

Abstract
Barrett's esophagus (BE) is defined as an incomplete intestinal metaplasia characterized generally by the presence of columnar and goblet cells in the formerly stratified squamous epithelium of the esophagus. BE is known as a precursor for esophageal adenocarcinoma. Currently, the cell of origin for human BE has yet to be clearly identified. Therefore, we investigated the role of Notch signaling in the initiation of BE metaplasia. Affymetrix gene expression microarray revealed that BE samples express decreased levels of Notch receptors (NOTCH2 and NOTCH3) and one of the the ligands (JAG1). Furthermore, BE tissue microarray showed decreased expression of NOTCH1 and its downstream target HES1. Therefore, Notch signaling was inhibited in human esophageal epithelial cells by expression of dominant-negative-Mastermind-like (dnMAML), in concert with MYC and CDX1 overexpression. Cell transdifferentiation was then assessed by 3D organotypic culture and evaluation of BE-lineage specific gene expression. Notch inhibition promoted transdifferentiation of esophageal epithelial cells toward columnar-like cells as demonstrated by increased expression of columnar keratins (K8, K18, K19, K20) and glandular mucins (MUC2, MUC3B, MUC5B, MUC17) and decreased expression of squamous keratins (K5, K13, K14). In 3D culture, elongated cells were observed in the basal layer of the epithelium with Notch inhibition. Furthermore, we observed increased expression of KLF4, a potential driver of the changes observed by Notch inhibition. Interestingly, knockdown of KLF4 reversed the effects of Notch inhibition on BE-like metaplasia. Overall, Notch signaling inhibition promotes transdifferentiation of esophageal cells toward BE-like metaplasia in part via upregulation of KLF4. These results support a novel mechanism through which esophageal epithelial transdifferentiation promotes the evolution of BE.
Keywords: Barrett's esophagus, KLF4, metaplasia, Notch signaling, transdifferentiation
Abbreviations
- BE
Barrett's esophagus; CDX1, caudal type homeobox 1; dnMAML, dominant-negative Mastermind-like; DSC1, desmocollin 1; DSC3, desmocollin 3; EAC, esophageal adenocarcinoma; EGJ, esophago-gastric junction; ESC, embryonic stem cell; ESCC, esophageal squamous cell carcinoma; GERD, gastroesophageal reflux disease; GSI, gamma-secretase inhibitor; HES1, hairy and enhancer of split-1; ICN1, intracellular Notch 1; iPSC, inducible pluripotent stem cell; KLF4, Krüppel-like factor 4; JAG1, Jagged1; LOH, loss of heterozygosity; RBPJ (CSL), recombinant binding protein of hairless; TMA, tissue microarray
Introduction
Barrett's esophagus (BE) is defined as an incomplete intestinal metaplasia of the esophagus. BE is classically characterized by the presence of differentiated intestinal columnar cells and post-mitotic mucin-producing goblet cells, and is estimated to have a prevalence of 5–6% in the US.1,2 Gastroesophageal reflux disease (GERD), abdominal obesity, smoking and Helicobacter pylori infection eradication have been linked as factors associated with the development of BE. The local pro-inflammatory microenvironment plays a critical role in the development and maintenance of BE.1,3 BE can progress to low-grade dysplasia, high-grade dysplasia and culminates in the development of esophageal adenocarcinoma (EAC). It is estimated that 0.5% of BE patients will develop EAC, and this risk increases to 10% in BE patients with high-grade dysplasia.2
In 2013, an estimated 17,990 patients were diagnosed with esophageal cancer in the US, both EAC and esophageal squamous cell carcinoma (ESCC). Esophageal cancer is one of the deadliest cancers in the US with a 5-year survival rate of 17.3% in 2013.4 Therefore, studying the molecular mechanisms underlying the pathogenesis of BE could provide novel biomarkers or prognostic indicators for both BE and EAC patients. The study of BE has been historically limited to human biopsy samples, which have been used to analyze histopathological and genetic changes. Some of the known alterations occurring in BE are the methylation of p16 and loss of heterozygosity (LOH) of p16 and p53.5 A mosaic pattern of genetic alterations can be found in biopsy samples, complicating the identification of the initiating genetic changes that lead to BE. There have been several hypotheses proposed for the cell of origin of BE.6 These include: (1) transdifferentiation of cells from the esophageal basal layer or from the ducts of the esophageal submucosal glands, (2) migration of cells located at the esophago-gastric junction (EGJ) or in the gastric cardia7,8 and (3) bone marrow derived progenitor cells.9
Recent studies have undertaken a genetic approach to investigate the initiating events that lead to the development of BE. We have identified CDX1 and MYC (c-Myc) as having functional roles in the development of BE.10 CDX1 is part of the caudal homeobox family of transcription factors (CDX1 and CDX2), which are important in the development and differentiation of the small intestine and colon.11 In fact, conditional knockout of Cdx2 in mouse intestine results in squamous metaplasia.12 MYC is a transcription factor known to bind to E-box sequences and can activate 15% of all genes in the human genome.13,14 MYC is overexpressed in many cancers, including amplification in EAC.15,16 MYC is involved in the activation and regulation of a variety of cellular processes such as cell cycle progression, cell differentiation, energy metabolism, angiogenesis and DNA damage repair.14 Our microarray analysis of BE samples revealed that negative regulators of MYC, namely MXI1 and MXD1, were downregulated in BE human tissue. Conversely, MYC target genes ODC1 and CA2 were increased suggesting that MYC is active in BE.10 In addition, the microarray data showed increased CDX1 and CDX2 expression in BE.17 Our previous studies using a human esophageal epithelial cell line immortalized with hTERT (EPC2-hTERT) have allowed us to study human esophageal biology.17 Our previous data have shown that CDX1 overexpression together with MYC in EPC2-hTERT cells can lead to a partial change toward BE.10 This partial change toward BE suggests the need to explore additional gene changes in the context of MYC and CDX1.
The Notch signaling pathway regulates cell fate and differentiation through cell-cell communication. Interestingly, loss of Notch signaling is required for the differentiation of the goblet cell lineage in the small intestine.18,19 In addition, inhibition of Notch signaling in the small intestine by either γ-secretase inhibitor (GSI), or conditional knockout of Rbpj (CSL), can lead to goblet cell hyperplasia.18,20,21 The Notch signaling pathway is comprised of 4 homologous transmembrane Notch receptors (NOTCH1-NOTCH4) that can be activated by transmembrane ligands, Delta or Jagged, generally expressed by neighboring cells.22,23 Upon ligand binding, Notch receptors undergo cleavage by ADAM-family metalloproteases at the extracellular domain and by γ-secretase at the intracellular domain.22 These events lead to the release of the intracellular domain (ICN) allowing its nuclear translocation, binding to CSL/RBPJ and recruitment of co-activators from the Mastermind-like family (MAML). This leads to the activation of Notch target genes, such as HES1 and HES5.22,24 Interestingly, Notch signaling is known to be involved in cell fate decisions in several cell types including lymphocytes, neurons, skin and others.24
Notch signaling can be a negative regulator of Krüppel-like factor 4 (KLF4) expression in the small intestine. Indeed, it has been demonstrated previously that KLF4 promoter contains ICN-responsive elements, through which Notch can inhibit KLF4 expression.25,26 KLF4 is part of a family of DNA-binding transcription factors that has been shown to play a role in multiple processes such as proliferation, cell differentiation, inflammation and pluripotency. Furthermore, KLF4 is one of the key factors that can reprogram somatic cells to inducible pluripotent stem cells (iPSC).27 Recently, high KLF4 expression was reported in human BE biopsies; it was also shown that its promoter is activated by bile acid.28 In addition, KLF4 can increase the transcriptional activity of MUC2 and CDX2, suggesting a potential role in BE development.28
In this study, we utilize an innovative 3-dimensional (3D) organotypic culture model system to demonstrate that the cooperation of MYC, CDX1, and inhibition of Notch signaling promotes a switch of cell identity and lineage specification from the normal esophageal squamous epithelium to a BE-like metaplasia mediated through KLF4. Our data support the novel paradigm in which transdifferentiation of esophageal basal cells leads to initiation of BE.
Results
Notch signaling is downregulated in human Barrett's esophagus
In order to investigate the status of Notch signaling in Barrett's esophagus (BE), we performed a RNA microarray on human BE biopsies compared to adjacent normal squamous esophagus (GEO accession #GSE13083).10 A significant decrease in expression of NOTCH2 and NOTCH3 receptors (4- and fold2-, respectively) was observed in BE versus paired normal squamous esophagus. Expression of the Notch ligand Jagged1 (JAG1) was also decreased by fold3- in BE biopsies, thereby suggesting a downregulation of Notch signaling in BE (Fig. 1A). We next performed IHC of human BE and normal esophagus tissue microarray (TMA) for NOTCH1 receptor, the active form of NOTCH1, ICN1 (Intracellular NOTCH1) and its key downstream target, the transcription factor HES1 (Fig. 1B-C). We observed positive nuclear staining for HES1 and NOTCH1 restricted to the basal layer of normal esophageal epithelium, whereas ICN1 showed more diffuse cytoplasmic staining (Fig. 1B). In BE, we observed a loss of nuclear staining for HES1 and loss of nuclear and membranous staining for NOTCH1, and decreased staining for ICN1 (Fig. 1B). The TMA was scored for intensity of staining (Table S1). HES1, NOTCH1 and ICN1 protein expression is significantly decreased in BE when compared to normal esophagus (Fig. 1C). Thus, data from both the RNA microarray and TMA suggest that Notch signaling is downregulated in BE compared to the normal squamous esophageal epithelium. Therefore, Notch signaling may be required for the maintenance of normal esophageal squamous epithelium.
Figure 1.
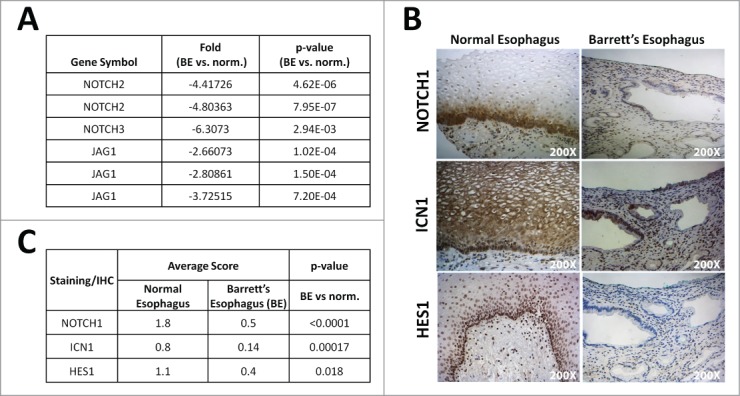
Notch signaling is decreased in human Barrett's esophagus. (A) Microarray analysis from 7 samples of Barrett's esophagus (BE) and their adjacent normal squamous esophagus for status of Notch signaling pathway, GEO accession Inhibition of Notch signaling in esophageal epithelial cells changes basal cell morphology in 3D cultures. #GSE13083. (B) Representative image of normal esophagus and Barrett's esophagus tissue from tissue microarray (TMA) stained for NOTCH1, ICN1 and HES1 (200X Magnification). (C) Average scoring for positive staining in the TMA and statistical analysis using Fisher's exact t-test.
Inhibition of Notch signaling induces morphological changes in esophageal epithelial cells
Our previous data have shown that CDX1 overexpression together with MYC in human immortalized esophageal epithelial EPC2-hTERT cells can initiate changes toward Barrett's esophagus.10 Yet, changes observed showed only partial transdifferentiation, suggesting additional genetic alterations may be required in the context of MYC and CDX1 overexpression. We therefore inhibited Notch signaling in the context of MYC and CDX1. We infected EPC2-hTERT-MYC-CDX1 (MYC-CDX1) cells with a construct encoding for a dominant-negative form of the co-activator Mastermind-like (dnMAML) to inhibit Notch signaling (MYC-CDX1-dnMAML cells). We confirmed expression of dnMAML-GFP tagged protein by Western blotting using a GFP specific antibody (Fig. 2A). To verify Notch signaling abolition following dnMAML overexpression, we used an 8X-CSL-Luciferase reporter construct, which upon expression of ICN1 activates luciferase expression. We observed a significant inhibition (9.98-fold) of luciferase activity in the presence of dnMAML (MYC-CDX1-dnMAML-ICN1) compared to MYC-CDX1-ICN1 cells (Fig. 2B). We also confirmed downregulation of Notch target genes expression by dnMAML via quantitative PCR (qPCR). Indeed, we observed a significant decrease of HES1 (3.fold3-) and HES5 (12.fold5-) expression in MYC-CDX1-dnMAML when compared to MYC-CDX1 cells (Fig. 2C). These results support that dnMAML overexpression is sufficient to inhibit Notch signaling.
Figure 2.

Inhibition of Notch signaling in esophageal epithelial cells changes basal cell morphology in 3D cultures. (A) Western blotting for GFP (dnMAML), MYC and CDX1 in EPC2-hTERT, MYC-CDX1 and MYC-CDX1-dnMAML cells. (B) Luciferase assay with Notch-responsive pGL3–8XCSL reporter vector in MYC-CDX1-ICN1 and MYC-CDX1-ICN1-dnMAML cells, graph represents mean ± SEM (n = 3). Student t-test was performed to determine significance, *P ≤ 0.05. (C) Quantitative PCR (qPCR) for Notch downstream targets HES1 and HES5 in MYC-CDX1 and MYC-CDX1-dnMAML cells. Graph represents mean ± SEM (n = 3) and student t-test was performed to determine significance, *P ≤ 0.05. (D) H&E staining of representative 3D organotypic cultures of MYC-CDX1 and MYC-CDX1-dnMAML cells, arrow indicates elongated cells, (200X Magnification). (E) Electron microscopy of MYC-CDX1 and MYC-CDX1-dnMAML 3D organotypic cultures, scale bars = 0.2 μm. (F) Graph represents relative height of MYC-CDX1 and MYC-CDX1-dnMAML basal layer cells mean ± SEM (n = 4). Student t-test was performed to determine significance, *P ≤ 0.0001.
We next used 3D organotypic cultures to analyze changes in cell differentiation and morphology.29 We observed that MYC-CDX1-dnMAML cells formed a thinner stratified epithelium than MYC-CDX1 cells, suggesting disruption of normal stratification and differentiation. We also noted that MYC-CDX1-dnMAML 3D cultures showed an altered cell morphology in the basal layer (Fig. 2D), when compared to MYC-CDX1 cells. In order to further characterize these changes in the basal layer, we performed electron microscopy of MYC-CDX1 and MYC-CDX1-dnMAML cultures (Fig. 2E). We observed an elongation of MYC-CDX1-dnMAML basal cells when compared to MYC-CDX1 cells, consistent with acquisition of columnar-like morphology. Indeed, basal cellular height was significantly increased (1.fold4-) in 3D cultures overexpressing dnMAML (Fig. 2F) compared to MYC-CDX1. These changes in cell morphology in the basal layer of MYC-CDX1-dnMAML cells suggest that the inhibition of Notch signaling promotes transdifferentiation of the normal esophageal squamous epithelium toward a more columnar-like epithelium.
Inhibition of Notch signaling induces a switch from squamous to columnar gene expression
We further analyzed our 3D cells to investigate if the morphological changes observed in MYC-CDX1-dnMAML cells reflect changes in cell lineages markers. We stained sections for the squamous keratin 13 (K13). In MYC-CDX1 cells, we observed strong staining for K13 in the suprabasal region, whereas staining was significantly reduced in MYC-CDX1-dnMAML cells. Conversely, we observed increased staining of columnar keratin 19 (K19) in both the basal and suprabasal compartment in MYC-CDX1-dnMAML cells compared to MYC-CDX1 cells (Fig. 3B).
Figure 3.
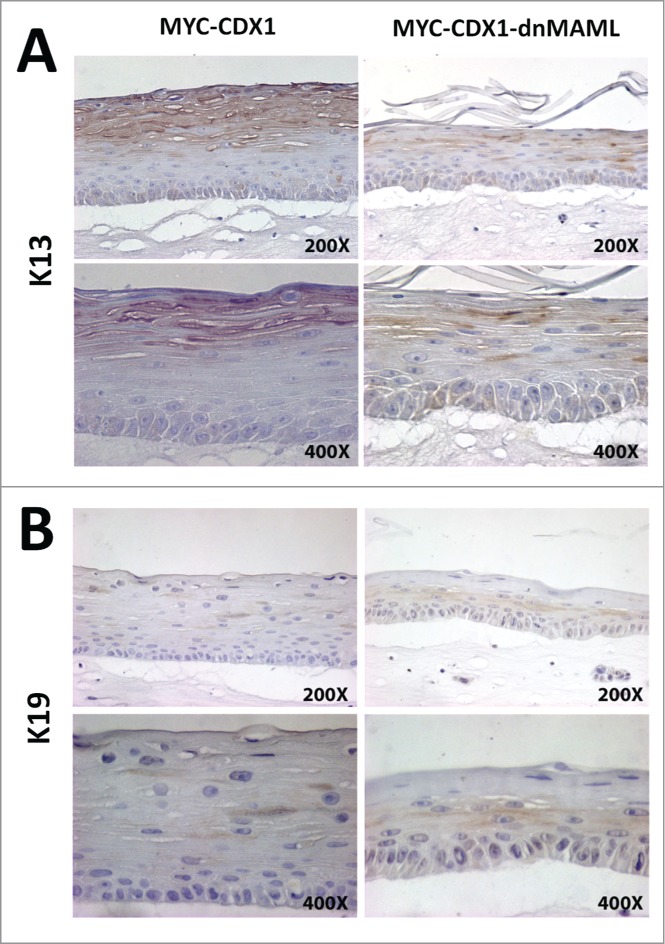
Inhibition of Notch signaling in esophageal epithelial cells decreases squamous K13+ cells and increases columnar K19+ cells in 3D organotypic culture. IHC staining of 3D organotypic cultures for squamous keratin K13 (A) and columnar keratin K19 (B) in MYC-CDX1 (left panel) and MYC-CDX1-dnMAML cultures (right panel) (200X and 400X Magnification).
We next used qPCR to evaluate additional squamous and columnar lineage keratins expression in MYC-CDX1-dnMAML cells. Prior to harvesting, cells were grown in the presence of calcium (0.6 mmol/L) for 48 hrs to allow squamous differentiation.30 We observed that MYC-CDX1-dnMAML cells expressed reduced levels of squamous keratins: K5 (fold5-), K13 (16.fold6-) and K14 (fold5-) (Fig. 4A). Furthermore, MYC-CDX1-dnMAML cells expressed higher levels of columnar keratins: K8 (2.fold2-), K18 (2.fold8-), K19 (1.fold9-) and K20 (2.fold8-) compared to MYC-CDX1 cells (Fig. 4B). These results suggest that inhibition of Notch signaling via dnMAML promotes a switch in gene expression from squamous to columnar keratins. Furthermore, since BE is often characterized by the presence of goblet cells in the esophageal epithelium, we investigated expression of mucins, the major protein family secreted by this cell type. Interestingly, we observed increased levels of MUC2 (10.fold4-), MUC3B (21.fold5-), MUC5B (305-fold) and MUC17 (116.fold3-) in MYC-CDX1-dnMAML cells when compared to MYC-CDX1 cells (Fig. 4C). Thus, inhibition of Notch signaling fosters expression of goblet cell lineage genes. Next, we quantified the expression of the squamous differentiation genes desmocollin1 and desmocollin3 (DSC1, DSC3). We observed that dnMAML overexpression in MYC-CDX1 cells decreased significantly DSC1 (fold20-) and DSC3 (3.fold7-) expression (Fig. 4D). Lastly, we investigated whether Notch signaling inhibition promoted the expression of CDX2. Previous studies have shown loss of Notch signaling can promote CDX2 and MUC2 expression in EAC cell lines.31 Interestingly, we observed significant increased CDX2 RNA expression in the MYC-CDX1-dnMAML cells, but we did not observe increased CDX2 protein expression by western blotting or IHC (data not shown). Taken together, these data support the premise that inhibition of Notch signaling in cooperation with MYC and CDX1 orchestrates a genetic switch from a squamous cell lineage to an intestinal columnar cell lineage.
Figure 4.:
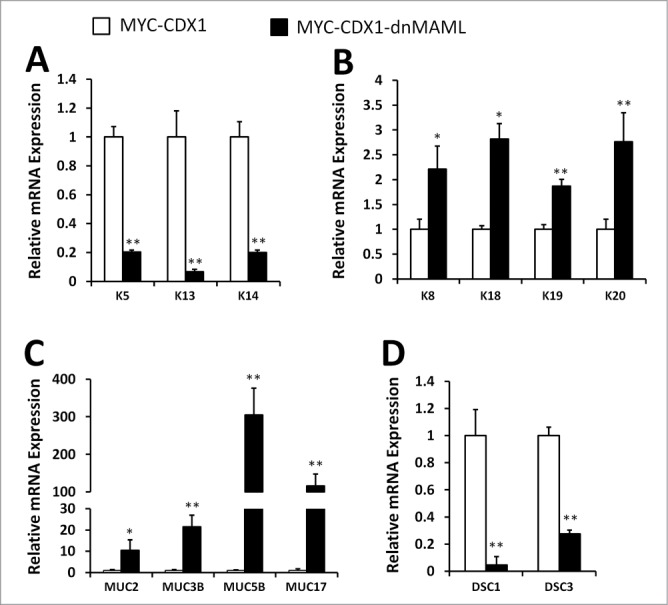
Inhibition of Notch signaling in esophageal epithelial cells promotes a switch from the squamous lineage to a columnar lineage. qPCR of (A) squamous keratins K5, K13 and K14; (B) columnar keratins K8, K18, K19 and K20; (C) mucin genes MUC2, MUC3B, MUC5B and MUC17; (D) and differentiation genes DSC1 and DSC3 in MYC-CDX1 and MYC-CDX1-dnMAML cells. Graph represents mean ± SEM (n = 6). Student t-test was performed to determine significance, *P ≤ 0.05, **P ≤ 0.001.
Recent studies show decreased expression of Notch signaling pathway in BE biopsies and BE-derived cell lines.31,32 Furthermore, previous studies have shown low expression of HES1 in the BE cell lines CP-A and CP-C.32 Therefore, we analyzed BE cell lines (CP-A, CP-B, CP-C and CP-D) for effectors of Notch signaling. Interestingly, we observed low expression of Notch target genes (HES1, HES5, HEY1 and HEY2) in all 4 BE cell lines (data not shown). Studies using the pharmacological inhibition of Notch signaling in EAC cell lines, through the use of a γ-secretase inhibitor (GSI), show increased expression of CDX2 and MUC2.31 Therefore, we inhibited Notch signaling in the BE cell line CP-A with a GSI. Interestingly, we observed that inhibition of Notch signaling caused a significant increase in the expression of the columnar keratins K18 and K19 and a significant increase of MUC3B (Figure S1B-C). We also observed a trend for increased expression of MUC2, MUC5AC and MUC17. These day imply that inhibition of Notch signaling in BE cells may drive further differentiation toward the secretory lineage (Figure S1C).
KLF4 knockdown reverses the morphological and transcriptional changes following Notch signaling inhibition
Classically, Notch signaling leads to activation of transcriptional factors such as HES1 and HES5. In the intestine, HES1 has been shown to be a negative regulator of HATH1, thereby promoting absorptive cell fate over secretory cell fate.33,34 Therefore, we investigated if downregulation of HES1 could mimic the results obtained with dnMAML overexpression in MYC-CDX1 cells. We performed stable HES1 knockdown with 2 independent shRNA constructs (shHES1 #1 and shHES1 #2) in MYC-CDX1 cells (Figure S2A). We did not observe any changes in squamous and columnar keratins or in mucin genes expression upon HES1 knockdown suggesting that HES1 alone is not sufficient to support transdifferentiation esophageal epithelial cells (Figure S2B-D). We observed a trend of upregulation of HATH1 expression validating the functional HES1 knockdown (Figure S2E). Thus, HES1 knockdown could not recapitulate the effects of Notch signaling inhibition, suggesting that Notch might act via other downstream targets to regulate transdifferentiation to BE.
Therefore, we next analyzed KLF4 as a potential downstream effector of inhibition of Notch signaling. Active Notch signaling mediated by ICN1 downregulates KLF4 expression in the intestinal epithelium.26 Conversely, inhibition of Notch signaling via GSI can cause upregulation of KLF4 expression.25 Interestingly, KLF4 expression is increased in MYC-CDX1-dnMAML cells when compared to MYC-CDX1 cells (Fig. 5A, 5B). Therefore, we investigated whether KLF4 knockdown in MYC-CDX1-dnMAML cells could reverse morphological changes observed following dnMAML overexpression. Using a stable lentiviral infection, we achieved significant KLF4 knockdown using 2 independent shRNA sequences in MYC-CDX1-dnMAML cells (Fig. 5C, 5D). We observed the highest degree of KLF4 knockdown in the MYC-CDX1-dnMAML-shKLF4 #3 cells (3.fold3- in RNA and 25-fold in protein). Stable KLF4 knockdown results in a decrease in elongated (columnar-like) cells observed with inhibition of Notch signaling in 3D culture (Fig. 5E). Furthermore, cells at the basal layer of the epithelium have a more cuboidal shape, suggesting that the inhibition of KLF4 in the MYC-CDX1-dnMAML cells can reverse morphological changes observed with inhibition of Notch signaling.
Figure 5.

KLF4 knockdown reverses partially the morphological changes induced by Notch signaling inhibition in 3D cultures. (A) qPCR of KLF4 expression in MYC-CDX1 and MYC-CDX1-dnMAML cells. (B) Western blotting for KLF4 in EPC2-hTERT, MYC-CDX1 and MYC-CDX1-dnMAML cells. (C) qPCR of KLF4 in MYC-CDX1-dnMAML-shScramble and MYC-CDX1-dnMAML-shKLF4 cells. (D) Western blotting for KLF4 in MYC-CDX1-dnMAML-shScramble and MYC-CDX1-dnMAML-shKLF4 cells. (E) H&E staining of MYC-CDX1-dnMAML-shScramble and MYC-CDX1-dnMAML-shKLF4 3D organotypic cultures (400X Magnification). Graph represents mean ± SEM (n = 6). Student t-test was performed to determine significance, *P ≤ 0.01.
Analysis of columnar keratins showed significantly decreased K8 and K20 expression, but no changes in K18 and K19 in MYC-CDX1-dnMAML-shKLF4 cells (Fig. 6A). We also observed a significant decrease in MUC2 and MUC5B expression, but no changes in MUC3B and MUC17 (Fig. 6B). Furthermore, we evaluated expression of the squamous keratins K5, K13 and K14 in MYC-CDX1-dnMAML-shKLF4 cells and MYC-CDX1-dnMAML-shScramble cells. KLF4 knockdown significantly increased expression of K5, K13 and K14; supporting the premise that KLF4 knockdown can partially reverse the switch from squamous to columnar keratins observed with Notch signaling inhibition (Fig. 6C). Moreover, expression of the squamous differentiation marker DSC1 is increased significantly in MYC-CDX1-dnMAML-shKLF4 cells (Fig. 6D). Overall, these results show that dnMAML-induced transdifferentiation is in part mediated by KLF4. Herein, we demonstrate a novel function for Notch signaling in BE development mediated by KLF4. Importantly, these data support the model in which esophageal epithelial basal cells might serve as the cell of origin for Barrett's esophagus metaplasia in our experimental system.
Figure 6.
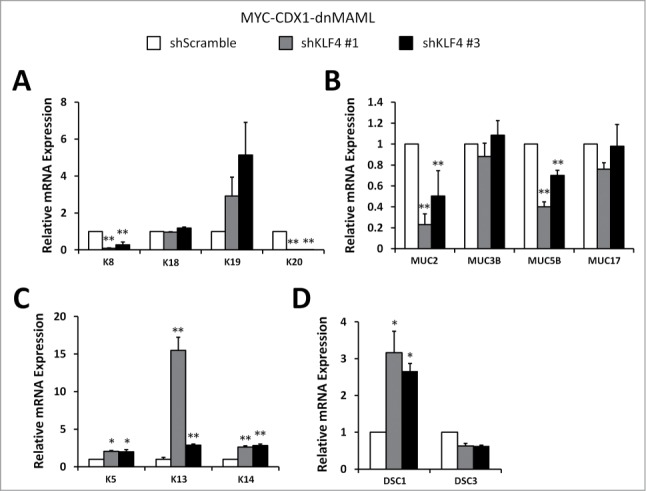
KLF4 knockdown reverses lineage changes induced by Notch signaling inhibition in esophageal epithelial cells. qPCR of (A) columnar keratins K8, K18, K19 and K20; (B) mucin genes MUC2, MUC3B, MUC5B and MUC17; (C) squamous keratins K5, K13 and K14; (D) and squamous differentiation markers DSC1 and DSC3 in MYC-CDX1-dnMAML-shScramble and MYC-CDX1-dnMAML-shKLF4 cells. Graph represents mean ± SEM (n = 6). Student t-test was performed to determine significance, *P ≤ 0.01, **P ≤ 0.001.
Discussion
Barrett's esophagus (BE) is defined as an incomplete intestinal metaplasia of the esophagus, and the biological mechanisms underlying its development remain to be clarified. Herein, we demonstrate that Notch signaling is downregulated in human BE, suggesting that development of intestinal metaplasia in the esophagus could require inhibition of Notch signaling. Indeed, inhibition of Notch promotes transdifferentiation in our model system. First, there is the appearance of elongated columnar-like cells in the basal layer of 3D organotypic cultures in response to Notch inhibition. Second, there is a gene expression switch that denotes the squamous vs. BE lineages, characterized by a robust diminution of squamous keratins and differentiation markers in favor of a strong induction of columnar keratins and mucins. These results suggest that the combination of Notch inhibition and MYC and CDX1 overexpression promotes transdifferentiation of esophageal epithelial cells toward a BE-like metaplasia state.
Transdifferentiation may be viewed as reprogramming as it involves the replacement of one cell type into another. The cell of origin for development of BE remains the subject of investigation, but transdifferentiation of esophageal stratified epithelium is one of the proposed models.6 One study of human BE biopsies has revealed that esophageal cells undergo a transition of expression of intestinal markers like CDX2 and MUC5AC.35 The Notch signaling pathway is known as important mediator of tissue development and homeostasis as well as identity of cell lineages and differentiation. Interestingly, NOTCH1 activation is observed at the onset of squamous differentiation of the esophageal epithelium. NOTCH1 and NOTCH3 orchestrate transcriptional regulation of early differentiation markers in a CSL-dependent manner. Perturbation of esophageal squamous differentiation is notably observed following loss of Notch signaling in the esophageal epithelium.36 Furthermore, loss of Notch signaling is required for the differentiated goblet cells and other secretory cell lineages in the small intestine.18,19 Herein, we demonstrate a potential role of Notch signaling in the identity of cell lineages. Our data show that loss of Notch signaling in the context of MYC and CDX1 can drive deregulation of the normal esophageal epithelium and the acquisition of expression of BE lineage markers.
We show that the combination of Notch inhibition with MYC and CDX1 overexpression leads to characteristic features of BE, namely the production of mucin by goblet-like cells and the presence of columnar-like cells. It was suggested previously that HATH1 induction by Notch inhibition induces MUC2 via CDX2 expression in esophageal cancer cell lines, although our data suggest that HATH1 may not be involved (data not shown).31 Moreover, goblet cell differentiation was induced by Notch inhibition in the L2-IL-1β mouse model of Barrett's-like metaplasia.8 Hence, Notch signaling inhibition could be necessary for the initiation of the BE metaplasia program by orchestrating transcriptional regulation of key genes implicated in goblet cell terminal differentiation.
KLF4 can be regulated negatively by Notch signaling.25 Herein, we demonstrate that inhibition of Notch signaling causes activation of KLF4 expression and that KLF4 knockdown reverses some of the genetic and morphological changes induced by Notch signaling inhibition. These data support KLF4 as a potential driver in the activation of intestinal cell lineage genes upon inhibition of Notch signaling, suggesting a new mechanism through which Notch signaling promotes BE initiation. Interestingly, KLF4 is strongly expressed in Barrett's esophagus and its expression is induced in response to bile acids. KLF4 and CDX2 also cooperate to induce production of MUC2.28 Interestingly, KLF4 has been linked to other models of transdifferentiation, including conversion of smooth muscle cells into osteogenic cells in the context of hyperphosphatemia and conversion of fibroblasts into neural progenitors or cardiomyocytes.37-39 KLF4 is one of the key factors (OCT4, SOX2, MYC, NANOG and KLF4) that can reprogram the fate of somatic cells into inducible pluripotent stem cells (iPSC). KLF4 is also recognized for its capacity to maintain the pluripotent state of embryonic stem cells (ESC).27 Therefore, KLF4 activation in response to Notch inhibition could facilitate transdifferentiation of esophageal squamous cells into intestinal-like cells by binding to promoters of columnar keratins and mucin genes to enhance their expression (Fig. 7).
Figure 7.
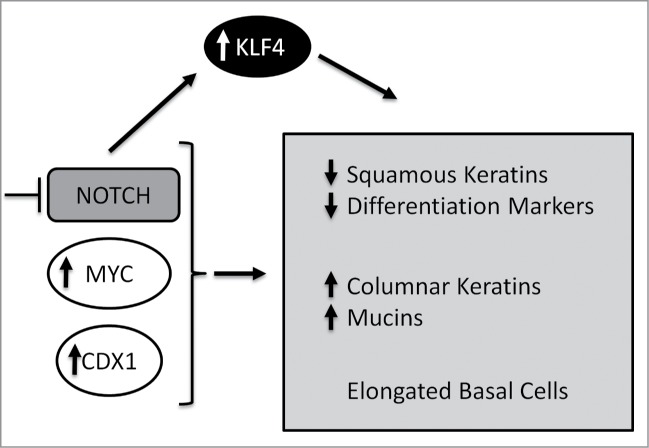
Model. Inhibition of Notch signaling in conjunction with MYC and CDX1 expression promotes increased expression of columnar keratins and mucin genes as well as decreased expression of squamous keratins and other markers of differentiation. Inhibition of Notch also triggers changes in cell morphology in the basal layer. Inhibition of Notch signaling promotes KLF4 expression and the initiation of a transdifferentiation program toward a BE-like metaplasia.
Herein, we provide evidence to support the notion that esophageal basal cells might serve as potential cells of origin for BE. Several models of BE cells of origin are proposed and they are not necessarily mutually exclusive and may be context dependent. One model suggests that cells may migrate from the esophago-gastric junction (EGJ) or from the gastric cardia.7 By lineage-labeling gastric cardia stem cells (LGR5+) in the L2-IL-1β mouse model of BE and EAC, it was demonstrated that migration of gastric cardia cells gives rise to BE tissue.8 It is also possible that bone marrow derived progenitor cells give rise to BE. Indeed, male to female bone marrow transplants in a severe reflux esophagitis rat model, showed that the developing BE epithelium was of male origin, suggesting that stem cells originating from the bone marrow can contribute to BE development.9
Metaplasia may represent an adaptive response to a stressful local environment, and involves a complex interplay between epigenetic and genetic alterations. It can occur in several tissues including the esophagus (BE), stomach, pancreas, lung, cervix and skin. Metaplasia may be reversible or irreversible, and may progress to dysplasia and cancer. For example, BE can progress to low-grade and high-grade dysplasia and culminates in esophageal adenocarcinoma (EAC).2 Interestingly, Notch signaling is highly context-dependent, specifically in its role in cancers. Recent genome-wide mutational analysis studies comparing ESCC and EAC showed inactivating mutations of Notch1 were found only in ESCC.40 Notably, Notch has been shown to act as both a tumor suppressor and oncogene in ESCC.36,41 By contrast, in EAC, our TMA (data not shown) and other studies show an increased expression of Notch signaling during the progression from BE to EAC.8,32 Thus, Notch may have oncogenic properties during the progression from BE metaplasia to esophageal adenocarcinoma.
Herein, we observe an initiation of transdifferentiation of esophageal epithelial cells to a BE-like metaplasia, involving a change in cell identity and adoption of a BE-like lineage. Our studies suggest cell autonomous mechanisms involving Notch signaling and pivotal transcription factors MYC, CDX1 and KLF4, which promote a partial reprogramming of the esophageal cells toward BE. However, the complete emergence of BE, and certainly progression to a dysplastic state and EAC, involves cell non-autonomous mechanisms, such as inflammation and activation of Hedgehog signaling and Wnt signaling.42-44 In summary, we now demonstrate key mechanisms underlying the initiation of BE, which hold the potential for future biomarker studies for patients at risk for progression to dysplasia and EAC.
Materials and Methods
Further detailed information about microarray, viral production, shRNA construction, RNA extraction and qPCR, luciferase assay and Western blotting are available in Supplemental Materials and Methods.
Cell lines
EPC2-hTERT-MYC-CDX1 cells and their derivatives: EPC2-hTERT-MYC-CDX1-dnMAML, EPC2-hTERT-MYC-CDX1-dnMAML-shKLF4, EPC2-hTERT-MYC-CDX1-dnMAML-shScramble, EPC2-hTERT-MYC-CDX1-shHES1 and EPC2-hTERT-MYC-CDX1-shScramble were grown in KSFM (Keratinocyte Serum Free Medium, Invitrogen) with Ca22+ and supplements: BPE (bovine pituitary extract), EGF and 1% Penicillin Streptomycin (Invitrogen), as described previously.17 Cells were treated with 0.06 mmol/L calcium chloride (Ca22+) to promote squamous differentiation for 48 hrs before harvesting RNA. Phoenix A cells were grown in DMEM (Invitrogen) with 10% FBS (Sigma) and 1% Penicillin Streptomycin. FEF3 cells (fetal embryonic fibroblasts) were grown in DMEM supplemented with 10% HyClone FBS (GE Healthcare Life Sciences) and 1% Penicillin Streptomycin, as described previously.45
Stable transduction
EPC2-hTERT-MYC-CDX1 (MYC-CDX1) cells were transduced with pBabe-puro, pBabe-dnMAML-GFP-puro, pBabe-zeo or pBabe-dnMAML-GFP-zeo virus. MYC-CDX1 cells were also transduced with pLKO.1 shScramble-puro or pLKO.1 TRC puro-shHES1 virus. EPC2-hTERT-MYC-CDX1-dnMAML-zeo (MYC-CDX1-dnMAML) cells were transduced with pLKO.1 shScramble-puro or pLKO.1 shKLF4 virus. Transduced cells were selected with 1 μg/ml puromycin or 10 μg/ml zeocin for 7 d
3D Organotypic culture
MYC-CDX1 cells and their derivatives were grown using the 3D organotypic culture system as described previously.29 Cultures were fixed overnight in 10% buffered formalin phosphate (Fisher) before paraffin embedding and sectioning.
Histology and immunohistochemistry
Hematoxylin and eosin (H&E) staining as well as immunohistochemistry (IHC) were performed as described previously.10 The following antibodies were used for IHC: K13 (Abcam) 1:500, K19 (BioLegend) 1:100, HES1 (Abcam) 1:500, NOTCH1 (Epitomics) 1:100 and ICN1 (Cell Signaling) 1:200. Biotinylated secondary antibodies (Jackson Immunoresearch Laboratories Inc..) and ABC avidin-biotin-DAB detection kit (Vector Labs) were used for detection and visualization, according to the manufacturer's protocol.
Cell height measurement
Quantification of cell height at the basal layer of MYC-CDX1 and MYC-CDX1-dnMAML cells grown in 3D organotypic cultures was performed by measuring 15 cells per HPF (high power field) of H&E (n = 360). We measured 4 independent 3D organotypic cultures for each cell line. Statistical analysis for significance was determined by student t-test with P ≤ 0.05 as statistically significant.
Tissue microarray
Tissue microarray (TMA) of human biopsies of Barrett's esophagus (n = 15–23), normal esophagus (n = 25–27) and liver control was stained for status of Notch signaling. IHC staining of TMA was performed using the following antibodies: HES1, NOTCH1 and ICN1. Scoring for positive staining was analyzed by quantitative evaluation of staining intensity with a scale of 0–2 (0 = none to 2 = strong), by a pathologist (AJK-S) in a blinded fashion.
Statistical analysis
For gene expression changes in qPCR studies, statistical significance of comparisons between MYC-CDX1 and MYC-CDX1-dnMAML cells and between MYC-CDX1-dnMAML-shScramble and MYC-CDX1-dnMAML-shKLF4 cells was determined by the student t-test with P ≤ 0.05 as statistically significant. Graph represents the mean ± SEM (standard error of the mean) from at least 3 independent experiments. Scoring data of TMA were analyzed by the Fisher's exact test with P ≤ 0.05 as statistically significant.
Disclosures of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We are grateful to: Molecular Pathology and Imaging Core, Molecular Biology Core, Cell Culture Core and Electron Microscopy Resource Laboratory. We are thankful to Dr. Phyllis Gimotty and Anne Blair for assistance with statistical analysis. Finally, we are grateful to Kelly A. Whelan, Marie-Pier Tétreault, Jianping Kong, Apple Long, Kathryn E. Hamilton and Tatiana Karakasheva for scientific discussions.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- 1. Fitzgerald RC. Molecular basis of barrett's oesophagus and oesophageal adenocarcinoma. Gut 2006; 55:1810-20; PMID:17124160; http://dx.doi.org/ 10.1136/gut.2005.089144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hayeck TJ, Kong CY, Spechler SJ, Gazelle GS, Hur C. The prevalence of barrett's esophagus in the US: Estimates from a simulation model confirmed by SEER data. Dis Esophagus 2010; 23:451-7; PMID:20353441; http://dx.doi.org/ 10.1111/j.1442-2050.2010.01054.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Reid BJ, Li X, Galipeau PC, Vaughan TL. Barrett's oesophagus and oesophageal adenocarcinoma: Time for a new synthesis. Nat Rev Cancer 2010; 10:87-101; PMID:20094044; http://dx.doi.org/ 10.1038/nrc2773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Howlader N, Noone A, Krapcho M, Garshell J, Neyman N, Altekruse S, Kosary C, Yu M, Ruhl J, Tatalovich Z, et al. . SEER cancer statistics factsheets: Esophageal cancer. April, 2013; 2014. [Google Scholar]
- 5. Graham TA, McDonald SA. Genetic diversity during the development of barrett's oesophagus-associated adenocarcinoma: How, when and why? Biochem Soc Trans 2010; 38:374-9; PMID:20298186; http://dx.doi.org/ 10.1042/BST0380374 [DOI] [PubMed] [Google Scholar]
- 6. DeVault K, McMahon BP, Celebi A, Costamagna G, Marchese M, Clarke JO, Hejazi RA, McCallum RW, Savarino V, Zentilin P, et al. . Defining esophageal landmarks, gastroesophageal reflux disease, and barrett's esophagus. Ann N Y Acad Sci 2013; 1300:278-95; PMID:24117649; http://dx.doi.org/ 10.1111/nyas.12253 [DOI] [PubMed] [Google Scholar]
- 7. Quante M, Abrams JA, Lee Y, Wang TC. Barrett esophagus: What a mouse model can teach us about human disease. Cell Cycle 2012; 11:4328-38; PMID:23095673; http://dx.doi.org/ 10.4161/cc.22485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Quante M, Bhagat G, Abrams JA, Marache F, Good P, Lee MD, Lee Y, Friedman R, Asfaha S, Dubeykovskaya Z, et al. . Bile acid and inflammation activate gastric cardia stem cells in a mouse model of barrett-like metaplasia. Cancer Cell 2012; 21:36-51; PMID:22264787; http://dx.doi.org/ 10.1016/j.ccr.2011.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sarosi G, Brown G, Jaiswal K, Feagins LA, Lee E, Crook TW, Souza RF, Zou YS, Shay JW, Spechler SJ. Bone marrow progenitor cells contribute to esophageal regeneration and metaplasia in a rat model of barrett's esophagus. Dis Esophagus 2008; 21:43-50; PMID:18197938; http://dx.doi.org/ 10.1111/j.1442-2050.2007.00744.x [DOI] [PubMed] [Google Scholar]
- 10. Stairs DB, Nakagawa H, Klein-Szanto A, Mitchell SD, Silberg DG, Tobias JW, Lynch JP, Rustgi AK. Cdx1 and c-myc foster the initiation of transdifferentiation of the normal esophageal squamous epithelium toward barrett's esophagus. PLoS One 2008; 3:e3534; PMID:18953412; http://dx.doi.org/ 10.1371/journal.pone.0003534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Guo RJ, Suh ER, Lynch JP. The role of cdx proteins in intestinal development and cancer. Cancer Biol Ther 2004; 3:593-601; PMID:15136761 [DOI] [PubMed] [Google Scholar]
- 12. Gao N, White P, Kaestner KH. Establishment of intestinal identity and epithelial-mesenchymal signaling by Cdx2. Dev Cell 2009; 16:588-99; PMID:19386267; http://dx.doi.org/ 10.1016/j.devcel.2009.02.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Herold S, Herkert B, Eilers M. Facilitating replication under stress: An oncogenic function of MYC? Nat Rev Cancer 2009; 9:441-4; PMID:19461668; http://dx.doi.org/ 10.1038/nrc2640 [DOI] [PubMed] [Google Scholar]
- 14. Meyer N, Penn LZ. Reflecting on 25 years with MYC. Nat Rev Cancer 2008; 8:976-90; PMID:19029958; http://dx.doi.org/ 10.1038/nrc2231 [DOI] [PubMed] [Google Scholar]
- 15. Ruggero D. The role of myc-induced protein synthesis in cancer. Cancer Res 2009; 69:8839-43; PMID:19934336; http://dx.doi.org/ 10.1158/0008-5472.CAN-09-1970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. von Rahden BH, Stein HJ, Puhringer-Oppermann F, Sarbia M. c-myc amplification is frequent in esophageal adenocarcinoma and correlated with the upregulation of VEGF-A expression. Neoplasia 2006; 8:702-7; PMID:16984727; http://dx.doi.org/ 10.1593/neo.06277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Harada H, Nakagawa H, Oyama K, Takaoka M, Andl CD, Jacobmeier B, von Werder A, Enders GH, Opitz OG, Rustgi AK. Telomerase induces immortalization of human esophageal keratinocytes without p16INK4a inactivation. Mol Cancer Res 2003; 1:729-38; PMID:12939398 [PubMed] [Google Scholar]
- 18. van Es JH, van Gijn ME, Riccio O, van den Born M, Vooijs M, Begthel H, Cozijnsen M, Robine S, Winton DJ, Radtke F, et al. . Notch/gamma-secretase inhibition turns proliferative cells in intestinal crypts and adenomas into goblet cells. Nature 2005; 435:959-63; PMID:15959515; http://dx.doi.org/ 10.1038/nature03659 [DOI] [PubMed] [Google Scholar]
- 19. Fre S, Huyghe M, Mourikis P, Robine S, Louvard D, Artavanis-Tsakonas S. Notch signals control the fate of immature progenitor cells in the intestine. Nature 2005; 435:964-8; PMID:15959516; http://dx.doi.org/ 10.1038/nature03589 [DOI] [PubMed] [Google Scholar]
- 20. Zecchini V, Domaschenz R, Winton D, Jones P. Notch signaling regulates the differentiation of post-mitotic intestinal epithelial cells. Genes Dev 2005; 19:1686-91; PMID:16024658; http://dx.doi.org/ 10.1101/gad.341705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Menke V, van Es JH, de Lau W, van den Born M, Kuipers EJ, Siersema PD, de Bruin RW, Kusters JG, Clevers H. Conversion of metaplastic barrett's epithelium into post-mitotic goblet cells by gamma-secretase inhibition. Dis Model Mech 2010; 3:104-10; PMID:20075383; http://dx.doi.org/ 10.1242/dmm.003012 [DOI] [PubMed] [Google Scholar]
- 22. LaVoie MJ, Selkoe DJ. The notch ligands, jagged and delta, are sequentially processed by α-secretase and presenilin/gamma-secretase and release signaling fragments. J Biol Chem 2003; 278:34427-37; PMID:12826675; http://dx.doi.org/ 10.1074/jbc.M302659200 [DOI] [PubMed] [Google Scholar]
- 23. Benedito R, Roca C, Sorensen I, Adams S, Gossler A, Fruttiger M, Adams RH. The notch ligands Dll4 and Jagged1 have opposing effects on angiogenesis. Cell 2009; 137:1124-35; PMID:19524514; http://dx.doi.org/ 10.1016/j.cell.2009.03.025 [DOI] [PubMed] [Google Scholar]
- 24. Weinmaster G. The ins and outs of notch signaling. Mol Cell Neurosci 1997; 9:91-102; PMID:9245493; http://dx.doi.org/ 10.1006/mcne.1997.0612 [DOI] [PubMed] [Google Scholar]
- 25. Zheng H, Pritchard DM, Yang X, Bennett E, Liu G, Liu C, Ai W. KLF4 gene expression is inhibited by the notch signaling pathway that controls goblet cell differentiation in mouse gastrointestinal tract. Am J Physiol Gastrointest Liver Physiol 2009; 296:G490-8; PMID:19109406; http://dx.doi.org/ 10.1152/ajpgi.90393.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ghaleb AM, Aggarwal G, Bialkowska AB, Nandan MO, Yang VW. Notch inhibits expression of the kruppel-like factor 4 tumor suppressor in the intestinal epithelium. Mol Cancer Res 2008; 6:1920-7; PMID:19074836; http://dx.doi.org/ 10.1158/1541-7786.MCR-08-0224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. McConnell BB, Yang VW. Mammalian kruppel-like factors in health and diseases. Physiol Rev 2010; 90:1337-81; PMID:20959618; http://dx.doi.org/ 10.1152/physrev.00058.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kazumori H, Ishihara S, Takahashi Y, Amano Y, Kinoshita Y. Roles of kruppel-like factor 4 in oesophageal epithelial cells in barrett's epithelium development. Gut 2011; 60:608-17; PMID:21193454; http://dx.doi.org/ 10.1136/gut.2010.221648 [DOI] [PubMed] [Google Scholar]
- 29. Kalabis J, Wong GS, Vega ME, Natsuizaka M, Robertson ES, Herlyn M, Nakagawa H, Rustgi AK. Isolation and characterization of mouse and human esophageal epithelial cells in 3D organotypic culture. Nat Protoc 2012; 7:235-46; PMID:22240585; http://dx.doi.org/ 10.1038/nprot.2011.437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ohashi S, Natsuizaka M, Wong GS, Michaylira CZ, Grugan KD, Stairs DB, Kalabis J, Vega ME, Kalman RA, Nakagawa M, et al. . Epidermal growth factor receptor and mutant p53 expand an esophageal cellular subpopulation capable of epithelial-to-mesenchymal transition through ZEB transcription factors. Cancer Res 2010; 70:4174-84; PMID:20424117; http://dx.doi.org/ 10.1158/0008-5472.CAN-09-4614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tamagawa Y, Ishimura N, Uno G, Yuki T, Kazumori H, Ishihara S, Amano Y, Kinoshita Y. Notch signaling pathway and Cdx2 expression in the development of barrett's esophagus. Lab Invest 2012; 92:896-909; PMID:22449796; http://dx.doi.org/ 10.1038/labinvest.2012.56 [DOI] [PubMed] [Google Scholar]
- 32. Mendelson J, Song S, Li Y, Maru DM, Mishra B, Davila M, Hofstetter WL, Mishra L. Dysfunctional transforming growth factor-β signaling with constitutively active notch signaling in barrett's esophageal adenocarcinoma. Cancer 2011; 117:3691-702; PMID:21305538; http://dx.doi.org/ 10.1002/cncr.25861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zheng JL, Shou J, Guillemot F, Kageyama R, Gao WQ. Hes1 is a negative regulator of inner ear hair cell differentiation. Development 2000; 127:4551-60; PMID:11023859 [DOI] [PubMed] [Google Scholar]
- 34. Kazanjian A, Noah T, Brown D, Burkart J, Shroyer NF. Atonal homolog 1 is required for growth and differentiation effects of notch/gamma-secretase inhibitors on normal and cancerous intestinal epithelial cells. Gastroenterology 2010; 139:918,28, 928.e1-6; PMID:20621629; http://dx.doi.org/ 10.1053/j.gastro.2010.05.081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hahn HP, Blount PL, Ayub K, Das KM, Souza R, Spechler S, Odze RD. Intestinal differentiation in metaplastic, nongoblet columnar epithelium in the esophagus. Am J Surg Pathol 2009; 33:1006-15; PMID:19363439; http://dx.doi.org/ 10.1097/PAS.0b013e31819f57e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ohashi S, Natsuizaka M, Yashiro-Ohtani Y, Kalman RA, Nakagawa M, Wu L, Klein-Szanto AJ, Herlyn M, Diehl JA, Katz JP, et al. . NOTCH1 and NOTCH3 coordinate esophageal squamous differentiation through a CSL-dependent transcriptional network. Gastroenterology 2010; 139:2113-23; PMID:20801121; http://dx.doi.org/ 10.1053/j.gastro.2010.08.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yoshida T, Yamashita M, Hayashi M. Kruppel-like factor 4 contributes to high phosphate-induced phenotypic switching of vascular smooth muscle cells into osteogenic cells. J Biol Chem 2012; 287:25706-14; PMID:22679022; http://dx.doi.org/ 10.1074/jbc.M112.361360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kim J, Efe JA, Zhu S, Talantova M, Yuan X, Wang S, Lipton SA, Zhang K, Ding S. Direct reprogramming of mouse fibroblasts to neural progenitors. Proc Natl Acad Sci U S A 2011; 108:7838-43; PMID:21521790; http://dx.doi.org/ 10.1073/pnas.1103113108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Efe JA, Hilcove S, Kim J, Zhou H, Ouyang K, Wang G, Chen J, Ding S. Conversion of mouse fibroblasts into cardiomyocytes using a direct reprogramming strategy. Nat Cell Biol 2011; 13:215-22; PMID:21278734; http://dx.doi.org/ 10.1038/ncb2164 [DOI] [PubMed] [Google Scholar]
- 40. Agrawal N, Jiao Y, Bettegowda C, Hutfless SM, Wang Y, David S, Cheng Y, Twaddell WS, Latt NL, Shin EJ, et al. . Comparative genomic analysis of esophageal adenocarcinoma and squamous cell carcinoma. Cancer Discov 2012; 2:899-905; PMID:22877736; http://dx.doi.org/ 10.1158/2159-8290.CD-12-0189 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kagawa S, Natsuizaka M, Whelan KA, Facompre N, Naganuma S, Ohashi S, Kinugasa H, Egloff AM, Basu D, Gimotty PA, et al. . Cellular senescence checkpoint function determines differential Notch1-dependent oncogenic and tumor-suppressor activities. Oncogene 2014; PMID:24931169; http://dx.doi.org/ 10.1038/onc.2014.169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Fang Y, Chen X, Bajpai M, Verma A, Das KM, Souza RF, Garman KS, Donohoe CL, O'Farrell NJ, Reynolds JV, et al. . Cellular origins and molecular mechanisms of barrett's esophagus and esophageal adenocarcinoma. Ann N Y Acad Sci 2013; 1300:187-99; PMID:24117642; http://dx.doi.org/ 10.1111/nyas.12249 [DOI] [PubMed] [Google Scholar]
- 43. Clement G, Guilleret I, He B, Yagui-Beltran A, Lin YC, You L, Xu Z, Shi Y, Okamoto J, Benhattar J, et al. . Epigenetic alteration of the wnt inhibitory factor-1 promoter occurs early in the carcinogenesis of barrett's esophagus. Cancer Sci 2008; 99:46-53; PMID:18005197; http://dx.doi.org/ 10.1111/j.1349-7006.2007.00663.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yang L, Wang LS, Chen XL, Gatalica Z, Qiu S, Liu Z, Stoner G, Zhang H, Weiss H, Xie J. Hedgehog signaling activation in the development of squamous cell carcinoma and adenocarcinoma of esophagus. Int J Biochem Mol Biol 2012; 3:46-57; PMID:22509480 [PMC free article] [PubMed] [Google Scholar]
- 45. Grugan KD, Miller CG, Yao Y, Michaylira CZ, Ohashi S, Klein-Szanto AJ, Diehl JA, Herlyn M, Han M, Nakagawa H, et al. . Fibroblast-secreted hepatocyte growth factor plays a functional role in esophageal squamous cell carcinoma invasion. Proc Natl Acad Sci U S A 2010; 107:11026-31; PMID:20534479; http://dx.doi.org/ 10.1073/pnas.0914295107 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.