

Abstract
Unlike other organisms that have evolved distinct H2A variants for different functions, Drosophila melanogaster has just one variant which is capable of filling many roles. This protein, H2A.V, combines the features of the conserved variants H2A.Z and H2A.X in transcriptional control/heterochromatin assembly and DNA damage response, respectively. Here we show that mutations in the gene encoding H2A.V affect chromatin compaction and perturb chromosome segregation in Drosophila mitotic cells. A microtubule (MT) regrowth assay after cold exposure revealed that loss of H2A.V impairs the formation of kinetochore-driven (k) fibers, which can account for defects in chromosome segregation. All defects are rescued by a transgene encoding H2A.V that lacks the H2A.X function in the DNA damage response, suggesting that the H2A.Z (but not H2A.X) functionality of H2A.V is required for chromosome segregation. We also found that loss of H2A.V weakens HP1 localization, specifically at the pericentric heterochromatin of metaphase chromosomes. Interestingly, loss of HP1 yielded not only telomeric fusions but also mitotic defects similar to those seen in H2A.V null mutants, suggesting a role for HP1 in chromosome segregation. We also show that H2A.V precipitates HP1 from larval brain extracts indicating that both proteins are part of the same complex. Moreover, we found that the overexpression of HP1 rescues chromosome missegregation and defects in the kinetochore-driven k-fiber regrowth of H2A.V mutants indicating that both phenotypes are influenced by unbalanced levels of HP1. Collectively, our results suggest that H2A.V and HP1 work in concert to ensure kinetochore-driven MT growth.
Keywords: chromosome segregation, Drosophila, H2A.V, HP1, mitosis
Introduction
Eukaryotic chromatin contains basic units called nucleosomes, consisting of octamers of 4 core histones with 146 base pair stretches of DNA wrapped around them.1,2 The chromatin's function and structure are determined by histone modifications such as acetylation, methylation, phosphorylation, and ubiquitylation,3,4 and the subsequent binding of specific factors to modified histones promotes further changes in chromatin organization.5 In addition to histone post-translational modifications, histone variants have recently emerged as important determinants of chromatin composition.6 Variants of H3 and H2A histones are to date the best characterized. Among them, H2A.Z, the highly evolutionarily conserved variant of the canonical histone H2A,7 is unique in representing the only variant required for viability and development in several organisms.8–11 Mounting evidence has demonstrated that H2A.Z is implicated in multiple cellular pathways such as maintenance of chromosome stability and segregation,12-14 modulation of transcription (for a review see Refs15,16), as well as regulation of heterochromatin spreading.17-19 However, though H2A.Z plays a role in different pathways, its functional mechanisms have not been fully elucidated.
Drosophila melanogaster has a single H2A.Z variant, H2A.V that represents 5–10% of cellular H2A proteins. In addition to sharing structural similarities with metazoan H2A.Z, H2A.V also harbors a 4 amino acid stretch at the end of its extended tail domain that resembles the canonical phosphorylation motif found in the H2A.X variant histones (for a recent review see Ref 20). This site contains a serine residue (S137), which is phosphorylated by ATM and ATR kinases following DNA break and repair, indicating that H2A.V, like its counterpart H2A.X, acts as a DNA damage sensor.21,22 Similar to mammalian H2A.Z, H2A.V nonrandomly localizes to heterochromatin regions and many euchromatin regions on polytene chromosomes.23 Genome-wide chromatin immunoprecipitation studies revealed that H2A.V is associated with constitutively expressed genes as well as developmental silenced loci. H2A.V levels appeared reduced at the promoters of induced heat shock genes indicating that, like its human H2A.Z ortholog, this variant is depleted from chromatin upon activation of transcription.24,25 Finally, it has been reported that H2A.V behaves genetically as a PcG gene and that mutations in H2A.V suppress position effect variegation. In addition H2A.V mutants exhibit reduced H3 lysine 9 (H3K9) methylation and reduced binding of HP1 and Pc suggesting that this H2A histone variant is required for euchromatic silencing and heterochromatin formation.26 Thus, it appears that Drosophila H2A.V fulfills several distinct functions and combines the features of H2A.Z in transcriptional control with the functions of H2A.X in DNA damage response.20
Here we demonstrate that loss of H2A.V causes missegregation defects in Drosophila neuroblast cells highlighting an unanticipated function of H2A.Z, but not of H2A.X, in Drosophila cell division. We found that H2A.V mutant chromosomes fail to align at the metaphase plate and show reduced HP1 localization as well as a delayed kinetochore-driven microtubule re-growth after cold treatment. Consistently, we provide evidence that HP1 mutant neuroblasts demonstrate defects in kinetochore-driven MT growth, similar to the effect observed in H2A.V mutants. We furthermore show that H2A.V forms a complex with HP1 and that overexpression of HP1 substantially reduces the cell division defects of H2A.V mutants. These results suggest that H2A.V stabilizes HP1 binding to chromatin. Thus, our findings indicate that in Drosophila H2A.V and HP1 cooperate to regulate chromosome segregation and reveal a new role for HP1 in the assembly of mitotic spindle.
Results
Loss of H2A.V affects chromosome segregation in Drosophila mitotic cells
To investigate whether H2A.V regulates heterochromatin organization in mitotic chromosomes, we conducted a cytological analysis of metaphase spreads from the H2A.V810 null mutant.23,26 Colchicine-treated H2A.V810 homozygous and H2A.V810/Df(3R)TI-P hemizygous larval brains exhibited a plethora of mitotic abnormalities, which are depicted in Figure 1A. In a significant portion of mutant mitotic cells (∼10%; n = 850), centromeric chromatin appeared poorly condensed giving rise to evident DAPI staining discontinuities which have the appearance of chromatin gaps (Fig. 1Ab, B). In addition, ∼30% of mutant cells exhibited irregular condensation of both euchromatic and heterochromatic regions of chromosomes. We also observed cells with overcondensed chromosomes (∼7%), precocious sister chromatid separation and rare chromosome breaks (Fig. 1Ac,d, B).Taken together with recent studies showing that H2A.V may participate in chromatin remodeling,27 these results indicate that H2A.V plays a global role in the organization of chromatin. To assess whether loss of H2A.V affected cell cycle progression, we measured the Mitotic Index (MI) and the Anaphases Frequency (AF) in no-colchicine treated DAPI-stained H2A.V810 homozygous and H2A.V810/Df(3R)TI-P hemizygous larval brains. During this characterization, we found that H2A.V810 mutant brains, in addition to distinct prometaphase, metaphase and anaphase cells, contained unique cells (14%, n = 900) with chromosomes and/or chromatids scattered along the major axis of the cell. This result suggests that either chromosome congression or segregation, or both, are influenced during mitosis (Fig. 1B,C; Supplementary Fig. 1A). Yet, as we could not unambiguously define these figures as anaphases (see also below), we included them only in the total number of dividing cells. When we calculated the mitotic parameters we found that in the H2A.V810 mutant brains the MI was indistinguishable from wild type, whereas the frequency of anaphases showed a mild, but statistically significant (∼20%), reduction compared to wild-type cells (n = 900; Fig. 1C). Thus, although H2A.V depleted cells can normally enter mitosis, a small percentage of mutant cells either fail to complete mitosis or exhibit a delayed anaphase onset. All H2A.V810 mutant mitotic abnormalities were rescued by a wild-type H2A.V-GFP transgene9 indicating that they were indeed caused by an H2A.V depletion. Interestingly, the H2A.V810 mutant phenotypes were also rescued by a transgene that encodes H2A.V with a C-terminal truncation and therefore lacking of H2A.X function9 (Fig. 1B, C and E). This suggests that it is the loss of H2A.Z function, not H2A.X, that affects chromosome behavior.
Figure 1.
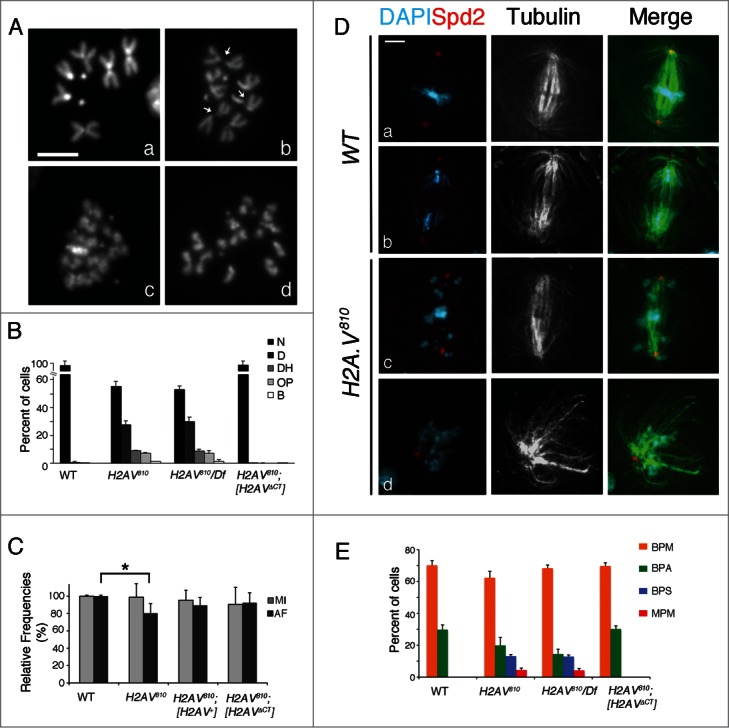
H2A.V is required for proper chromosome condensation and segregation. (A) DAPI stained colchicine-treated (Aa–d) mitotic cells from control (Aa), H2A.V810 (Ab) and H2A.V810/Df(3R)TI-P (Ac–d) mutant brains. H2A.V mutant metaphases exhibit chromosomes with poorly condensed centromeric chromatin (arrows in Ab), overcondensed chromosomes (Ac), or chromosomes with precocious sister chromatid separation (Ad). (B) Quantification of mitotic defects in H2AV mutants and in H2A.V mutants also bearing a H2A.V transgene with a C-terminal truncation. See text for further details. N = Normal cells; D = Cells with poorly condensed chromosomes; DH = cells with chromosomes exhibiting decondensation at the pericentric region; OP = Cells showing either overcondensed chromosomes or precocious sister chromatid separation; B = Cells with chromosome breaks. (C) Mitotic Index (MI) and Frequency of Anaphases (AF) in WT, H2A.V810, H2A.V810 [H2A.V+] and H2A.V810 [H2A.VΔCT] larval brain cells (*P < 0.01, t Student's Test). (D) Mitotic spindles of wild type (Da,b) and H2A.V mutant (Dc,d) stained for tubulin (green), Spd2 (red) and DAPI. (Da) Wild type metaphase; (Db) wild type anaphase; (Dc) a H2A.V mutant cell with scattered chromosomes; (Dd) a rare example of H2A.V mutant cell with a monopolar spindle. Bar = 5 μm. E) Quantification of spindle defects in WT, H2A.V810 and H2A.V810 [H2A.VΔCT]. BPM = prometaphases/metaphases with normal bipolar spindles; BPA = ana/telophases with normal bipolar spindles; BPS = cells with dispersed chromosomes and normal bipolar spindles; MPM = metaphases and ana/telophases with monopolar spindles.
H2A.V mutants exhibit defects in the organization of kinetochore-driven MTs
To assess whether chromosome segregation defects were due to defective spindles, we immunostained H2A.V810 mutant neuroblasts with anti-α tubulin and anti-Spd2 antibodies, which recognize spindle fibers and centrosomes, respectively.28 This analysis confirmed the presence of cells (∼15%; n = 300, Fig. 1C–E) with chromosomes scattered across the mitotic spindles. We have also found that a small percentage of dividing H2A.V810 mutant cells displayed monopolar spindles (∼5%; n = 300; Fig. 1Dd, E) indicating that the absence of H2A.V leads to a mild spindle disorganization. However, nearly every cell with dispersed chromosomes contained normal bipolar spindles (Fig. 1D, E), suggesting that chromosome misalignment in H2A.V810 mutants is not due to spindle defects. To test whether this defective chromosome behavior was due to a reduction of MTs, we measured the density of MTs in wild-type and mutant metaphases and found that it did not generally vary between H2A.V810 mutant and wild-type cells. However the H2A.V810 mutant cells with dispersed chromosomes represented an exception, as they exhibited a reduction of MT density (Fig. 1Dc; Fig. S1B), though this reduction was mild. To check whether the decreased MT density was related to a specific sub-set of MTs, we compared the spindle MT density with that of centrosomal MTs and found that MT density in mutant cells decreased mainly within the spindle (Fig. S1B). This last result suggests that a reduced number of kinetochore MTs could account for the chromosome misalignment seen in these mutant cells.
To better understand the mitotic phase of cells with misaligned chromosomes, we immunostained H2A.V810 mutant cells for the spindle assembly checkpoint (SAC) proteins BubR1 and Zw10. This analysis revealed that all cells with misaligned chromosomes from homozygous or hemizygous H2A.V810 mutant brains retain a robust centromeric localization of both BubR1 and Zw10 (Fig. 2). This indicates that in these cells the SAC is not satisfied thus preventing anaphase onset. Consistently, H2A.V810 mutant neuroblasts with misaligned chromosomes exhibit high levels of cyclin B (Fig. S2), which is normally degraded at the metaphase-to-anaphase transition. Altogether, these data confirm that H2A.V810 abnormal figures are in a pro/metaphase (rather than ana/telophase)-like stage, although most of them exhibit spindle length similar to that of anaphases. Nonetheless, for the sake of clarity, we will refer to them as pseudo-anaphases (PAs), a term used in previous studies characterizing similar effects.29,30 We have also observed that in most mutant cells Zw10 (40–80%; n = 40), although strongly associated with kinetochores, failed to migrate toward the spindle poles along the microtubules, as it normally does in control metaphases31,32 (Fig. 2B, C). As it is known that Zw10 streaming is due to the association of the SAC component to k-MTs, we hypothesized that loss of Zw10 streaming reflected an improper organization of kinetochore fibers (k-fibers).
Figure 2.
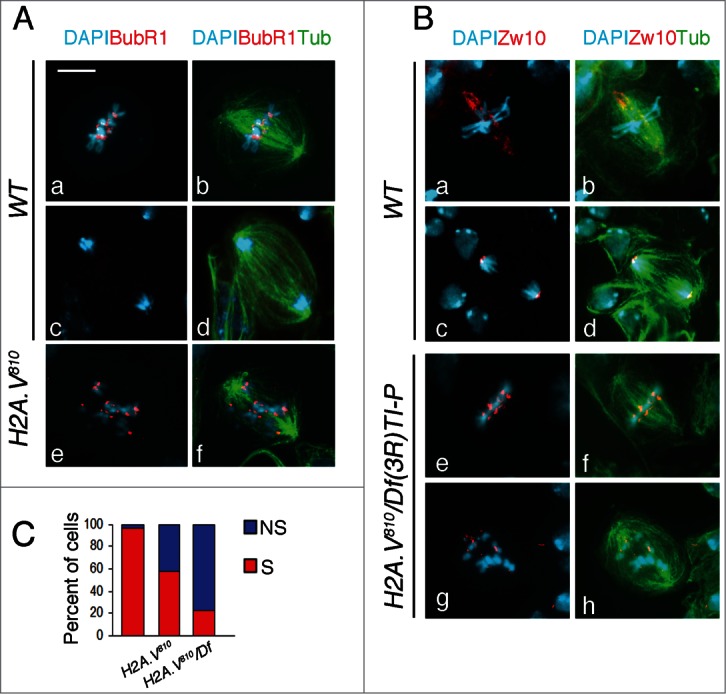
H2A.V810 mutant cells fail to proceed through mitosis. (A) Localization of BubR1 (red) in wild-type (Aa–d) and H2A.V810 mutant (Ae,f) cells. (B) Localization of Zw10 (red) in wild-type (Ba–d) and H2A.V mutant (Be–h) cells. Note that in H2A.V mutants, Zw10 does not stream toward the spindle poles. Bar = 5 μm. (C) Quantification of cells with (S) or without (NS) streaming of Zw10.
To confirm these results, we immunostained H2A.V810 mutant cells with the augmin component Dgt6, a well-established marker for k-fibers in Drosophila cells.29,33 Consistent with a recent work,29 we found that in control cells Dgt6 associates mainly with k-fibers (Fig. 3A). Interestingly, H2A.V810 mutant cells exhibited a dramatic reduction of Dgt6 localization during both metaphase and anaphase (Fig. 3A, B) indicating that the reduction of Dgt6 in the mutant neuroblasts is likely due to defects in the k-fiber organization.
Figure 3.
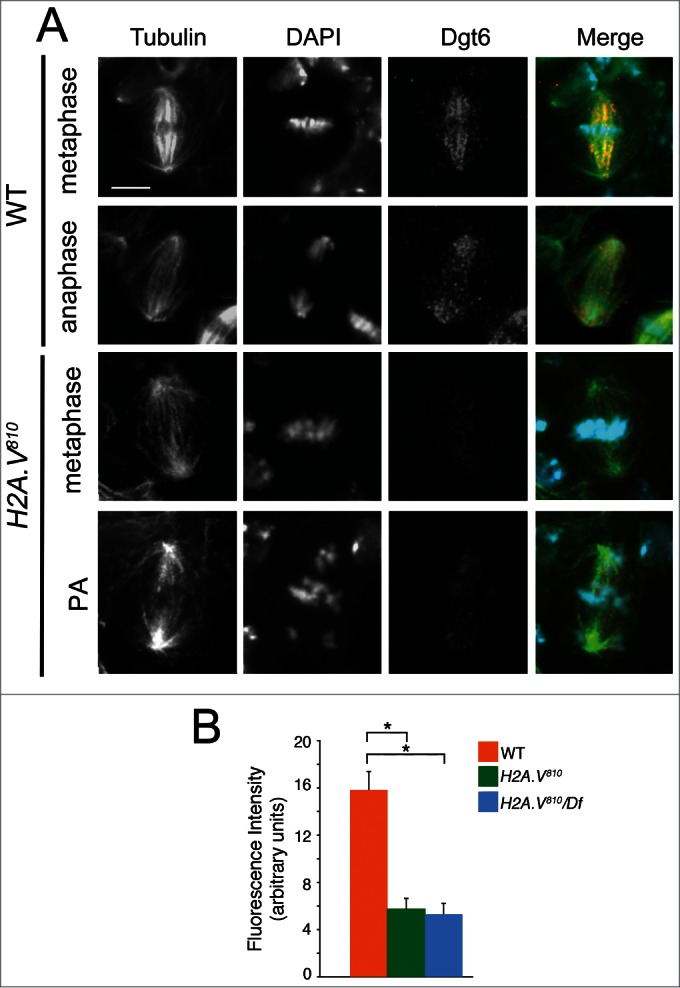
Dgt6 fails to localize at MTs of H2A.V810 mutant cells. (A) Localization of Dgt6 in a wild-type NB metaphase (Aa), wild-type anaphase (Ab), in H2A.V mutant metaphase (Ac) and PA (Ad). (B) Quantification of Dgt6 localization (bars indicate SEM; 50 cells analyzed for each genotype, *P < 0.01, t Student's Test).
To verify whether k-fiber organization was impaired in H2A.V810 mutants, we employed a spindle microtubule re-growth assay following cold-induced MT depolymerization in mitotic neuroblasts34 and analyzed the MT growth from kinetochores. Consistent with published data,29,34 we found that the formation of kinetochore-associated MT bundles precedes MT re-growth form the centrosomes. In particular, after 50 min of cold treatment MTs were completely depolymerized in a large proportion of both wild type and mutant brain cells that displayed only an array of short MTs associated to centrosomes (0 min; Fig. 4A, C). After returning to room temperature, differences between wild type and mutant cells emerged. In wild-type cells 1 min after returning to room temperature, we observed ∼30% of cells (n = 250) with MTs associated to centromeric regions, ∼55% with MTs associated to both chromosomes and centrosomes and ∼10% only to centrosomes. After 2, min we found the majority of cells (65%) elicited MTs associated with both chromosomes and centrosomes. Finally, after 5 min almost all wild type cells displayed a completely organized mitotic spindle, which appeared indistinguishable from that of untreated cells (Fig. 4A, C).
Figure 4.
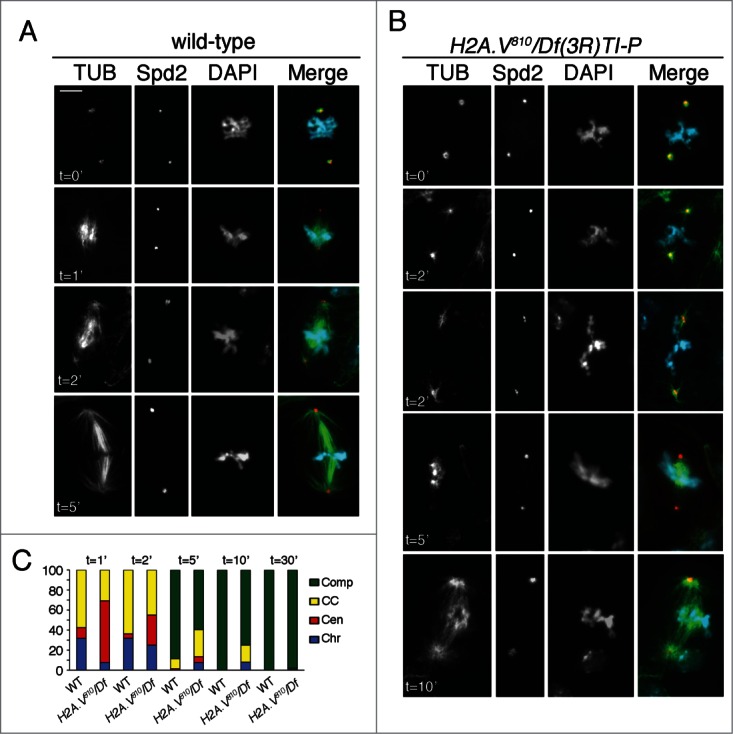
H2A.V is required for chromosome-driven MT growth. (A) MT re-growth after cold depolymerization in wild type and H2A.V neuroblast metaphases. Cells were stained for Spd2 (red), tubulin (green) and DNA (blue). In both wild-type and H2A.V brains, spindle MTs are completely depolymerized after 50 min of cold treatment, except for some very short centrosome-associated MTs (0 minutes). In wild-type cells, MTs regrow mainly near the chromosomes, or at both the chromosomes and the centrosomes (1 and 2 minutes); at 5, min most spindles are fully assembled. (B) In both metaphase and PM H2A.V mutant cells, MT nucleation near the chromatin is delayed, as it occurs only after 2 min and persists up to 5 min; at 5 and 10 min, a significant proportion of H2A.V810 cells still exhibits MT regrowth from both chromosomes and centrosomes. Bar = 5 μm. (C) Frequencies of wild-type and H2A.V810 metaphases showing MT nucleation from the chromosomes only (Chr), from the centrosomes only (Cen), or from both the chromosomes and the centrosomes (CC). Comp = fully assembled spindles.
In 60% of H2A.V810 mutant cells (n = 70) that were recovered for 1 min at room temperature, MTs re-grew only from centrosomes whereas only a small percentage (6%) of mutant neuroblasts displayed MTs associated to chromosomes (Fig. 4B, C). This MT re-growth pattern was observed in cells with normal and irregular (PA) chromosome congression. After 2 min, the number of cells with MT re-growth from centrosomes halved (from 60% to 30%), whereas the percentage of cells displaying MTs associated to chromosomes increased. Interestingly, few cells (∼7%) with only kinetochore-associated MTs were found also after 5 and 10 min suggesting that H2A.V810 mutant neuroblasts experienced a delay in the kinetochore-driven MT formation (Fig. 4B, C). As consequence, all mutant cells displayed fully developed spindles only after 30 minwhile in wild-type completely formed spindles were observed after 5 min(Fig. 4C).
H2A.V forms a complex with HP1
Given the defects in the organization of k-fibers observed upon loss of H2A.V, we hypothesized that this effect was precipitated by an improper recruitment of Drosophila centromere/kinetochore proteins. To test this, we verified the localization of representative centromere factors (Cid/Cenp-A, Cenp-C), and inner (Mis12, Nls1), or outer (Ndc80/Hec1) plate components. Immunofluorescence experiments showed that all tested centromere/kinetochore proteins localize normally in H2A.V810 mutant PA chromosomes (Fig. S3), as well as in normally condensed mutant chromosomes, indicating that chromosome missegregation is not a consequence of loss of these centromere/kinetochore proteins.
Previous studies demonstrated that depletion of H2A.Z in mammalian cells disrupts Heterochromatin Protein 1α (HP1α)-chromatin interactions at chromosomal arms and pericentric regions.13 In addition Drosophila H2A.V was shown to be important for recruiting HP1 (the Drosophila ortholog of mammalian HP1α) on polytene chromosome chromocenter.26 Thus, we wanted to investigate whether an H2A.V-HP1 functional relationship could account for the cytological phenotypes observed in H2A.V810 mutant cells.
By immunostaining wild-type larval brain cells with the mouse monoclonal anti-HP1 antibody C1A9, we found that HP1 is strongly enriched at heterochromatin regions (chromocenter) of interphase nuclei. HP1 strongly localizes also to pericentric regions of mitotic chromosomes from prometaphase to anaphase (Fig. 5A-C). In particular, we found that HP1 staining at heterochromatin regions peaks during anaphase, likely as a consequence of a physiological centromere clustering (Fig. 5H). In addition, HP1 staining is also found along all chromosome arms, including chromosome ends, although this localization pattern is not as intense as at heterochromatin regions. HP1 immunostaining in H2A.V mutant brain cells revealed a severe reduction of HP1 localization at both heterochromatic and euchromatic regions of H2A.V810 homozygous (75% of cells; n = 250) and hemizygous (70% of cells; n = 150) mitotic chromosomes with respect to wild type (15% of cells; n = 200; Fig. 5D-G). In particular, the intensity of HP1 staining decreased mainly during metaphase and anaphase (Fig. 5H), but not in prophase and interphase. This indicates that the reduction of HP1 localization at centromeres upon loss of H2A.V occurs preferentially during mitosis.
Figure 5.
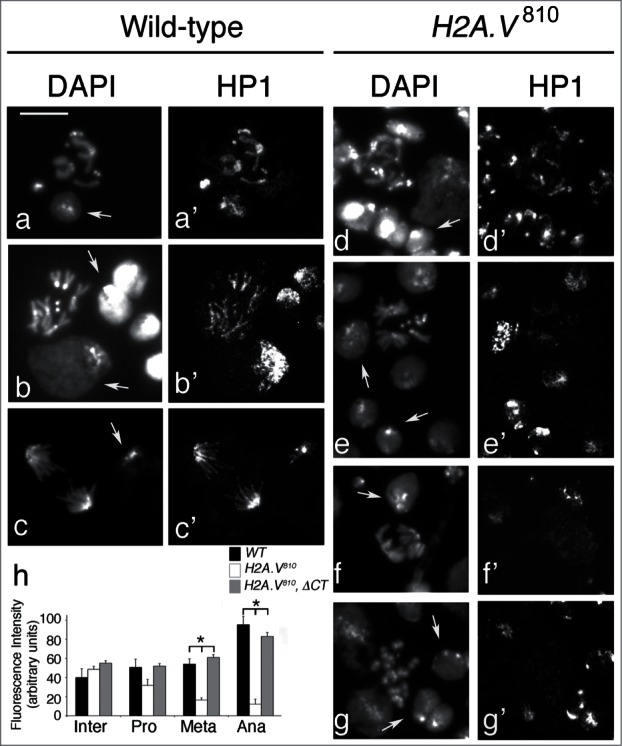
Loss of H2A.V reduces HP1 accumulation during mitosis. HP1 localization in wild-type (a-c) and H2A.V810 mutant (d-g) cells. Arrows indicate examples of interphase cells. Note that HP1 staining is reduced on H2A.V810 mutant metaphase and anaphase chromosomes (e′-f′) but not at the interphase chromocenters. Bar = 5 μm. (h) Quantification of HP1 intensity in interphase (Inter) and during prometaphase (Pro), metaphase (Meta) and anaphase (Ana) (*P < 0.01, t Student's Test)
Based on our anti-HP1 immunofluorescence analyses, we then asked whether H2A.V and HP1 could interact. GFP-trap mediated immunoprecipitation of H2A.V from H2A.V -GFP expressing brain extracts revealed that H2A.V is able to precipitate HP1, suggesting that both proteins are part of the same complex (Fig. 6A). The HP1 protein consists of an N-terminal Chromo Domain (CD) separated from a related C-terminal Chromo Shadow Domain (CSD) by a hinge region (H). All three domains are required for specific and different HP1 functions and interactions35 (Fig. 6B). Interestingly, pulldown experiments using a GST-tagged HP1 (GST-HP1) and HP1 truncations (namely, GST-HP1ΔCD, -HP1ΔH and -HP1ΔCSD, which lack of the N-terminal CD, hinge or C-terminal CSD domains, respectively) revealed that H2A.V is not precipitated by HP1 when it lacks the CD domain, indicating that the HP1-H2A.V interaction is mediated directly or indirectly by the HP1 N-terminal domain (Fig. 6C).
Figure 6.
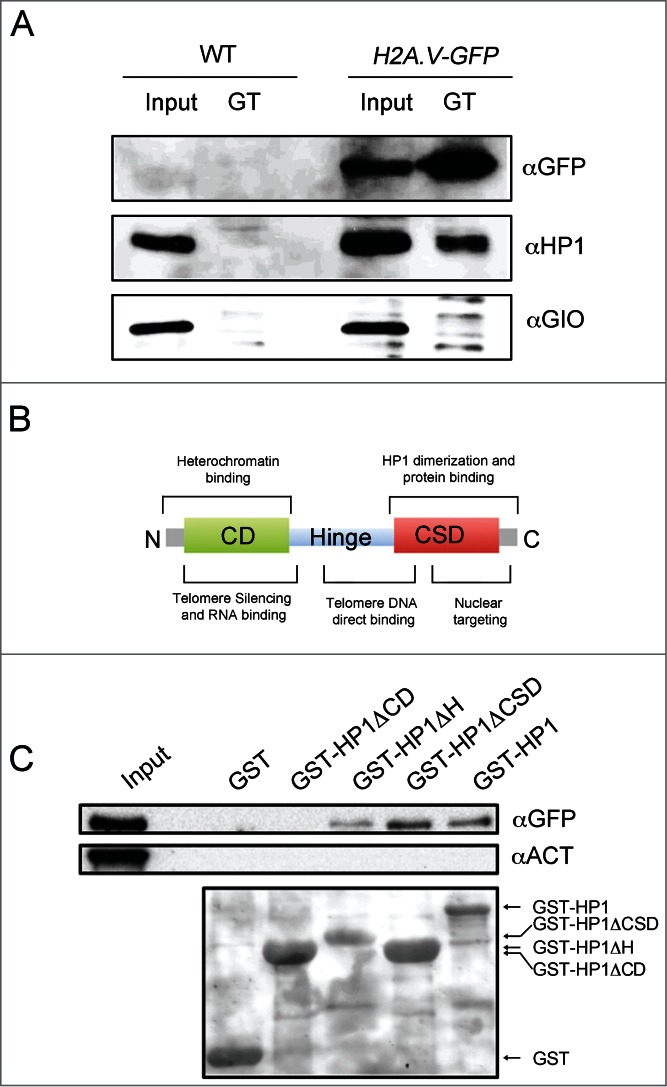
H2A.V interacts with HP1. (A) GFP-Trap immunoprecipitation (GT) from wild type and H2A.V-GFP expressing brain extracts. Giotto (GIO)55 has been used as loading control. (B) The HP1 protein domains (adapted from Fanti and Pimpinelli (2008)35). (C) GST-pulldown of H2A.V-GFP expressing brain extracts with GST-tagged HP1 (GST-HP1) and HP1 truncations in which either the Chromo domain (GST-HP1ΔCD), the Hinge (GST-HP1ΔH) or the Chromo Shadow domain (GST-HP1ΔCSD) has been deleted.
Loss of HP1 impairs kinetochore-driven MT formation
Our analysis of the functional relationship between H2A.V and HP1 led us to speculate that HP1 may also be involved in the organization of kinetochore-fibers. We thus asked whether loss of HP1 could phenocopy the cell division defects seen in H2A.V810 mutants. We performed a cytological characterization of mitosis in Su(var)205/HP1 mutants and checked for the presence of PAs and/or metaphases with defects in kinetochore-driven MT formation. It is well known that null mutations in the Su(var)205/HP1 gene cause frequent telomere fusions in Drosophila larval brain cells, leading to the formation of trains of chromosomes.36 These multiple telomeric associations give rise to frequent chromosome bridges and segregation defects during anaphase leading to hyperploid and polyploid cells with either fused or broken chromosomes.36 This would mask the analysis of a potential centromere/kinetochore dysfunction. To circumvent this problem, we looked for mitotic defects in a UAS HP1 RNAi line that behaved as a hypomorphic HP1 mutant allele (see Materials and Methods). By crossing UAS HP1 RNAi to the 69B GAL4-bearing flies, we drew HP1 depletion specifically in brains and observed a ∼60% reduction of HP1 levels and a frequency of telomeric associations lower than that observed for Su(var)205 null alleles (data not shown). The cytological characterization of DAPI-stained no-colchicine treated larval brains revealed that the MI and the AF in the HP1 RNAi neuroblasts are comparable to wild-type (data not shown). This indicates that, consistent with published data36,37 and unlike H2A.V810 mutants, cells without HP1 are still able to complete mitosis. To determine whether cells lacking HP1 demonstrated the PA phenotype observed in H2A.V810 mutants, we analyzed a sample of cells with no telomeric fusions. Of these, 5% (n = 200) had chromosomes scattered across the major axis of the cytoplasm, reminiscent of the PAs observed in H2A.V810 mutants. It is also worth noting that this frequency may not be representative of the overall rate of PAs, because cells with telomeric fusions were excluded from the count because the fusion of chromosomes impedes the observation of PAs.
Anti-α tubulin and anti-Spd2 immunostainings revealed that ∼75% (n = 140) of dividing cells exhibited normal bipolar spindles, with the remaining cells displaying either multipolar (∼8%) or monopolar (∼17%) spindle. While multipolar spindles were associated with hyperploid/polyploid cells, the presence of few monopolar spindles suggests that loss of HP1 might affect spindle assembly (Fig. S4A). This result is important in that it demonstrates phenotypic similarities with H2A.V810 PAs, which also exhibited normal bipolar spindles (Fig. 7A, C). It also lends support to the hypothesis that PAs do not arise from spindle alteration. We also found that HP1 mutant PA chromosomes show a normal localization of centromeric antigens, a robust localization of the SAC protein BubR1 and high levels of cyclin B. This confirms that, like H2A.V mutant cells, these mutants never move into metaphase (Fig. S4B, C). Moreover, Dgt6 localization was significantly reduced in the majority of mutant dividing cells (Fig. 7A, B), suggesting that HP1 depleted cells, alike H2A.V810 mutant cells, may experience defects in kinetochore-driven MT formation.
Figure 7.
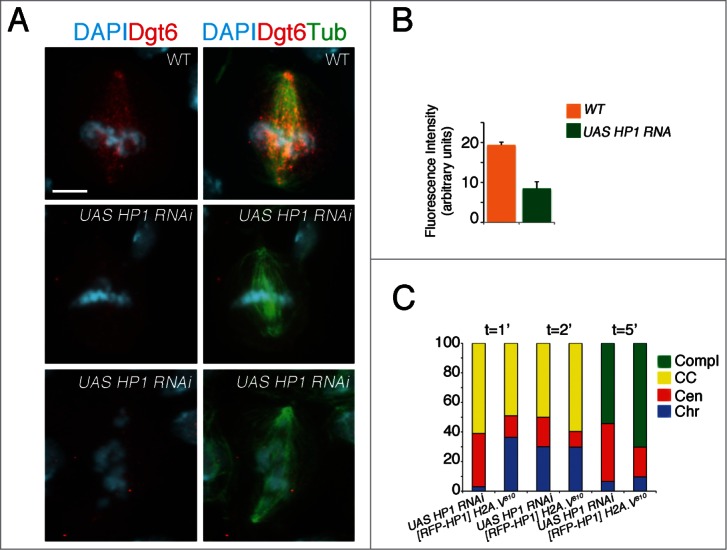
HP1 is required for cell division. (A) Dgt6 fails to localize at MTs of HP1 mutant metaphases (central panel) and PAs (bottom). (B) Quantification of Dgt6 localization (bars indicate SEM). A total number of 50 (WT) and 100 (HP1 mutant) cells were analyzed (C) Frequencies of HP1 mutant cells and H2A.V810 mutant cells expressing the [RFP-HP1] transgene showing MT regrowth after cold. Loss of HP1 delays MT regrowth from kinetochores as observed in H2A.V810 mutants. Note that HP1 overexpression increases the number of H2A.V810 mutant cells with MT nucleation form chromosomes (Chr) and from both chromosomes and centrosomes (CC; see Fig. 4 for comparison).
To confirm these observations, we performed a MT regrowth assay after cold-treatment. We found that HP1 depleted cells exhibit a scarce MT re-growth from chromosomes after 1 min recovery, with the majority of cells showing MT re-growth from centrosomes (Fig. 7E; see Fig. 4C for comparison). Even after 2 min recovery, MTs were found to re-grow mainly from centrosomes (∼30%) and very few (∼2%) from chromosomes (n = 40). Finally, after 5 min similarly to the H2A.V810 mutants, only ∼40% of HP1 mutant cells (n = 35) completed spindle formation, whereas a full developed spindle is already seen in almost all control cells (Fig. 7C; see Fig. 4C for comparison). Thus, we can conclude that, in addition to protecting telomeres from fusion events and regulating gene expression, HP1, like H2A.V, is required for establishing a correct kinetochore-driven MT formation during mitosis.
Taken together, our data suggest that chromosome segregation defects seen upon depletion of H2A.V are mainly caused by the loss of HP1 from centromeres. To test this hypothesis, we overexpressed HP1 in H2A.V810 mutants. We found that the expression of a [RFP-HP1] transgene yields a 2-fold increase of HP1 levels (data not shown) and, when expressed in H2A.V810 mutant background, is able to reduce the frequency of PAs from 20% to 5% (n = 130). This confirms our hypothesis that chromosome misalignments in H2A.V810 mutant cells are mainly due to reduced HP1 levels. MT re-growth analysis shows that 1 and 2 min after cold treatment, the proportion of the HP1-expressing H2A.V810 mutant neuroblasts showing microtubule re-growth from kinetochore and from both kineotochore and centrosomes is higher than that seen in H2A.V810 mutants and very similar to wild-type (Fig. 7C; see also Fig. 4C for comparison), indicating that HP1 transgene rescues defects in the kinetochore-driven MT formation. As consequence after 5 min of recovery from cold, fully formed mitotic spindles are much more frequent in [RFP-HP1] H2A.V810 recombinants (∼80% of cells; n = 30) compared to H2A.V810 mutants (Fig. 7C and Fig. 4C for a direct comparison) confirming that additional copies of HP1 ameliorate spindle assembly when H2A.V is lost. However, a significant fraction of cells showing microtubule growth from centrosomes or kinetochores is still present in the [RFP-HP1] H2A.V810 recombinant brains, so HP1 overexpression alone is not sufficient to fully restore functional spindles. Thus, a simultaneous presence of H2A.V and HP1 is required to fulfill a proper spindle behavior.
Discussion
Here, we provide compelling evidence that H2A.V, the Drosophila histone H2A variant, plays an important and unanticipated role during Drosophila mitosis. The cytological characterization of H2A.V810 mutant larval brain chromosomes revealed that loss of H2A.V has an impact on chromosome organization and cell proliferation (Fig. 1), which is consistent with previous results on a role of this histone variant in chromatin remodeling and heterochromatin organization.20,26,27 We also demonstrate that a significant proportion of H2A.V mutant cells fails to complete mitosis and contains chromosomes that remain scattered across the spindle (pseudo anaphase or PA) due to failed metaphase plate alignment. Similar effects have been previously described in Drosophila S2 cells depleted by RNAi of either kinetochore proteins, augmin components or splicing factors.29,30 However unlike those S2 interfered cells, which exhibit PAs with long spindles, H2A.V810 mutant cells have PA spindles that appear similar to wild type anaphases. The reason why H2A.V mutant cells are not elongated is unclear, but it may depend on the different cellular systems employed in the different studies. Intriguingly, the presence of PAs in H2A.V810 mutants indicates for the first time that Drosophila H2A.V is also necessary for chromosome segregation.
Interestingly, our results from both Dgt6 immunolocalization and spindle microtubule re-growth assay following cold-induced MT depolymerization in mitotic neuroblasts reveal that H2A.V might be involved in the organization of kinetochore-driven k-fibers. However, we believe that defects in the organization of k-fibers are not a consequence of the reduction of Dgt6. Recent studies demonstrated that Wac, a newly discovered component of Augmin complex, is required for spindle formation in S2 cells but is dispensable for somatic mitosis. In fact, a wac deletion mutant was viable and displayed only weak defects in brain cell divisions, suggesting that the components of Augmin complex (including Dgt6) might have non essential roles in spindle assembly.38
It has been previously reported that defective k-fiber formation and elongation disrupt chromosome segregation and spindle formation in Drosophila cells.29 Our results, which are in line with this finding, indicate that a specific chromatin organization is also necessary to ensure a proper spindle assembly. We speculate that the observed PAs are a result of improper organization of k-fibers, and that PAs fail to complete mitosis, thus reducing in part the frequency of anaphases in H2A.V810 mutants. It is also plausible that persistent chromosome misalignment leads to a mitotic arrest of these cells, which in turn could explain the presence of H2A.V810 mutant cells with overcondensed chromosomes (Fig. 1). However, while in Bucciarelli et al (2009) the presence of PAs was always associated to a strong increase of mitotic index (MI), in our mutants the MI did not change. One explanation is that the reduction of anaphase frequency in H2A.V810 mutants (20%) is not as dramatic as that reported for Dgt6-depleted S2 cells (50%)29 and therefore it unlikely affects mitotic progression. An alternative explanation is that loss of H2A.V might affect the regulation of G2-M and/or M-A cell cycle checkpoints thus preventing a metaphase arrest. Further investigations are required to verify this hypothesis. It is worth noting that, although a role for H2A.Z in chromosome segregation has been previously documented in human and yeast cells,12,13,39-42 our data provide the first evidence of an potential involvement of H2A.Z in the organization of k-fibers.
We also provide unanticipated molecular evidence that H2A.V interacts directly or indirectly with HP1, confirming that both proteins are part of same complex (see also Ref 27). It is intriguing that the H2A.V-HP1 interaction depends on the HP1 CD domain, which also binds H3K9me2/3 and mediates heterochromatin formation.43 This supports the existence of a cascade of events that requires the recruitment of H2A.V and different histone modifications for the establishment of heterochromatin.26 Yet, the reason why depletion of H2A.V causes a direct loss of HP1 and particularly during mitosis is unclear. Nonetheless, as HP1 overexpression in H2A.V mutant cells prevents PA formation, we speculate that a H2A.V-dependent stabilization/localization of HP1 at centromeric region is essential to ensure proper chromosomal behavior.
Previous studies have shown that H2A.Z alters the nucleosomal surface, thus enabling preferential binding of HP1a to condensed higher chromatin structures.44 It is conceivable then that the H2A.V-HP1 interaction is favored by the condensation of pericentric chromatin fiber in metaphase. Alternatively, these interactions may be encouraged by metaphase-specific posttranslational modifications of H2A.V, HP1 or other interacting proteins. Indeed, it has been proposed the mechanism underpinning HP1 recruitment on mitotic chromosomes might be different from that in interphase.45 Still, little is known about the factors required for specific localization of HP1 at mitotic centromeres save for a few discoveries. Human HP1α binding to INCENP, for instance, has been demonstrated as necessary for HP1α targeting to mitotic centromeres. We believe that H2A.V may play a similar role in mediating HP1 binding,46-48 but how this takes place remains to be seen.
Our functional characterization of H2A.V has also unveiled the role of Drosophila HP1a in the assembly of the mitotic spindle. Our results indicate that the loss of HP1 yields defects in the kinetochore-driven k-fiber organization, which can in turn compromise chromosome segregation thus generating PAs. Past studies have shown that HP1a contributes to chromosome segregation and centromere stability in a variety of organisms including mammals,49,50 but the mechanism is still not completely understood. HP1 is known to interact with components of the centromere and the kinetochore complex,47,51 providing targets to begin understanding how downregulation or mislocalization of HP1 result in mitotic defects.51–53 It has also been reported that in contrast to Swi6 in S. pombe, the correct localization of HP1 is not required for the recruitment of cohesins to centromeric regions in mammals.54 Yet, HP1a seems to help in protecting cohesins from degradation by recruiting the Shugoshin protein.45 Here, we have highlighted an additional function of HP1 during chromosome segregation, one which depends on interaction with H2A.V and is required to regulate k-fiber organization. These results thus provide further evidence of a functional versatility of HP1 that is likely conserved also in mammals.
Materials and Methods
Drosophila strains
The l(3)H2A.V810 mutant line, Df(3R)TI-P deficiency that uncovers H2A.V, the P [RFP-HP1] stock, and the Su(var)205HMS00278 (HP1 RNAi) line were obtained from the Bloomington Stock Center. The H2A.V-gfp transgene was obtained from R. Saint and the C-terminally truncated (H2A.VΔCT) rescue constructs from Y. Rong (NCI, NHI, Bethesda, MD). Both lines were previously described.9 The 69B GAL4 driver, which expresses GAL4 in brains, was obtained from the Vienna Drosophila RNAi center (VDRC). The [RFP-HP1] H2A.V810 bearing chromosome were obtained by recombination and balanced over TM6c. Information on the genetic markers and balancers used in this study is available at Flybase (http://flybase.bio.indiana.edu/). Stocks were maintained and crosses were made on standard Drosophila medium at 25°C.
Chromosome cytology and immunostaining
DAPI-stained brain squashes were obtained according to published methods.56 Microtubule regrowth after cold was performed as previously described.34 HP1 immunostaining of mitotic chromosomes was carried out according to Fanti et al. (1998).36 Brain squashes for H2A.V immunostaining were prepared as previously indicated.57 For the other immunostaining experiments, brains from third instar larvae were dissected and fixed as previously described.58 After several rinses in phosphate buffered saline 0.1% Triton (PBS-T) brain preparations were incubated overnight at 4°C with primary antibodies diluted in PBS. After two rinses in PBS these primary antibodies were detected by incubation for 1 h with the appropriate secondary antibody. We used the following primary antibodies: anti-α tubulin monoclonal (1:1000, Sigma), rabbit anti-Spd2 (1:3500),28 rabbit anti-ZW10 (1:100),32 rabbit anti-cyclin B (1:2000) and rabbit anti-CENP-C (1:100) (gifts of Christian Lehner, Zurich, Switzerland), Chicken anti-CID (1:5000),59 rabbit anti-BubR1 (1:500), rabbit anti- Mis12 (1:100) and rabbit anti Nsl-1(1:100) (gifts from David Glover University of Cambridge, England), and rabbit anti-Ncd80 (1:50) (gifts of M. Goldberg, Cornell University Ithaca NY) rabbit-anti-Dgt6 (1:100),29 rabbit anti-H2AV (1:50; gift of V. Corces; Johns Hopkins University, Baltimore, MD), mouse monoclonal anti-HP1 (C1A9; 1:20, developed by L.Wallrath, University of Iowa, Iowa City, IA and obtained from the Developmental Studies Hybridoma Bank, created by the NICHD of the NIH and maintained at The University of Iowa, Department of Biology, Iowa City, (IA). The secondary antibodies were FITC-conjugated anti-mouse (1:100), Cy3-conjugated anti-rabbit (1:300), or Cy3-conjugated anti-chicken IgGs (1:100), all from Jackson Laboratories. All preparations are mounted in Vectashield H-1200 with DAPI (Vector Laboratories) to stain the chromosomes and examined with a Zeiss Axioplan microscope, equipped with an HBO100W mercury lamp and a cooled charged-coupled device (CCD camera; Photometrics CoolSnap HQ). Grayscale images were collected separately, converted to Photoshop (Adobe Systems), pseudocolored and merged.
To estimate the MT density, the Dgt6 and the HP1 fluorescence in wild-type and mutant cells, we used the ImageJ software (rsb.info.nih.gov/ij/). Mean pixel intensities of fluorescence were measured within a fixed region and the fluorescence intensity of an area adjacent to the region of interest was used for background subtraction. In particular, to quantify MT fluorescence intensity we considered a spindle area near (but not including) the chromosomes and an identical spindle area near the poles. Dgt6 was quantified considering a spindle area near (but not including) the chromosomes. To measure HP1 fluorescence intensity, we considered an area including the chromocenter in interphase nuclei, and an area including the pericentromeric regions of prophase, metaphase and anaphase chromosomes.
Production and purification of recombinant proteins
To obtain GST-HP1, – HP1ΔCD, -HP1ΔH and -HP1ΔCSD fusion proteins, the corresponding cDNAs were cloned in the pGEX-6P1 vectors as described previously.60 Bacterially expressed GST fusion proteins were purified by incubating crude lysates with glutathione sepharose beads (QIAGENE) as recommended by the manufacturer.
Protein extract preparation, GST pulldown and Western Blot
To obtain extracts for GST-pulldown, GFP-Trap and Western Blot analysis, dissected third instar larval brains were lysed in an ice-cold buffer containing 20 mM Hepes KOH pH 7.9, 1.5 mM MgCl2, 10 mM KCl, 420 mM NaCl, 30 mM NaF, 0.2 mM Na3Vo4, 25 mM BGP, 0.5 M PMSF, 0.1% NP40, 1ċ protease inhibitor cocktail (Roche).
For GST-pulldown and GFP-trap assays, protein extracts from ∼50 brains were incubated with 2 μg of each GST fusion protein bound to sepharose beads in a buffer containing 20 mM Hepes KOH, 20 mM NaF and 0.8% NP40 for 1 h at 4°C. Sepharose-bound GST proteins were collected by centrifugation and washed several times with 20 mM Hepes KOH, 20 mM NaF and 1.8% NP40.
For immunoblotting, protein samples were resuspended in 1X Laemmli Buffer, run into SDS polyacrilammide gels and electro-blotted on a nitrocellulose membrane (Bio-Rad) in a phosphate buffer containing 390 mM NaH2PO4H2O and 610 mM Na2HPO42H2O. After blocking with 5% low-fat dry milk, the membrane was probed with appropriate primary antibody. The blots were developed by the ECL or ECL Plus method (Amersham Biosciences) and signals detected with the ChemiDoc scanning system (BioRad). We used the following primary antibodies: monoclonal mouse anti-HP1 (C1A9; 1:500), rabbit anti-GFP (1:1000; a gift from G. Cestra, CNR, Rome, IT), anti-actin HRP conjugated (1:10000; Roche). Secondary antibodies were: sheep anti-mouse IgG HRP-conjugated (1:5000), or donkey anti-rabbit IgG HRP-conjugated (1:5000) (both from Amersham Biosciences).
Quantification of band intensities was obtained by using the densitometry software ImageJ.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank V. Corces, ML. Goldberg, G. Cestra, Y. Rong, C. Lehner, D. Glover and M. Gatti for providing reagents. We thank M.Gatti, P. Somma, J. Blum and members of Cenci lab for critical reading of the manuscript.
Funding
This work was funded by grants of Italian Association for Cancer Research (AIRC, IG 12749) to G.C and Progetto di Ateneo (Sapienza University of Rome) to F.V and G.C.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- 1.Kornberg RD, Thomas JO. Chromatin structure; oligomers of the histones. Science 1974; 184:865-8; PMID:4825888; http://dx.doi.org/10.1126/science.184.4139.865 [DOI] [PubMed] [Google Scholar]
- 2.Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 1997; 389:251-60; PMID:9305837; http://dx.doi.org/10.1038/38444 [DOI] [PubMed] [Google Scholar]
- 3.Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res 2011; 21:381-95; PMID:21321607; http://dx.doi.org/10.1038/cr.2011.22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kouzarides T. Chromatin modifications and their function. Cell 2007; 128:693-705; PMID:17320507; http://dx.doi.org/10.1016/j.cell.2007.02.005 [DOI] [PubMed] [Google Scholar]
- 5.Ruthenburg AJ, Li H, Patel DJ, Allis CD. Multivalent engagement of chromatin modifications by linked binding modules. Nat Rev Mol Cell Biol 2007; 8:983-94; PMID:18037899; http://dx.doi.org/10.1038/nrm2298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Henikoff S, Ahmad K. Assembly of variant histones into chromatin. Ann Rev Cell Dev Biol 2005; 21:133-53; PMID:16212490; http://dx.doi.org/10.1146/annurev.cellbio.21.012704.133518 [DOI] [PubMed] [Google Scholar]
- 7.Malik HS, Henikoff S. Phylogenomics of the nucleosome. Nat Struct Biol 2003; 10:882-91; PMID:14583738; http://dx.doi.org/10.1038/nsb996 [DOI] [PubMed] [Google Scholar]
- 8.Liu X, Li B, GorovskyMa. Essential and nonessential histone H2A variants in Tetrahymena thermophila. Mol Cell Biol 1996; 16:4305-11; PMID:8754831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Clarkson MJ, Wells JR, Gibson F, Saint R, Tremethick DJ. Regions of variant histone His2AvD required for Drosophila development. Nature 1999; 399:694-7; PMID:10385122; http://dx.doi.org/10.1038/21436 [DOI] [PubMed] [Google Scholar]
- 10.Ridgway P, Brown KD, Rangasamy D, Svensson U, Tremethick DJ. Unique residues on the H2A.Z containing nucleosome surface are important for Xenopus laevis development. J Biol Chem 2004; 279:43815-20; PMID:15299007; http://dx.doi.org/10.1074/jbc.M408409200 [DOI] [PubMed] [Google Scholar]
- 11.Faast R, Thonglairoam V, Schulz TC, Beall J, Wells JR, Taylor H, Matthaei K, Rathjen PD, Tremethick DJ, Lyons I. Histone variant H2A.Z is required for early mammalian development. Curr Biol 2001; 11:1183-7; PMID:11516949; http://dx.doi.org/10.1016/S0960-9822(01)00329-3 [DOI] [PubMed] [Google Scholar]
- 12.Ahmed S, Dul B, Qiu X, Walworth NC. Msc1 acts through histone H2A.Z to promote chromosome stability in Schizosaccharomyces pombe. Genetics 2007; 177:1487-97; PMID:17947424; http://dx.doi.org/10.1534/genetics.107.078691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rangasamy D, Greaves I, Tremethick DJ. RNA interference demonstrates a novel role for H2A.Z in chromosome segregation. Nat Struct Mol Biol 2004; 11:650-5; PMID:15195148; http://dx.doi.org/10.1038/nsmb786 [DOI] [PubMed] [Google Scholar]
- 14.Hou H, Wang Y, Kallgren SP, Thompson J, Yates JR, 3rd, Jia S. Histone variant H2A.Z regulates centromere silencing and chromosome segregation in fission yeast. J Biol Chem 2010; 285:1909-18; PMID:19910462; http://dx.doi.org/10.1074/jbc.M109.058487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Draker R, Cheung P. Transcriptional and epigenetic functions of histone variant H2A.Z. Biochem Cell Biol 2009; 87:19-25; PMID:19234520; http://dx.doi.org/10.1139/O08-117 [DOI] [PubMed] [Google Scholar]
- 16.Guillemette B, Gaudreau L. Reuniting the contrasting functions of H2A.Z. Biochem Cell Biol 2006; 84:528-35; PMID:16936825; http://dx.doi.org/10.1139/o06-077 [DOI] [PubMed] [Google Scholar]
- 17.Greaves IK, Rangasamy D, Devoy M, Marshall Graves JA, Tremethick DJ. The X and Y chromosomes assemble into H2A.Z-containing [corrected] facultative heterochromatin [corrected] following meiosis. Mol Cell Biol 2006; 26:5394-405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Meneghini MD, Wu M, Madhani HD. Conserved histone variant H2A.Z protects euchromatin from the ectopic spread of silent heterochromatin. Cell 2003; 112:725-36; PMID:12628191; http://dx.doi.org/10.1016/S0092-8674(03)00123-5 [DOI] [PubMed] [Google Scholar]
- 19.Sarcinella E, Zuzarte PC, Lau PN, Draker R, Cheung P. Monoubiquitylation of H2A.Z distinguishes its association with euchromatin or facultative heterochromatin. Mol Cell Biol 2007; 27:6457-68; PMID:17636032; http://dx.doi.org/10.1128/MCB.00241-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baldi S, Becker PB. The variant histone H2A.V of Drosophila–three roles, two guises. Chromosoma 2013; 122:245-58; PMID:23553272; http://dx.doi.org/10.1007/s00412-013-0409-x [DOI] [PubMed] [Google Scholar]
- 21.Joyce EF, Pedersen M, Tiong S, White-Brown SK, Paul A, Campbell SD, McKim KS. Drosophila ATM and ATR have distinct activities in the regulation of meiotic DNA damage and repair. J Cell Biol 2011; 195:359-67; PMID:22024169; http://dx.doi.org/10.1083/jcb.201104121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Madigan JP, Chotkowski HL, Glaser RL. DNA double-strand break-induced phosphorylation of Drosophila histone variant H2Av helps prevent radiation-induced apoptosis. Nucleic Acids Res 2002; 30:3698-705; PMID:12202754; http://dx.doi.org/10.1093/nar/gkf496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.van Daal A, Elgin SC. A histone variant, H2AvD, is essential in Drosophila melanogaster. Mol Biol Cell 1992; 3:593-602; PMID:1498368; http://dx.doi.org/10.1091/mbc.3.6.593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hardy S, Jacques PE, Gevry N, Forest A, Fortin ME, Laflamme L, Gaudreau L, Robert F. The euchromatic and heterochromatic landscapes are shaped by antagonizing effects of transcription on H2A.Z deposition. PLoS Genet 2009; 5:e1000687; PMID:19834540; http://dx.doi.org/10.1371/journal.pgen.1000687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Leach TJ, Mazzeo M, Chotkowski HL, Madigan JP, Wotring MG, Glaser RL. Histone H2A.Z is widely but nonrandomly distributed in chromosomes of Drosophila melanogaster. J Biol Chem 2000; 275:23267-72; PMID:10801889; http://dx.doi.org/10.1074/jbc.M910206199 [DOI] [PubMed] [Google Scholar]
- 26.Swaminathan J, Baxter EM, Corces VG. The role of histone H2Av variant replacement and histone H4 acetylation in the establishment of Drosophila heterochromatin. Genes Dev 2005; 19:65-76; PMID:15630020; http://dx.doi.org/10.1101/gad.1259105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Messina G, Damia E, Fanti L, Atterrato MT, Celauro E, Mariotti FR, Accardo MC, Walther M, Verni F, Picchioni D, et al. Yeti, an essential Drosophila melanogaster gene, encodes a protein required for chromatin organization. J Cell Sci 2014; 127:2577-88; PMID:24652835; http://dx.doi.org/10.1242/jcs.150243 [DOI] [PubMed] [Google Scholar]
- 28.Giansanti MG, Bucciarelli E, Bonaccorsi S, Gatti M. Drosophila SPD-2 is an essential centriole component required for PCM recruitment and astral-microtubule nucleation. Curr Biol 2008; 18:303-9; PMID:18291647; http://dx.doi.org/10.1016/j.cub.2008.01.058 [DOI] [PubMed] [Google Scholar]
- 29.Bucciarelli E, Pellacani C, Naim V, Palena A, Gatti M, Somma MP. Drosophila Dgt6 interacts with Ndc80, Msps/XMAP215, and gamma-tubulin to promote kinetochore-driven MT formation. Curr Biol 2009; 19:1839-45; PMID:19836241; http://dx.doi.org/10.1016/j.cub.2009.09.043 [DOI] [PubMed] [Google Scholar]
- 30.Somma MP, Fasulo B, Cenci G, Cundari E, Gatti M. Molecular dissection of cytokinesis by RNA interference in Drosophila cultured cells. Mol Biol Cell 2002; 13:2448-60; PMID:12134082; http://dx.doi.org/10.1091/mbc.01-12-0589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Karess R. Rod-Zw10-Zwilch: a key player in the spindle checkpoint. Trends Cell Biol 2005; 15:386-92; PMID:15922598; http://dx.doi.org/10.1016/j.tcb.2005.05.003 [DOI] [PubMed] [Google Scholar]
- 32.Williams BC, Karr TL, Montgomery JM, Goldberg ML. The Drosophila l(1)zw10 gene product, required for accurate mitotic chromosome segregation, is redistributed at anaphase onset. J Cell Biol 1992; 118:759-73; PMID:1339459; http://dx.doi.org/10.1083/jcb.118.4.759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Goshima G, Mayer M, Zhang N, Stuurman N, Vale RD. Augmin: a protein complex required for centrosome-independent microtubule generation within the spindle. J Cell Biol 2008; 181:421-9; PMID:18443220; http://dx.doi.org/10.1083/jcb.200711053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mottier-Pavie V, Cenci G, Verni F, Gatti M, Bonaccorsi S. Phenotypic analysis of misato function reveals roles of noncentrosomal microtubules in Drosophila spindle formation. J Cell Sci 2011; 124:706-17; PMID:21285248; http://dx.doi.org/10.1242/jcs.072348 [DOI] [PubMed] [Google Scholar]
- 35.Fanti L, Pimpinelli S. HP1: a functionally multifaceted protein. Curr Opin Genet Dev 2008; 18:169-74; PMID:18329871; http://dx.doi.org/10.1016/j.gde.2008.01.009 [DOI] [PubMed] [Google Scholar]
- 36.Fanti L, Giovinazzo G, Berloco M, Pimpinelli S. The heterochromatin protein 1 prevents telomere fusions in Drosophila. Mol Cell 1998; 2:527-38; PMID:9844626; http://dx.doi.org/10.1016/S1097-2765(00)80152-5 [DOI] [PubMed] [Google Scholar]
- 37.Musaro M, Ciapponi L, Fasulo B, Gatti M, Cenci G. Unprotected Drosophila melanogaster telomeres activate the spindle assembly checkpoint. Nat Genet 2008; 40:362-6; PMID:18246067; http://dx.doi.org/10.1038/ng.2007.64 [DOI] [PubMed] [Google Scholar]
- 38.Meireles AM, Fisher KH, Colombie N, Wakefield JG, Ohkura H. Wac: a new Augmin subunit required for chromosome alignment but not for acentrosomal microtubule assembly in female meiosis. J Cell Biol 2009; 184:777-84; PMID:19289792; http://dx.doi.org/10.1083/jcb.200811102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Carr AM, Dorrington SM, Hindley J, Phear GA, Aves SJ, Nurse P. Analysis of a histone H2A variant from fission yeast: evidence for a role in chromosome stability. Mol Gen Genet 1994; 245:628-35; PMID:NOT_FOUND; http://dx.doi.org/10.1007/BF00282226 [DOI] [PubMed] [Google Scholar]
- 40.Kim HS, Vanoosthuyse V, Fillingham J, Roguev A, Watt S, Kislinger T, Treyer A, Carpenter LR, Bennett CS, Emili A, et al. An acetylated form of histone H2A.Z regulates chromosome architecture in Schizosaccharomyces pombe. Nat Struct Mol Biol 2009; 16:1286-93; PMID:19915592; http://dx.doi.org/10.1038/nsmb.1688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rangasamy D, Berven L, Ridgway P, Tremethick DJ. Pericentric heterochromatin becomes enriched with H2A.Z during early mammalian development. EMBO J 2003; 22:1599-607; PMID:12660166; http://dx.doi.org/10.1093/emboj/cdg160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sharma U, Stefanova D, Holmes SG. Histone variant H2A.Z functions in sister chromatid cohesion in Saccharomyces cerevisiae. Mol Cell Biol 2013; 33:3473-81; PMID:23816883; http://dx.doi.org/10.1128/MCB.00162-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jacobs SA, Taverna SD, Zhang Y, Briggs SD, Li J, Eissenberg JC, Allis CD, Khorasanizadeh S. Specificity of the HP1 chromo domain for the methylated N-terminus of histone H3. EMBO J 2001; 20:5232-41; PMID:11566886; http://dx.doi.org/10.1093/emboj/20.18.5232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fan JY, Rangasamy D, Luger K, Tremethick DJ. H2A.Z alters the nucleosome surface to promote HP1alpha-mediated chromatin fiber folding. Mol Cell 2004; 16:655-61; PMID:15546624; http://dx.doi.org/10.1016/j.molcel.2004.10.023 [DOI] [PubMed] [Google Scholar]
- 45.Yamagishi Y, Sakuno T, Shimura M, Watanabe Y. Heterochromatin links to centromeric protection by recruiting shugoshin. Nature 2008; 455:251-5; PMID:18716626; http://dx.doi.org/10.1038/nature07217 [DOI] [PubMed] [Google Scholar]
- 46.Kang J, Chaudhary J, Dong H, Kim S, Brautigam CA, Yu HT. Mitotic centromeric targeting of HP1 and its binding to Sgo1 are dispensable for sister-chromatid cohesion in human cells. Mol Biol Cell 2011; 22:1181-90; PMID:21346195; http://dx.doi.org/10.1091/mbc.E11-01-0009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ainsztein AM, Kandels-Lewis SE, Mackay AM, Earnshaw WC. INCENP centromere and spindle targeting: Identification of essential conserved motifs and involvement of heterochromatin protein HP1. J Cell Biol 1998; 143:1763-74; PMID:9864353; http://dx.doi.org/10.1083/jcb.143.7.1763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nozawa RS, Nagao K, Masuda HT, Iwasaki O, Hirota T, Nozaki N, Kimura H, Obuse C. Human POGZ modulates dissociation of HP1 alpha from mitotic chromosome arms through Aurora B activation. Nat Cell Biol 2010; 12:719-U212; PMID:20562864; http://dx.doi.org/10.1038/ncb2075 [DOI] [PubMed] [Google Scholar]
- 49.Canzio D, Larson A, Narlikar GJ. Mechanisms of functional promiscuity by HP1 proteins. Trends Cell Biol 2014; 24:377-86; PMID:24618358; http://dx.doi.org/10.1016/j.tcb.2014.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Eissenberg JC, Elgin SC. HP1a: a structural chromosomal protein regulating transcription. Trends Genet 2014; 30:103-10; PMID:24555990; http://dx.doi.org/10.1016/j.tig.2014.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Obuse C, Iwasaki O, Kiyomitsu T, Goshima G, Toyoda Y, Yanagida M. A conserved Mis12 centromere complex is linked to heterochromatic HP1 and outer kinetochore protein Zwint-1. Nat Cell Biol 2004; 6:1135-U37; PMID:15502821; http://dx.doi.org/10.1038/ncb1187 [DOI] [PubMed] [Google Scholar]
- 52.Auth T, Kunkel E, Grummt F. Interaction between HP1alpha and replication proteins in mammalian cells. Exp Cell Res 2006; 312:3349-59; PMID:16950245; http://dx.doi.org/10.1016/j.yexcr.2006.07.014 [DOI] [PubMed] [Google Scholar]
- 53.Guenatri M, Bailly D, Maison C, Almouzni G. Mouse centric and pericentric satellite repeats form distinct functional heterochromatin. J Cell Biol 2004; 166:493-505; PMID:15302854; http://dx.doi.org/10.1083/jcb.200403109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Koch B, Kueng S, Ruckenbauer C, Wendt KS, Peters JM. The Suv39h-HP1 histone methylation pathway is dispensable for enrichment and protection of cohesin at centromeres in mammalian cells. Chromosoma 2008; 117:199-210; PMID:18075750; http://dx.doi.org/10.1007/s00412-007-0139-z [DOI] [PubMed] [Google Scholar]
- 55.Giansanti MG, Bonaccorsi S, Kurek R, Farkas RM, Dimitri P, Fuller MT, Gatti M. The class I PITP giotto is required for Drosophila cytokinesis. Curr Biol 2006; 16:195-201; PMID:16431372; http://dx.doi.org/10.1016/j.cub.2005.12.011 [DOI] [PubMed] [Google Scholar]
- 56.Cenci G, Rawson RB, Belloni G, Castrillon DH, Tudor M, Petrucci R, Goldberg ML, Wasserman SA, Gatti M. UbcD1, a Drosophila ubiquitin-conjugating enzyme required for proper telomere behavior. Genes Dev 1997; 11:863-75; PMID:9106658; http://dx.doi.org/10.1101/gad.11.7.863 [DOI] [PubMed] [Google Scholar]
- 57.Ciapponi L, Cenci G, Gatti M. The Drosophila Nbs protein functions in multiple pathways for the maintenance of genome stability. Genetics 2006; 173:1447-54; PMID:16648644; http://dx.doi.org/10.1534/genetics.106.058081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bonaccorsi S, Giansanti MG, Gatti M. Spindle assembly in Drosophila neuroblasts and ganglion mother cells. Nat Cell Biol 2000; 2:54-6; PMID:10620808; http://dx.doi.org/10.1038/71378 [DOI] [PubMed] [Google Scholar]
- 59.Blower MD, Karpen GH. The role of Drosophila CID in kinetochore formation, cell-cycle progression and heterochromatin interactions. N Cell Biol 2001; 3:730-9; PMID:11483958; http://dx.doi.org/10.1038/35087045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Raffa GD, Siriaco G, Cugusi S, Ciapponi L, Cenci G, Wojcik E, Gatti M. The Drosophila modigliani (moi) gene encodes a HOAP-interacting protein required for telomere protection. Proc Natl Acad Sci U S A 2009; 106:2271-6; PMID:19181850; http://dx.doi.org/10.1073/pnas.0812702106 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.