LB. HDAC 1/4-mediated silencing of microRNA-200b promotes chemoresistance in human lung adenocarcinoma cells. Oncotarget 2014; 5:3333-49; PMID:24830600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Apontes P, Leontieva OV, Demidenko ZN, Li F, Blagosklonny MV. Exploring long-term protection of normal human fibroblasts and epithelial cells from chemotherapy in cell culture. Oncotarget 2011; 2:222-33; PMID:21447859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Darzynkiewicz Z. Novel strategies of protecting non-cancer cells during chemotherapy: are they ready for clinical testing? Oncotarget 2011; 2:107-8; PMID:21487161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. van Leeuwen IM, Lain S. Pharmacological manipulation of the cell cycle and metabolism to protect normal tissues against conventional anticancer drugs. Oncotarget 2011; 2:274-6; PMID:21512204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Enns LC, Pettan-Brewer C, Ladiges W. Protein kinase A is a target for aging and the aging heart. Aging (Albany NY) 2010; 2:238-43; PMID:20448293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Glass DB, Lundquist LJ, Katz BM, Walsh DA. Protein kinase inhibitor-(6-22)-amide peptide analogs with standard and nonstandard amino acid substitutions for phenylalanine 10. Inhibition of cAMP-dependent protein kinase. J Biol Chem 1989; 264:14579-84; PMID:2760075 [PubMed] [Google Scholar]
- 61. Singhal R, Kandel ES. The response to PAK1 inhibitor IPA3 distinguishes between cancer cells with mutations in BRAF and Ras oncogenes. Oncotarget 2012; 3:700-8; PMID:22869096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kandel ES, Skeen J, Majewski N, Di Cristofano A, Pandolfi PP, Feliciano CS, Gartel A, Hay N. Activation of Akt/protein kinase B overcomes a G(2)/m cell cycle checkpoint induced by DNA damage. Mol Cell Biol 2002; 22:7831-41; PMID:12391152; http://dx.doi.org/ 10.1128/MCB.22.22.7831-7841.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]